
Abstract
In intensive agricultural landscapes, plant species previously relying on semi-natural habitats may persist as metapopulations within landscape linear elements. Maintenance of populations’ connectivity through pollen and seed dispersal is a key factor in species persistence in the face of substantial habitat loss. The goals of this study were to investigate the potential corridor role of ditches and to identify the landscape components that significantly impact patterns of gene flow among remnant populations. Using microsatellite loci, we explored the spatial genetic structure of two hydrochorous wetland plants exhibiting contrasting local abundance and different habitat requirements: the rare and regionally protected Oenanthe aquatica and the more commonly distributed Lycopus europaeus, in an 83 km2 agricultural lowland located in northern France. Both species exhibited a significant spatial genetic structure, along with substantial levels of genetic differentiation, especially for L. europaeus, which also expressed high levels of inbreeding. Isolation-by-distance analysis revealed enhanced gene flow along ditches, indicating their key role in effective seed and pollen dispersal. Our data also suggested that the configuration of the ditch network and the landscape elements significantly affected population genetic structure, with (i) species-specific scale effects on the genetic neighborhood and (ii) detrimental impact of human ditch management on genetic diversity, especially for O. aquatica. Altogether, these findings highlighted the key role of ditches in the maintenance of plant biodiversity in intensive agricultural landscapes with few remnant wetland habitats.
Similar content being viewed by others

Introduction
The intensification of agricultural practices in the second half of the 20th century has led to the fragmentation of former semi-natural habitats in agricultural landscapes (Stoate et al., 2001). Reduction of available habitat area is likely to adversely affect species richness and population sizes (Fahrig, 2003; Liira et al., 2008).
Dispersal among fragmented populations is a key parameter that reduces the negative effects of habitat loss by providing a demographic and genetic ‘rescue effect’ (Frankham et al., 2010). These effects may alleviate inbreeding and increase levels of genetic diversity, thus reducing extinction risks (Brown and Kodric-Brown, 1977; Slatkin, 1985; Ellstrand and Elam, 1993). In turn, efficient dispersal is strongly dependant on the spatial arrangement of landscape features that determine landscape connectivity, that is, the degree to which landscape facilitates or impedes movement between habitat patches (Taylor et al., 1993; Aavik et al., 2013). Plants are sessile organisms: gene flow among populations is therefore mediated through pollen and seed dispersal vectors (Heywood, 1991; De Cauwer et al., 2010; Choo et al., 2012). Landscape characteristics can act upon seed and pollen dispersers, driving the population genetic structure by either facilitating or impeding levels of gene flow (Aavik et al., 2014). In intensive agricultural landscapes, connectivity reduction due to the resulting habitat fragmentation and decline in pollinators negatively impacts effective pollen and seed dispersal and, consequently, gene flow among populations (McConkey et al., 2012).
Landscape properties influence functional connectivity, that is, the actual dispersal of individuals or genes among populations. Landscape linear elements (LLEs) like ditches, hedgerows or road verges, may constitute efficient refuge habitats for many plant species, and facilitate propagule dispersal within the agricultural matrix (Smart et al., 2002). The moisture gradient found along ditch banks provides habitat for various species, especially for wetland taxa, which often suffer greatly from the massive destruction of wetland habitats (Blomqvist et al., 2003; Herzon and Helenius, 2008). Furthermore, the presence of water allows hydrochorous seed dispersal to occur along ditches (Soomers et al., 2010). Hydrochory is an efficient dispersal mode for plant species that may be restricted along small water courses, and enables occasional long-distance gene flow (Kudoh and Whigham, 1997; Pollux et al., 2009; Leys et al., 2014). Therefore, in facilitating connectivity among populations, ditches have the potential to contribute to the persistence of hydrochorous plant species in agricultural landscapes.
However, potential facilitating effect of suitable linear elements for efficient gene flow is not straightforward. The response of a given species may not necessarily be correlated to the spatial configuration and composition of the landscape. For instance, Aavik et al. (2014) did not find significant relationships between patterns of genetic differentiation and structural connectivity measures based on the topology of habitat networks allowing gene flow to occur. Surprisingly, pollen dispersal may also be restricted above a density threshold of hedgerow network, which means that a network of potentially suitable habitats does not always ensure functional connectivity in terms of gene flow (Campagne et al., 2009).
In this regard, beyond ditch presence per se, landscape connectivity may also be dependent on water availability, on the timing of flow and drying and on current orientation that in turn might impact patterns of gene flow for plant species located within or next to ditches. Kudoh and Whigham (1997) and Schleuning et al. (2011), for example, documented higher levels of gene flow and lower genetic differentiation among populations connected via flowing waters in a wetland macrophyte and a clonal herb, respectively. Most studies about the role of watercourses on patterns of gene flow focused on rivers and streams with defined current orientation, although hydrochorous seed dispersal can still occur in slow-flowing ditches for various wetland plant species (Gornall et al., 1998; Soomers et al., 2010). Such stagnant networks may form complex associations of watercourses with and without current orientation, leading to partially oriented networks were hydraulic connectivity estimations may be quite different from classical dendritic hydrologic networks. In addition, connectivity may also be impacted by the ditch network structure and the presence of particular elements, like culverts, that may be barriers to seed dispersal (Soomers et al., 2010). In heterogenous habitats like ditches, adjacent land-use and management may further impact micro-habitat quality, which in turn influences species-specific metapopulation dynamics and patterns of genetic structure along linear habitats (Geertsema et al., 2002; Geertsema, 2005). To understand the patterns of genetic structure of remnant wetland species, investigating the role of the ditch network orientation and configuration, along with adjacent land-use features, is of crucial importance. To our knowledge, studies dealing with wetland ditch plant species representative of contrasting hydrologic conditions and micro-habitat requirements have yet to be completed.
Examining whether landscape structural connectivity translate into functional connectivity can be achieved by examining the relationship between levels of genetic differentiation and various landscape structure measurements (Storfer et al., 2007; Aavik et al., 2014). The goal of this study was to investigate the spatial genetic structure in hydrochorous plant species within an intensive agricultural landscape located in northern France. This area was chosen because of its high ditch network density, with ditches representing the main remnant habitat for wetland species. A previous study had led to a characterization of ditch plant metacommunities in this study site (Favre-Bac et al., 2014a). Among a set of species of interest, we choose two diploid species with contrasting local abundances to develop suitable molecular markers for population genetic analyses (Favre-Bac et al., 2014b). The studied species were the fine-leaved water Dropwort, Oenanthe aquatica (L.) Poir. (Apiaceae), which is scarce in the area and regionally protected, and the more widespread Gypsywort, Lycopus europaeus L. (Lamiaceae). Both species are insect-pollinated plants though to be mainly outcrossed (Kühn et al., 2004; Westberg and Kadereit, 2014). Such species may be particularly vulnerable to habitat fragmentation (Aavik et al. 2014). Besides, O. aquatica is a colonizer species experiencing localized extinction/recolonization events (Westberg et al., 2010; Westberg and Kadereit, 2014; L. Favre-Bac, pers. obs.), which may lead to founder effects and higher levels of genetic differentiation as compared with the more common L. europaeus. Finally, owing to the scarcity of remnant semi-natural habitats in the study area, both species were restricted to the ditch network and provided the unique opportunity to examine the effect of landscape linear elements on patterns of genetic structure. Using recently isolated nuclear microsatellite markers (Favre-Bac et al., 2014b) and GIS tools to characterize the landscape configuration of the study area (Favre-Bac et al., 2014a), we asked the following questions:
-
1
What are the levels of genetic diversity and the levels of genetic differentiation among populations within this intensive agricultural area with few remnant wetland habitats? Owing to a stronger effect of genetic drift, we hypothesized a lower level of genetic diversity and a higher genetic differentiation in the rare and vulnerable O. aquatica as compared with the more widespread L. europaeus.
-
2
Second, we evaluated whether isolation-by-distance occurred through spatially restricted migration and whether the ditch network facilitated gene flow events among populations. In both species we hypothesized that extents of gene flow would decrease with increasing geographical distance among populations. We further expected that stagnant waterbodies may reduce levels of gene flow, whereas running waters may, in contrast, facilitate gene flow events among distant populations. As O. aquatica is mainly located in stagnant water ditches, we thus expected that the spatial scale of the genetic neighborhood would be reduced as compared with L. europaeus, which is mainly located along running water ditches.
-
3
Finally, we examined whether population genetic features depended on the adjacent habitat composition and on the ditch network configuration in the neighborhood of local populations. We expected that adjacent land-use composition, which may change ditch habitat quality owing to differential management (particularly mowing and nutrient, herbicides and pesticides inputs), would be more likely to impact O. aquatica owing to its scarcity. In addition, this species should also be more sensitive to the degree of connectivity of the neighboring ditch network around surveyed populations due to its localization within stagnant water ditches.
Materials and methods
Study area
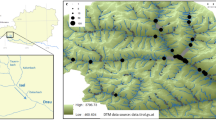
The study area is located in the ‘Nord-Pas de Calais’ district, situated in northern France and characterized by an important cover of agricultural and urban areas (range: 50° 38′ 36.72′′ N, 2° 46′ 28.23′′ E and 50° 32′ 50.09′′ N, 2° 35′ 40.70′′ E). This 83 km2 study area is dominated by large agricultural fields (62.4%), with few remnant semi-natural areas comprised of grasslands, woodlands and fallow lands (15.3%), all of which are scarce and highly fragmented. However, a dense and well-connected ditch network still occurs, totaling 642 km of linear wetland habitat within the study area (Figure 1). Owing to its flat topography, this area was initially largely covered in wetlands, and early large-scale drainage is thought to have begun during the XIIIth century. About 1/5th (20.6%) of this network consists of large oriented flowing-water ditches with higher water levels. The remaining 79.4% of the network are stagnant ditches with little or no consistent unidirectional flow. The Lawe River, which flows from south to north, crosses the middle of the study area (Figure 1). Overall, the ditch network acts as refuge non-crop habitat for many plant species, and hosts some regionally rare or declining species such as Butomus umbellatus, Hottonia palustris or O. aquatica (Favre-Bac et al., 2014a). It has further been shown that ditch network adjacent land-use composition and connectivity influenced hydrochorous as well as non-hydrochorous plant metacommunities (Favre-Bac et al., 2014a).

Geographical localization of O. aquatica (blue dots) and L. europaeus (green dots) populations in the study area (northern France). Rivers (bold blue lines), flowing-water ditches (dark blue lines) and stagnant ditches (gray lines) are also represented.
Studied species
The fine-leaved water Dropwort O. aquatica (L.) Poir. (Apiaceae) and the Gypsywort L. europaeus L. (Lamiaceae) are two insect-pollinated, hygrophilous, plant species restricted to ditches in the study area. According to the Biolflor database, both species are reported to be facultative autogamous with a mixed-mating system (Kühn et al., 2004). These species are hydrochorous (Kühn et al., 2004; Kleyer et al., 2008) and have been found within the propagule pool dispersed at the water surface of ditches in the study area (unpublished results).
O. aquatica is a colonizer species growing in disturbed sites on ditches, ponds and shallow pools in naturally nutrient-rich habitats with unpredictable fluctuations of water levels (Hroudová et al., 1992; Jensch and Poschlod, 2008; Westberg and Kadereit, 2014). Its geographical distribution encompasses most of Europe. Because of habitat destruction, this species is declining in some parts of its range, including northern France where O. aquatica is protected. The species is scarce in our study area, where stagnant ditches are the only substantive favorable habitat. This species may be either an annual or biannual (Hroudová et al., 1992).
The Gypsyworth L. europaeus (Lamiaceae) is a perennial species living on the banks of various wetland habitats (ditches, ponds and streams), with a range extending from most of Europe to Eastern Asia (China, Japan) and North Africa. In our study area, this species is more commonly distributed than O. aquatica, although it is also almost exclusively restricted to the ditch network. Most populations were found growing along the banks of flowing-water ditches. This species may reproduce vegetatively (Klimeš and Klimešová, 1999), and exhibits long viability (12–15 months) of seeds after floating (Delisle et al., 2003).
Ditch network composition and connectivity characterization
Landscape cover and linear elements were digitized from 2009 aerial photographs obtained from the ‘Institut Géographique National’, using ArcGIS 10.1 (ESRI, Redlands, CA, USA). We distinguished eight land-cover categories: crops, built areas, fallow lands and woods, grasslands, grassy strips, roads and stretches of water. With regard to the linear elements, rivers and ditches were also digitized, along with culverts located within the ditch network.
Adjacent land-use composition and network connectivity were characterized in ‘catchment areas’, which are defined here as including the surrounding ditches connected to sampled populations within a threshold distance. Catchment areas were calculated using the Network Analyst extension for ArcGIS 10.1 (ESRI). This GIS tool implements current direction for ditches with one-way (clearly-defined unidirectional current orientation) and two-way (absence of identified unidirectional flow in stagnant ditches) orientations in the network data set. We calculated the length of connected ditches upstream from each sampled ditch within a given threshold distance, taking into account the potential current orientation of surrounding ditches. Three catchment threshold distances were considered: 100, 500 and 1000 m. A previous study realised on the same study area showed that the composition of land-use elements adjacent to the network influenced ditch plant metacommunity structure, possibly through their indirect effect on ditch habitat quality due to differential ditch bank management (Favre-Bac et al., 2014a). We measured catchment area composition as the proportion of ditches adjacent to each of the eight digitized land-use elements. In addition, three variables characterizing catchment area connectivity were computed: (i) the total length of ditches within the catchment area, that is, the length of connected ditches around sampled populations, (ii) the number of intersections per network kilometer, which represents an estimate of the network’s density and (iii) the proportion of the catchment area ditch network composed of culverts, which may represent partial barriers to hydrochorous propagule dispersal within ditches (Soomers et al., 2010). These variables were recognized as potentially impacting the dispersal of hydrochorous plant species in ditches (Favre-Bac et al., 2014a).
Plant sampling and genotyping
Seventeen and 31 populations were sampled for O. aquatica and L. europaeus, respectively. For O. aquatica, all known putative populations in the surveyed study area were included, resulting in an exhaustive sampling of populations for this species. Leaf tissue was collected for genotyping and sample sizes ranged from four to 30 individuals for O. aquatica (mean=16.2±7.1), and from six to 40 for L. europaeus (mean=15.6±6.2), for a total of 274 and 472 individuals, respectively. Each individual was geo-referenced. Leaves were dried for 3 days at 50 °C, and were further conserved with silica gel dessicant at ambient temperature prior to DNA extraction.
Genomic DNA from L. europaeus and O. aquatica was extracted using the NucleoSpin 96 plant II kit (MACHEREY-NAGEL, Duren, Germany). Fifteen and 16 recently isolated microsatellite loci were amplified for L. europaeus and O. aquatica, respectively, as described in Favre-Bac et al. 2014a, b. PCR products were electrophoresed using a 3130 XL DNA Sequencer (Applied Biosystems, Saint Aubin, France) and allele size scoring was completed with GENEMAPPER V4.0 (Applied Biosystems).
Linkage disequilibrium, basic parameters of genetic diversity and estimation of genetic differentiation
Clonal individuals were removed prior to other analyses. Genotypic disequilibrium tests were performed for all locus pairs with a log-likelihood ratio G-statistic on contingency tables, using FSTAT V2.9.3 (Goudet 1995). Significances of P-values were assessed after Bonferroni correction (Rice, 1989). Standard population genetic parameters, calculated using SPAGeDi V1.4 (Hardy and Vekemans, 2002) and FSTAT V2.9.3, included the total number of alleles (NA), the mean number of alleles (MNA), the allelic richness corrected for smallest sample size (Ar), the observed heterozygosity (H0) and the expected heterozygosity (He). Apportionment of genetic diversity was estimated through Wright’s F-statistics following the analysis of variance procedure described in Weir and Cockerham (1984) and using FSTAT V2.9.3. Significance of single and multilocus FIS and FIT estimators (measuring departures from panmixia within and overall sampling locations) was tested using 10 000 random permutations of alleles among individuals within populations and for the total population, respectively. It remains unclear whether classical estimates of FIS reflect true levels of inbreeding because they can be influenced by the presence of null alleles. To circumvent this drawback and to obtain corrected FIS estimates (FIS corr) accounting for the probability of null alleles occurrence (an), we used the Bayesian individual inbreeding model described in Chybicki and Burczyk (2009) and implemented in the software INEST V.2.0 available at http://genetyka.ukw.edu.pl. We used the full model accounting for null alleles, inbreeding and genotyping failures and the number of MCMC replicates was set to 500 000 with a burn-in of 50 000 cycles. Genetic differentiation was quantified by the FST index and tested for significance using a G-test by randomly permuting (10 000 permutations) multilocus genotypes among populations, as appropriate when it is suspected that populations do not meet Hardy–Weinberg requirements (Goudet et al., 1996). For estimating population genetic differentiation, we only used population samples with at least seven genotyped individuals. For such populations characterized by this minimal sample size, the sampling was exhaustive (two populations for both species). Corrections for multiple testing were conducted following Rice (1989). Genetic divergence among pairwise populations was also quantified from nuclear genetic data using the chord distance (DCE) based on allelic frequencies (Cavalli-Sforza and Edwards, 1967).
Selfing rates were estimated using the classical relationship (s=2FIS/(1+FIS)) expected for a mixed-mating species with populations at genetic equilibrium (Hartl and Clark, 1997). Both classical multilocus FIS estimates and corrected FIS corr values robust to occurrence of null alleles were used. We additionally derived selfing rates independent of FIS from the multilocus correlation structure of population sample, using RMES (David et al., 2007). Maximum likelihood estimates of selfing rates based on the observed distribution of multilocus heterozygosity, unlike those derived from FIS, have also been shown to be insensitive to technical artefacts such as null alleles (David et al., 2007).
Population memberships and genetic affinities among populations
To test whether the genetic differentiation among sampled locations mirrored the occurrence of distinct gene pools, Bayesian genetic clustering was performed using STRUCTURE V2.3.4 (Pritchard et al., 2000). Each run consisted of a burn-in period of 100 000 iterations followed by 2 × 106 Markov chain Monte Carlo replications without any prior information on the putative population affiliation of individuals. Thirty runs were performed for each tested K-value, ranging from 1 to 31 for O. aquatica and 1 to 17 for L. europaeus. The ad hoc statistic ΔK was calculated for each species to assess the most accurate number of K clusters (Evanno et al., 2005). For the optimal K-value, similarity coefficients between replicated runs and average matrices of individual membership proportions were analyzed using CLUMP V1.2 that accounts for label switching, and identifies potentially distinct solutions across replicates (Jakobsson and Rosenberg, 2007). Finally, the most likely clustering solution was displayed using DISTRUCT V1.1 (Rosenberg, 2003).
Spatial principal component analyses (sPCA) were also performed, which make no genetic assumptions with regard to mating system, population structure or allele frequency models (Jombart et al., 2008). sPCA summarizes allelic data sets into a few uncorrelated principal components that optimize the genetic variance among populations, taking into account the spatial autocorrelation between sampling locations. The overall significance of global and local structure was assessed using permutation tests as described in Jombart et al. (2008).
Spatial genetic structure
Spatial genetic structuring was assessed by two complementary approaches. First, to test for isolation-by-distance, pairwise DCE genetic distances were regressed against various geographical distances (described below) using the normalized Mantel statistic rz. Statistical significance of rz values was tested using PASSAGE V2 (Rosenberg and Anderson, 2011).
To identify the scale over which a spatial genetic structure occurred, we also used pairwise kinship coefficients Fij among individuals (Loiselle et al., 1995). Fij describes pairwise changes in relatedness among individuals over spatial distances and allows for the depiction of isolation-by-distance due to geographically restricted gene flow. Standard errors of Fij were estimated using a jackknifing procedure among loci. Distance classes were based on an even distribution of pairwise individuals within 20 (for Euclidean and oriented network distances) or 10 (for resistance distances) spatial scales. Confidence intervals (95%) were generated using 10 000 permutations of individual locations to test whether average kinship coefficients significantly departed from zero. The spatial scale of positive autocorrelation, defining genetic neighborhood in the broad sense, was considered as the distance value for which Fij coefficients dropped under zero.
Four different geographical distances were considered as descriptors of population’s connectedness: (i) the simple Euclidean distances among individuals, (ii) the geographical shortest path distances among individuals recalculated along the oriented ditch network, (iii) resistance-based distances incorporating all possible pathways for gene flow to occur through the ditch network (McRae, 2006) and (iv) geographical distances recalculated along a set of different network models.
Geographical Euclidean distances were computed using PASSAGE V2 (Rosenberg and Anderson, 2011). Shortest path distances along the oriented ditch network were obtained using the Network Analyst Extension for ArcGIS 10.1. In addition, four network types (reviewed in Storfer et al., 2007) described in Table 1 were generated using PASSAGE V2. Distance-based networks, that is, geographical distances recalculated through the above-mentioned networks, were retained if they included a sufficient proportion of pairwise populations.
Resistance distances, metrics capitalizing on circuit theory, are based on resistance maps that illustrate the way in which landscape elements facilitate or impede the dispersal of the target species (McRae, 2006; McRae et al., 2008). We built a raster resistance map using ArcGIS V10.1 where the cells corresponding to the ditch network were assigned a cost of 1, and all the remaining landscape was considered as unsuitable for dispersal, including rivers. This enabled us to calculate resistance distances based on the network density and the degree of connections between sampled locations. Resistance distances were computed using CIRCUITSCAPE V4.0 (Shah and McRae, 2008).
Analysis of the relationships between genetic diversity and network composition/configuration
Allelic richness (Ar), expected heterozygosity (He) and intra-population fixation index (FIS corr) were related to landscape variables estimated for the three catchment area scales (100, 500 and 1000 m) using Generalized Linear Models. The error distribution model was defined as Gaussian. We applied a multi-model inference to identify the optimal set of adjacent land-use composition and connectivity variables for each genetic factor (Burnham and Anderson, 2002). This approach involved ranking all the potential models from best to worst, then averaging the top models to obtain an approximate average ‘best’ model. For each scale and each genetic variable, standardized linear average regression coefficients, weighted by small sample AIC, were computed across all models that exhibited a ΔAIC<2 (Hurvich and Tsai, 1989; Burnham and Anderson, 2002). Analyses were performed using RV.2.14.0 with the ADE4 and MUMIN libraries.
Results
Genetic diversity of O. aquatica and L. europaeus
For O. aquatica and L. europaeus, respectively, 1 and 40 clonal individuals were identified and subsequently removed from the data set, leaving a total of 259 and 422 individuals successfully genotyped and retained for analysis. For O. aquatica, there was no significant genotypic linkage between microsatellite loci after Bonferroni correction. Within each L. europaeus population, exact tests for genotypic linkage disequilibrium only depicted 25 significant unbiased P-values out of 2940 comparisons after Bonferroni corrections. Altogether, these results suggested no genotypic disequilibrium in the studied populations for both species.
For O. aquatica, levels of allelic richness per locus ranged from 1.13 to 5.91, and the expected heterozygosity ranged from 0.047 to 0.815 (Supplementary Table S1). When analyzed over all populations, 9 out of 16 loci showed a significant (P<0.05) deviation from Hardy–Weinberg expectations, with mean single-locus FIS values ranging from 0.025 to 0.467 (Supplementary Table S1). The mean multilocus FIS value was 0.109 and of 0.076 for corrected (FIS corr) estimates, suggesting moderate inbreeding in this mixed-mating species (see Table 2). Estimated selfing rates were 0.19 for FIS and 0.14 for FIS corr, respectively. The RMES estimates of s ranged from 0 to 0.365 with a mean of 0.11 (Table 2). Single-locus FST values were all significant and ranged from 0.060 to 0.153 with a mean multilocus estimate of 0.089 (P<0.001, Supplementary Table S1). Among the 105 comparisons, mean pairwise FST between populations ranged from 0.002 to 0.225 and 95.2% were found to be significant at P<0.05 after Bonferroni correction (see Supplementary Figure S1A). Populations of O. aquatica thus exhibited a substantial pattern of genetic differentiation despite the small scale of sampling.
For L. europaeus, allelic richness per locus ranged from 3.16 to 5.25 (Supplementary Table S1). Expected heterozygosity ranged from 0.470 to 0.695. In contrast to O. aquatica, all loci significantly deviated from Hardy–Weinberg expectations: single-locus FIS values were high but demonstrated low between-value variance, ranging from 0.314 to 0.387 with a mean multilocus estimate of 0.347 and 0.331 for corrected FIS estimate (P<0.001, see Table 2 and Supplementary Table S1). Selfing rates were of 0.51 for FIS and 0.49 for FIS corr. Using RMES, selfing rates ranged from 0 to 0.69 with a mean of 0.43 (Table 2). As would be expected for a species with high propensity for selfing, single-locus FST estimates were high (twice the O. aquatica FST estimate) and ranged from 0.133 to 0.232 with a mean multilocus estimate of 0.173 (all at P<0.05, Supplementary Table S1). Among the 756 comparisons, mean pairwise FST between populations ranged from –0.030 to 0.787 (84.6% significant at P<0.05) (see Supplementary Figure S1B).
Genetic affinities among populations
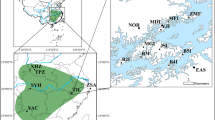
For O. aquatica, The ΔK statistic showed a modal K=4: non-spatially explicit Bayesian clustering analysis, therefore classified the whole data set into four genetically distinct O. aquatica population groups (Figure 2a, Supplementary Figure S1A). The K vs ΔK distribution was multimodal with another mode at K=10, suggesting a further hierarchical structure with more subtle genetic structuring (Supplementary Figure S2A). The isolated population located in the western portion of the study area was clearly separated from the central section, which contained the greatest number of populations (Figure 2b). This central area was divided into three clusters, with a northern and southern genetic cleavage, and an isolated population with probability membership that departed from the other two clusters. Three remaining populations, located on the east side of the Lawe River, appeared genetically admixed between the different clusters.

Bayesian clustering results for O. aquatica (a, b) and L. europaeus (c, d). (a, c) Assignment probabilities of individual memberships of individuals into the first modal K values for O. aquatica (a, K=4) and L. europaeus (c, K=3). Each individual is represented by a thin horizontal line (y axis) partitioned into a number of colored segments that represent the individual’s estimated membership fraction to the best number of inferred cluster in each species. (b, d) Map of mean population membership probabilities for O. aquatica (b, K=4) and L. europaeus (d, K=3). Some population localizations have been adjusted slightly in order to avoid superposition.
sPCA analysis based on the best connection network (neighborhood by distance with a maximum distance of 2500 m) failed to show a significant global structure (P=0.082) but results were clearly in accordance with the Bayesian clustering. Only the two first sPCA axes accounted for the spatial genetic pattern in the data (I=0.813, var=0.157 and I=0.224, var=0.139, respectively). The first axis separated populations located on both sides of the river, whereas the second axis isolated the most western population from other O. aquatica populations (Supplementary Figure S3B and C). sPCA analysis thus emphasized the separation between populations located in the central part of the study area, which hosts the main group of O. aquatica individuals, and the two peripheral population groups, as shown in Supplementary Figure S4A.
For L. europaeus, the most likely number of clusters was identified as K=3 (Figure 2d, Supplementary Figure S2B). Mapping of mean population membership probabilities then revealed the occurrence of two genetically distinct population groups in the central study area, once again on either side of the Lawe River (Figure 2e). The remaining cluster included all other populations, some of which were admixed with the first two distinct groups of populations depicted.
sPCA analysis revealed a significant global structure (P=0.045) that indicated a more subtle genetic structuring when compared with Bayesian clustering. The first axis of the sPCA (I=0.847, var=0.382) captured 32.3% of the global spatial pattern. Scores from this first principal component depicted a genetic discontinuity separating populations located on both west and east sides of the Lawe River. (Supplementary Figure S3E). The second axis (I=0.756, var=0.222) split the first group including the south–west and north–east populations, proximal to the largest cluster identified by Bayesian clustering, and a second group covering the central populations, including both central clusters depicted by Bayesian clustering (Supplementary Figure S3F). The color plot summarizing the first three axes of the sPCA clearly individualized these two central groups from the remaining populations (Supplementary Figure S4B).
Spatial genetic structure
Overall, we found a general process of isolation-by-distance due to spatially restricted gene flow for both species. For O. aquatica, Mantel tests of association revealed a significant relationship between all tested geographical distances and pairwise DCE genetic distances, except for resistance distances (Table 1). The neighborhood, to a distance of 2500 m, exhibited the highest correlation with our genetic data (rz=0.74, P<10–3). Similarly, spatial autocorrelation analysis suggested isolation-by-distance, regardless of the geographical distance used. Nonetheless, average kinship coefficients among individuals dropped to zero differentially; spatial autocorrelation threshold distances were at least twice as large when considering distances along ditches (1285 m) as compared with Euclidean distances (540 m) (Figures 3a and c). In addition, spatial autocorrelation was observed at <203 resistance distance units (Figure 3e). Mapping of connections among populations using the along ditches distance threshold revealed a pattern of genetic connectivity concordant with Bayesian clustering results: populations included in the western and southern central clusters of the central area appeared more closely connected (Supplementary Figures S5A and B). Based on the threshold resistance distance, the western and southern clusters of the central area were also connected, but not the different populations forming the northern cluster, except those located very close to each other (Supplementary Figure S5C).

Spatial autocorrelograms depicting the relationship between pairwise kinship coefficients (Loiselle et al., 1995) among individuals and geographical distances among sampled individuals for O. aquatica (a, c, e) and L. europaeus (b, d, f). (a, b) Euclidean distances. (c, d) geographical distances along the oriented ditch network. (e, f) Resistance distances based on circuit theory, taking into account all possible pathways connecting populations along the ditch network. Dotted lines represent the 95% confidence intervals bracketing the null hypothesis of no significant spatial genetic structure obtained from 9999 permutations. Black dots: P<0.05; gray dots: non significant.
For L. europaeus, only geographical distances along the oriented ditch network, the resistance distances and the distances along the 1500 m connection network were significantly correlated with pairwise DCE genetic distances (Table 2). Spatial correlograms revealed the presence of a positive spatial genetic structure for distances largely exceeding those observed for O. aquatica, up to 2200 m for euclidean distances and 4250 m along the ditch network when taking into account the current orientation (Figures 3b and d). In contrast, the threshold resistance distance was almost the same as for O. aquatica (Figure 3f), and connections based on this distance linked only close populations, when compared with those based on distances along the ditch network (Supplementary Figure S5F).
Influence of local and landscape-derived variables on genetic diversity
For O. aquatica, levels of genetic diversity were found to be significantly lower in populations located on road verge ditches as compared with populations situated on ditches located apart from roads (Figure 4a). No significant effect of the adjacent composition and connectivity of the surrounding ditch network was found on FIS corr values, suggesting these parameters did not influence the genotypic structure in O. aquatica populations (Table 3). However, levels of allelic richness (Ar) and expected heterozygosity (He) were significantly and positively impacted by the surrounding ditch network within the 500 and 1000 m catchment areas (Table 3 and Figure 4b). The proportion of adjacent crops and the number of intersections within the 500 m catchment area also increased Ar and He for O. aquatica. Genetic diversity was reduced in relation to the proportion of culverts and adjacent roads within the 500 m catchment areas. A rising amount of connected ditches surrounding sampled populations increased their genetic diversity within the 500 m catchment area. In addition, levels of Ar increased with the proportion of grasslands in the 1000 m catchment area (Table 3). Mapping the geographical distribution of mean allelic richness across populations highlighted the higher genetic diversity observed in the central group, either in the northern or southern cluster, as determined by Bayesian clustering (Supplementary Figure S6A).

Relationships between genetic diversity and ditch network properties for O. aquatica (a, b) and L. europaeus (c, d). (a) Boxplot illustrating the variation of mean levels of allelic richness in O. aquatica in road verge ditches (RV) as compared with ditches located apart from roads (O). P-values from the Wilcoxon tests are shown on the graphs. (b) Linear regression of expected heterozygosity (He) with respect to ditch length within the 500 m catchment areas. Correlation coefficients R2 are shown on the graphs, along with the significance of the variable in the approximate average ‘best’ GLM model: *P<0.05; ***P<0.001. (c) Boxplot illustrating the variation of mean levels of allelic richness in L. europaeus in flowing-water ditches (R) as compared with stagnant ditches (S). (d) Linear regression of mean levels of FIS corr in L. europaeus with respect to the proportion of roads within the 500 m catchment area surrounding the sampled populations.
For L. europaeus, mean levels of genetic diversity were higher in populations located along ditches with flowing water when compared with populations located on stagnant ditches (Figure 4c). Mean FIS corr estimates were negatively impacted by a higher number of ditches adjacent to roads in the 500 and 1000 m catchment areas (Figure 4d). An increasing number of culverts within both the 500 and 1000 m catchment areas also negatively influenced L. europaeus genetic diversity metrics (Ar and He). In addition, an increased ditch length in the 1000 m catchment area was associated with a rise in He, whereas an increased number of intersections had the opposite effect within the 100 and 1000 m areas (Table 3). The map representing the geographical repartition of allelic richness over populations illustrates the highest levels of genetic diversity within the largest cluster identified by both Bayesian clustering and sPCA, as compared with both central, smaller clusters (Supplementary Figure S6B).
Discussion
Genetic structure of O. aquatica and L. europaeus populations
We showed a substantial genetic differentiation and a significant spatial genetic structure in both O. aquatica and L. europaeus located along narrow linear habitats in an intensive agricultural area. We initially hypothesized a lower level of genetic diversity and a higher genetic differentiation in the rare and vulnerable O. aquatica. However, in contrast to what was expected, both species displayed similar levels of genetic diversity and population genetic differentiation was more pronounced for L. europaeus.
O. aquatica populations were geographically organized around a ‘central’ group hosting the majority of the populations, and additional, more isolated satellite populations, as evidenced by Bayesian clustering and sPCA analysis. As the area hosting the central group appears isolated from the rest of the network due to the presence of running water ditches, this supports the inability of O. aquatica to use these ditches both as habitat and dispersal corridors. On a smaller scale, the central group itself was split into three genetically distinct entities, which was consistent with the spatial scale of positive kinship along the ditch network. Additional independent information on mating system, such as the use of progeny arrays, would allow a better understanding of the observed population genetic structure. However, independent estimates of selfing rates suggested a predominantly outcrossing mating system in O. aquatica. Thus, efficient pollen dispersal, though to be mostly mediated by insects, may mitigate drift effects due to low population densities. This may explain both the moderate, although significant, level of nuclear genetic differentiation and the relatively high level of genetic diversity observed for O. aquatica (Hamrick and Godt, 1996; Nybom, 2004).
In sharp contrast, whatever the estimate used, the genetic structure suggested high inbreeding rates due to self-fertilization in L. europaeus. The mating system is likely to influence mean levels of kinship among individuals, especially in anthropogenic, disturbed habitat where reproductive assurance by selfing may compensate for reduced outcrossing due to pollen limitation (Eckert et al., 2010). Self-fertilization may enhance both the genetic variance among separate populations and the potential for spatial isolation-by-distance because it decreases the average pollen dispersal distance and increases random genetic drift effects (Heywood, 1991; Vekemans and Hardy, 2004). The genotypic structure in L. europaeus could also be explained by the existence of short-distance vegetative propagation in this species, enhancing inbreeding levels. Concordantly, L. europaeus populations were highly genetically structured (mean FST=0.173) and displayed significant kinship structure, as expected for perennial species undergoing significant selfing (Hamrick and Godt, 1996). Finally, distal populations were clustered together with high membership probabilities. Human-mediated seed transfer due to agricultural machinery or sediment transport may explain this pattern (Strykstra et al., 1997; Auffret and Cousins, 2013).
Overall, both species are short-lived perennials (or a possible annual in the case of O. aquatica) and a short period of localized dispersal events would be sufficient for local spatial genetic structure to occur (for example, De Cauwer et al., 2010; Choo et al., 2012). A kinship structure was indeed observed for both species, featuring a striking isolation-by-distance process that emphasizes the predominant role of ditches as an avenue for efficient gene flow.
Ditches as dispersal corridors for hydrochorous plant species in agricultural landscapes
In both species we initially hypothesized that extents of gene flow would decrease with increasing geographical distance among populations with a facilitating effect of ditch network. We further expected that stagnant waterbodies may reduce levels of gene flow in O. aquatica. Although resistance distances based on circuit theory have been shown to be good predictors of gene flow among populations (McRae, 2006), resistance distances across the ditch network performed poorly when compared with shortest path-oriented distances along the network. Two recent studies found a significant effect of resistance distances calculated on linear stream networks (Blair et al., 2013; Trumbo et al., 2013). However, both studies were conducted on reptiles and amphibians, which have different dispersal patterns as compared with plant species. This lack of clear isolation-by-resistance may also be due to model over-simplification: we considered all ditch segments to be equivalent and did not take into account potential dispersal barriers like culverts. The impossibility to take current orientation into account in calculation of resistance distances is also likely to explain this result. This highlighted the importance of current orientation and the key corridor role had by ditches in shaping and facilitating gene flow patterns. Nonetheless, these effects were species-specific. As expected in our second hypothesis, we found larger genetic neighborhood distances and a greater effect of current orientation for L. europaeus as compared with O. aquatica. In both cases, taking into account the partially oriented character of this network appears to best reflect functional connectivity for these wetland species.
As semi-natural linear pathways within the agricultural matrix, native and introduced vegetation occurring on ditch banks may enhance pollinators’ movements among habitat patches, as shown for other linear landscape elements (Cranmer et al., 2012; Van Rossum and Triest, 2012). In addition, water flow is also expected to facilitate seed dispersal. In our case study, the two species showed different genetic patterns, with the local genetic neighborhood of O. aquatica extending to ~1200 m along the network, whereas the L. europaeus neighborhood went much further, up to 4 km. These differences presumably arose because L. europaeus occurred in large ditches with flowing water, while O. aquatica was generally restricted to stagnant ditches. In rivers and streams, the dispersal of propagules along a unidirectional lotic system is expected to cause downstream accumulation of genetic diversity (Ritland, 1989; Lundqvist and Andersson, 2001; Love et al., 2013). For L. europaeus, this phenomenon may explain the pivotal effect of distances along ditches, constrained by current orientation, and the lack of isolation-by-distance using Euclidean metric. In contrast to L. europaeus, O. aquatica was found almost exclusively in stagnant ditches, where wind may be the main driver of hydrochorous propagule dispersal. Several studies have shown that wind-driven hydrochory can be efficient, but with lower observed dispersal distances than observed in lotic systems (Soomers et al., 2010, 2013).
Influence of landscape features on population genetic structure
We initially expected that the local degree of connectivity and adjacent land-use composition would be more likely to impact O. aquatica owing to its scarcity, its localization within stagnant water ditches, and its sensitivity to mowing and weeding. Adjacent land-use and connectivity variables significantly impacted levels of genetic diversity in O. aquatica, as expected in our third hypothesis, but also impacted L. europaeus populations. For both species, an increase in the number of connected ditches within medium and/or large catchment areas caused a concurrent rise in genetic diversity. Larger catchment areas, with more connected ditches, thus appeared more favorable for hosting genetically sustainable plant populations. Conversely, a higher proportion of culverts in the surrounding ditch network resulted in a reduction of genetic diversity. This inverse relationship may occur because culverts convert open ditch segments into unsuitable habitats for plant species and might act as filters to seed dispersal for a large number of plant species (Soomers et al., 2010). In contrast, O. aquatica and L. europaeus responded differently to the amount of intersections within these catchment areas. For O. aquatica, genetic diversity increased in populations surrounded by networks with more intersections, probably due to increased dispersal possibilities in denser networks. The opposite effect was observed in L. europaeus. In this case, a dense network mostly reflects intersections with smaller ditches, which generally allow water to flow from smaller ditches to running water ditches, but not the opposite way.
Both local and landscape-scale characteristics of land-use elements adjacent to ditches were also found to impact levels of genetic diversity in O. aquatica and L. europaeus. The proportions of adjacent landscape elements mostly impacted genetic diversity in large catchment areas. This is consistent with the relatively large genetic neighborhood sizes depicted. This emphasizes the sensitivity of wetland species to structural elements of the ditch network over distances larger than the immediate neighborhood of a given population (for example, Schleuning et al., 2011).
O. aquatica populations had a reduced genetic diversity when they were located on road verges. Such ditches are mown frequently (2–3 times a year). Flowering individuals of O. aquatica are thus more likely to be cut by successive mowing. In addition, a negative effect of the proportion of ditches adjacent to road verges on levels of genetic diversity was observed up to 500 m around sampled populations. If mowing of suitable roadside habitat patches prevents or reduces seed production, such patches would appear as complete or partial sinks within the metapopulation (Pulliam, 1988; Kawecki, 2008). In contrast, for L. europaeus, higher proportions of ditches bordering roads up to 1 km around sampled populations were associated with reduced FIS corr values. This could be due both to enhanced seed and vegetative propagule dispersal along ditch banks by mowing machinery. The differences in response between these two species suggests the need for thoughtful evaluation with regard to management practices in roadside ditches, as a ‘one size fits all’ approach may prove counter-productive.
What makes ditches ‘good’ corridors for wetland plant species?
Our results suggested that, although ditches appeared as efficient corridors for wetland plant species, their effectiveness is species-specific, and particularly dependent on the abundance and habitat requirement of the species. Linear elements, therefore, cannot efficiently promote movement for all species, but favor certain dispersal traits (Von der Lippe and Kowarik, 2012; Van Dijk et al., 2014). Planning of management or restoration actions should include a preliminary reflection on which species group is likely to be favored for any given management activity.
In the case of rare species living in stagnant ditches, a modification of mowing practises on roadside ditches could enhance the reproduction of individuals located on road verge ditches, which may allow these populations to increase in size and enlarge the existing metapopulation network through increased seed dispersal. Our results further suggested that, when selecting sites for plant reintroduction, the surrounding network characteristics should be taken into account, especially its structure and the amount of connected ditches.
Conclusion
Our study highlighted the crucial role of ditches, often considered as marginal and remnant habitats, in the conservation of wetland plant species in agricultural landscapes. These ditches served as key corridors that differentially enhanced plant dispersal depending on species’ habitat requirement, which may be of great interest while investigating issues on the conservation of declining or rare species, or the invasive dynamics of exotic species (Delisle et al., 2003; Maheu-Giroux and De Blois, 2007). Our conclusions also support the idea that ditches, as landscape linear elements, may contribute to land-sharing of biodiversity with anthropogenic activities, including intensive agriculture, and that they are key elements of green veinings in agricultural landscapes. In addition, they might also condition ecosystem services provided by native plant species, for instance engineer species involved in bank consolidation or nutrient cycling, or species providing habitats for pollinators or pest predators (Herzon and Helenius, 2008).
Data archiving
DNA sequences: Genbank accessions KJ946205-KJ946220 (O. aquatica) and KJ946221-KJ946235 (L. europaeus). Coordinates of sample locations and nuclear genetic data are available in DRYAD: http://dx.doi.org/10.5061/dryad.s9n91.
References
Aavik T, Holderegger R, Bolliger J . (2014). The structural and functional connectivity of the grassland plant Lychnis flos-cuculi. Heredity 112: 471–478.
Aavik T, Holderegger R, Edwards PJ, Billeter R . (2013). Patterns of contemporary gene flow suggest low functional connectivity of grasslands in a fragmented agricultural landscape. J Appl Ecol 50: 395–403.
Auffret AG, Cousins SAO . (2013). Grassland connectivity by motor vehicles and grazing livestock. Ecography 36: 1150–1157.
Blair C, Jiménez Arcos VH, Mendez de la Cruz FR, Murphy RW . (2013). Landscape genetics of leaf-toed geckos in the tropical dry forest of northern Mexico. PloS One 8: e57433.
Blomqvist M, Vos P, Klinkhamer PG, ter Keurs W . (2003). Declining plant species richness of grassland ditch banks—a problem of colonisation or extinction? Biol Cons 109: 391–406.
Brown JH, Kodric-Brown A . (1977). Turnover rates in insular biogeography: effects of immigration on extinction. Ecology 58: 445–449.
Burnham KP, Anderson DR . (2002) Model selection and multimodel inference: a practical information-theoretic approach, 2nd edn. Springer, NY, USA.
Campagne P, Affre L, Baumel A, Roche P, Tatoni T . (2009). Fine-scale response to landscape structure in Primula vulgaris Huds.: does hedgerow network connectedness ensure connectivity through gene flow? Popul Ecol 51: 209–219.
Cavalli-Sforza LL, Edwards AWF . (1967). Phylogenetic analysis: models and estimation procedures. Am J Hum Genet 19: 233–257.
De Cauwer I, Dufay M, Cuguen J, Arnaud J-F . (2010). Effects of fine-scale genetic structure on male mating success in gynodioecious Beta vulgaris ssp. maritima. Mol Ecol 19: 1540–1558.
Choo J, Juenger TE, Simpson BB . (2012). Consequences of frugivore-mediated seed dispersal for the spatial and genetic structures of a neotropical palm. Mol Ecol 21: 1019–1031.
Chybicki IJ, Burczyk J . (2009). Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100: 106–113.
Cranmer L, McCollin D, Ollerton J . (2012). Landscape structure influences pollinator movements and directly affects plant reproductive success. Oikos 121: 562–568.
David P, Pujol B, Viard F, Castella V, Goudet J . (2007). Reliable selfing rate estimates from imperfect population genetic data. Mol Ecol 16: 2474–2487.
Delisle F, Lavoie C, Jean M, Lachance D . (2003). Reconstructing the spread of invasive plants: taking into account biases associated with herbarium specimens. J Biogeogr 30: 1033–1042.
Van Dijk WFA, van Ruijven J, Berendse F, de Snoo GR . (2014). The effectiveness of ditch banks as dispersal corridor for plants in agricultural landscapes depends on species’ dispersal traits. Biol Cons 171: 91–98.
Eckert CG, Kalisz S, Geber MA, Sargent R, Elle E, Cheptou PO et al. (2010). Plant mating systems in a changing world. Trends Ecol Evol 25: 35–43.
Ellstrand NC, Elam DR . (1993). Population genetic consequences of small population size: Implications for plant conservation. Annu Rev Ecol Syst 24: 217–242.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Fahrig L . (2003). Effects of Habitat Fragmentation on Biodiversity. Annu Rev Ecol Evol Syst 34: 487–515.
Favre-Bac L, Ernoult A, Mony C, Rantier Y, Nabucet J, Burel F . (2014a). Connectivity and propagule sources composition drive ditch plant metacommunity structure. Acta Oecologica 61: 57–64.
Favre-Bac L, Godé C, Arnaud J-F . (2014b). Characterization of polymorphic microsatellite markers for the fine-leaved water-Dropwort Oenanthe aquatica and the Gypsywort Lycopus europaeus, two farmland remnant wetland species. Cons Genet Resour 6: 995–998.
Frankham R, Ballou JD, Briscoe DA . (2010) Introduction to Conservation Genetics, 2nd edn. Cambridge University press: Cambridge, UK.
Geertsema W . (2005). Spatial dynamics of plant species in an agricultural landscape in the Netherlands. Plant Ecol 178: 237–247.
Geertsema W, Opdam P, Kropff MJ . (2002). Plant strategies and agricultural landscapes: survival in spatially and temporally fragmented habitat. Landscape Ecol 17: 263–279.
Gornall RJ, Hollingsworth PM, Christopher D . (1998). Evidence for spatial structure and directional gene flow in a population of an aquatic plant, Potamogeton coloratus. Heredity 80: 414–421.
Goudet J . (1995). Fstat version 1.2: a computer program to calculate Fstatistics. J Hered 86: 485–486.
Goudet J, Raymond M, De Meeüs T, Rousset F . (1996). Testing differentiation in diploid populations. Genetics 144: 1933–1940.
Hamrick JL, Godt MJW . (1996). Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc B 351: 1291–1298.
Hardy OJ, Vekemans X . (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Hartl DL, Clark AG . (1997). Principles of Population Genetics. 3rd edn. Sinauer Associates, Sunderland, MA, USA.
Herzon I, Helenius J . (2008). Agricultural drainage ditches, their biological importance and functioning. Biol Cons 141: 1171–1183.
Heywood JS . (1991). Spatial analysis of genetic variation in plant populations. Annu Rev Ecol Syst 22: 335–355.
Hroudová Z, Zákravský P, Hrouda L, Ostrý I . (1992). Oenanthe aquatica (L.)Poir.: Seed reproduction, population structure, habitat conditions and distribution in Czechoslovakia. Folia Geobotanica et Phytotaxonomica 27: 301–335.
Hurvich CM, Tsai C-L . (1989). Regression and time series model selection in small samples. Biometrika 76: 297–307.
Jakobsson M, Rosenberg NA . (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Jensch D, Poschlod P . (2008). Germination ecology of two closely related taxa in the genus Oenanthe: fine tuning for the habitat? Aquat Bot 89: 345–351.
Jombart T, Devillard S, Dufour A-B, Pontier D . (2008). Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101: 92–103.
Kawecki TJ . (2008). Adaptation to marginal habitats. Annu Rev Ecol Evol Syst 39: 321–342.
Kleyer M, Bekker RM, Knevel IC . (2008). The LEDA Traitbase: a database of life-history traits of the Northwest European flora. J Ecol 96: 1266–1274.
Klimeš L, Klimešová J . (1999). CLO-PLA2 - a database of clonal plants in central Europe. Plant Ecol 141: 9–19.
Kudoh H, Whigham DF . (1997). Microgeographic genetic structure and gene flow in Hibiscus moscheutos (Malvaceae) populations. Am J Bot 84: 1285–1293.
Kühn I, Durka W, Klotz S . (2004). BiolFlor — a new plant-trait database as a tool for plant invasion ecology. Divers Distrib 10: 363–365.
Leys M, Petit EJ, El-Bahloul Y, Liso C, Fournet S, Arnaud J-F . (2014). Spatial genetic structure in Beta vulgaris subsp. maritima and Beta macrocarpa reveals the effect of contrasting mating system, influence of marine currents, and footprints of postglacial recolonization routes. Ecol Evol 4: 1828–1852.
Liira J, Schmidt T, Aavik T, Arens P, Augenstein I, Bailey D et al. (2008). Plant functional group composition and large-scale species richness in European agricultural landscapes. J Veg Sci 19: 3–14.
Von der Lippe M, Kowarik I . (2012). Interactions between propagule pressure and seed traits shape human-mediated seed dispersal along roads. Perspect Plant Ecol Evol Syst 14: 123–130.
Loiselle BA, Sork VL, Nason J, Graham C . (1995). Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82: 1420–1425.
Love HM, Maggs CA, Murray TE, Provan J . (2013). Genetic evidence for predominantly hydrochoric gene flow in the invasive riparian plant Impatiens glandulifera (Himalayan balsam). Ann Bot 112: 1743–1750.
Lundqvist E, Andersson E . (2001). Genetic diversity in populations of plants with different breeding and dispersal strategies in a free-flowing boreal river system. Hereditas 135: 75–83.
Maheu-Giroux M, De Blois S . (2007). Landscape ecology of Phragmites australis invasion in networks of linear wetlands. Landscape Ecol 22: 285–301.
McConkey KR, Prasad S, Corlett RT, Campos-Arceiz A, Brodie JF, Rogers H et al. (2012). Seed dispersal in changing landscapes. Biol Cons 146: 1–13.
McRae BH . (2006). Isolation by resistance. Evolution 60: 1551–1561.
McRae BH, Dickson BG, Keitt TH, Shah VB . (2008). Using circuit theory to model connectivity in ecology, evolution, and conservation. Ecology 89: 2712–2724.
Nybom H . (2004). Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13: 1143–1155.
Pollux BJA, Luteijn A, van Groenendael JM, Ouborg NJ . (2009). Gene flow and genetic structure of the aquatic macrophyte Sparganium emersum in a linear unidirectional river. Freshwater Biol 54: 64–76.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Pulliam HR . (1988). Sources, sinks, and population regulation. Am Nat 132: 652–661.
Rice WR . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Ritland K . (1989). Genetic differenciation, diversity, and inbreeding in the moutain monkeyflower (Mimulus caespitosus of the Washington Cascades. Can J Bot 67: 2017–2024.
Rosenberg NA . (2003). Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Rosenberg MS, Anderson CD . (2011). PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Version 2. Methods Ecol Evol 2: 229–232.
Van Rossum F, Triest L . (2012). Stepping-stone populations in linear landscape elements increase pollen dispersal between urban forest fragments. Plant Ecol Evol 145: 332–340.
Schleuning M, Becker T, Vadiollo GP, Hahn T, Matthies D, Durka W . (2011). River dynamics shape clonal diversity and genetic structure of an Amazonian understorey herb. J Ecol 99: 373–382.
Shaffer M Minimum viable populations: coping with uncertainty Viable populations for conservation. In: Soulé ME (ed). Viable populations for conservation. Cambridge University press: Cambridge, UK, pp 69–86.
Shah VB, McRae BH . (2008). Circuitscape: a tool for landscape ecology. In: Proceedings of the 7th Python in Science Conference (SciPy 2008), pp. 62–65.
Slatkin M . (1985). Gene flow in natural populations. Annu Rev Ecol Syst 16: 393–430.
Smart SM, Bunce RGH, Firbank LG, Coward P . (2002). Do field boundaries act as refugia for grassland plant species diversity in intensively managed agricultural landscapes in Britain? Agri Ecosyst Environ 91: 73–87.
Soomers H, Karssenberg D, Soons MB, Verweij PA, Verhoeven JTA, Wassen MJ . (2013). Wind and water dispersal of wetland plants across fragmented landscapes. Ecosystems 16: 434–451.
Soomers H, Winkel DN, Du Y, Wassen MJ . (2010). The dispersal and deposition of hydrochorous plant seeds in drainage ditches. Freshwater Biol 55: 2032–2046.
Stoate C, Boatman N, Borralho R, Carvalho CR, de Snoo GR, Eden P et al. (2001). Ecological impacts of arable intensification in Europe. J Environ Manage 63: 337–365.
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF et al. (2007). Putting the ‘landscape’ in landscape genetics. Heredity 98: 128–142.
Strykstra RJ, Verweij GL, Bakker JP . (1997). Seed dispersal by mowing machinery in a Dutch brook valley system. Acta Botanica Neerlandica 46: 387–401.
Taylor PD, Fahrig L, Henein K, Merriam G . (1993). Connectivity is a vital element of landscape structure. Oikos 68: 571–573.
Trumbo DR, Spear SF, Baumsteiger J, Storfer A . (2013). Rangewide landscape genetics of an endemic Pacific northwestern salamander. Mol Ecol 22: 1250–1266.
Vekemans X, Hardy OJ . (2004). New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13: 921–935.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for analysis of population structure. Evolution 38: 1358–1370.
Westberg E, Kadereit JW . (2014). Genetic evidence for divergent selection on Oenanthe conioides and Oe. aquatica (Apiaceae), a candidate case for sympatric speciation. Biol J Linn Soc 113: 50–56.
Westberg E, Poppendieck H-H, Kadereit JW . (2010). Ecological differentiation and reproductive isolation of two closely related sympatric species of Oenanthe (Apiaceae). Biol J Linn Soc 101: 526–535.
Acknowledgements
We would like to express our gratitude to three anonymous referees and the associate editor for helpful comments that greatly improved the manuscript. We also thank M Cosquer, P Defives, R Demarle, F Bac for their help with sample collection, and A Gareil, S Mahé, G Seimandi-Corda and S Dreano for laboratory assistance. The present study was financially supported by the French Nord-Pas de Calais Region (CorEcol project).
Author contributions
CM, AE, FB designed the study; LFB, CM, AE, FB and JFA conducted the sampling and landscape characterization; LFB and JFA designed nuclear microsatellite loci; LFB performed molecular genotyping; LFB, CM, AE, FB and JFA analyzed the data and wrote the manuscript; all authors approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Favre-Bac, L., Mony, C., Ernoult, A. et al. Ditch network sustains functional connectivity and influences patterns of gene flow in an intensive agricultural landscape. Heredity 116, 200–212 (2016). https://doi.org/10.1038/hdy.2015.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.90
This article is cited by
-
Recruitment and migration patterns reveal a key role for seed banks in the meta-population dynamics of an aquatic plant
Scientific Reports (2023)
-
Getting lost in the matrix? On how the characteristics and arrangement of linear landscape elements influence ecological connectivity
Landscape Ecology (2022)
-
Effects of contemporary shifts of range margins on patterns of genetic structure and mating system in two coastal plant species
Heredity (2020)
-
Multi-species genetic connectivity in a terrestrial habitat network
Movement Ecology (2017)
-
The importance of small waterbodies for biodiversity and ecosystem services: implications for policy makers
Hydrobiologia (2017)
