
Abstract
Polyploidization, which is expected to trigger major genomic reorganizations, occurs much less commonly in animals than in plants, possibly because of constraints imposed by sex-determination systems. We investigated the origins and consequences of allopolyploidization in Palearctic green toads (Bufo viridis subgroup) from Central Asia, with three ploidy levels and different modes of genome transmission (sexual versus clonal), to (i) establish a topology for the reticulate phylogeny in a species-rich radiation involving several closely related lineages and (ii) explore processes of genomic reorganization that may follow polyploidization. Sibship analyses based on 30 cross-amplifying microsatellite markers substantiated the maternal origins and revealed the paternal origins and relationships of subgenomes in allopolyploids. Analyses of the synteny of linkage groups identified three markers affected by translocation events, which occurred only within the paternally inherited subgenomes of allopolyploid toads and exclusively affected the linkage group that determines sex in several diploid species of the green toad radiation. Recombination rates did not differ between diploid and polyploid toad species, and were overall much reduced in males, independent of linkage group and ploidy levels. Clonally transmitted subgenomes in allotriploid toads provided support for strong genetic drift, presumably resulting from recombination arrest. The Palearctic green toad radiation seems to offer unique opportunities to investigate the consequences of polyploidization and clonal transmission on the dynamics of genomes in vertebrates.
Similar content being viewed by others

Introduction
New species arise in several different ways, which can be classified in two main categories: on one hand, an ancestral species may give rise to two new daughter species (e.g., via allopatric, parapatric or sympatric speciation); on the other hand, two ancestral species may give rise to a new daughter species, via hybridization. The outcome of such hybridization processes partly depends on the level of divergence between the parental species (e.g., Chapman and Burke, 2007). If weakly differentiated, genomes might merge and lead to a diploid hybrid (homoploid hybrid speciation; Buerkle et al., 2000; Coyne and Orr, 2004; Mallet, 2007), with recombination taking place between homeologous chromosomes (i.e. partially homologous chromosomes derived from the same pair in the common ancestor). Alternatively, stronger differentiation may result in allopolyploidization (which may also occur between closely related species; Chapman and Burke, 2007): the two genomes will then coexist over evolutionary times within a tetraploid hybrid. Meiotic recombination will then take place between homologous and normally not between homeologous pairs of chromosomes (Otto and Whitton, 2000; Comai, 2005). The coexistence of homeologous pairs in allopolyploids generates a functional redundancy, which is expected to induce some re-diploidization via the elimination or silencing of redundant parts of the genome or their neo- or sub-functionalization (e.g., Ohno, 1970; Ferris and Whitt, 1977; Soltis and Soltis, 1999; Wendel, 2000; Ainouche and Jenczewski, 2010; Buggs et al., 2011; Braasch and Postlethwait, 2012; Roulin et al., 2012). In parallel to these changes in the function or expression of genes, chromosomal rearrangements, including deletions, inversions, intra- or inter-genomic translocations may occur rapidly and extensively after polyploidization (Soltis and Soltis, 1999; Adams, 2007; Gaeta et al., 2007). Comparative genetic mapping between polyploid species and their diploid relatives allows a better understanding of the evolution of genome structure and associated rearrangements. Linkage analyses have uncovered further genomic consequences of polyploidization, including increased rates of recombination in polyploid plants relative to diploid ancestors (e.g., Leflon et al., 2010; Pecinka et al., 2011; reviewed in Grandont et al., 2013) as well as homeologous (i.e., intergenomic) recombination, shown to occur in polyploid plants (Gaeta et al., 2007) and salamanders (Bi and Bogart, 2006, 2010). Thus, provided one may identify the parental species and timing of hybridization events, allopolyploid lineages represent unique opportunities to address important questions regarding the reorganization of genomes that is expected to follow hybridization, and more generally to gain crucial insights on the evolutionary dynamics of genomes.
Polyploidy is much rarer in animals than in plants (Otto and Whitton, 2000), a pattern attributed by Muller (1925) to the disruption of sex determination in gonochoristic animals. Nevertheless, polyploidization has been documented across a wide range of animal taxa, including turbellarians, annelids, mollusks, insects and crustaceans (Gregory and Mable, 2005), teleost fishes (Schultz, 1980; Le Comber and Smith, 2004), reptiles (Gregory and Mable, 2005), and amphibians (Bogart, 1980). Natural polyploids are especially frequent in the latter class (Bogart, 1980; Schmid, 1980; Kawamura, 1984); they have evolved independently in multiple families (Mable et al., 2011), with 50 polyploid anuran species and six polyploid salamander species described (Evans et al., 2012). Palearctic green toads offer a particularly interesting system, forming a monophyletic radiation of at least 12 major mitochondrial DNA (mtDNA) haplotype groups (Stöck et al., 2006) with several cases of range overlap and interactions through hybridization and polyploidization (Colliard et al., 2010; Dufresnes et al., 2014). This radiation includes in particular bisexually reproducing species of three ploidy levels (2n, 3n and 4n; Stöck et al., 2010).
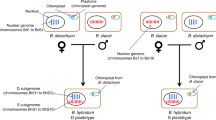
Here, we focused on five green-toad species of a diploid-polyploid complex from Central Asia, of which phylogenetic relationships are provided in Figure 1, based on mtDNA d-loop sequences. The diploid Bufo latastii (Boulenger, 1882; LL in Figure 2), spread in eastern Kashmir and Ladakh, constitutes an early-diverged lineage. The tetraploid B. pewzowi (Bedriaga, 1898), found in eastern Central Asia (from Uzbekistan to western Mongolia), presents two sets of chromosomes (PmPm; termed ‘TT’ in Stöck et al., 2010) from a maternal ancestor that, according to the mtDNA d-loop and three nuclear sequence markers (Stöck et al., 2010), have been assumed to be related to the geographically proximate B. turanensis (Hemmer et al., 1978; TT in Figure 1) whose range is restricted to the lowlands of Central Asia (from Eastern Iran to Kazahkstan). Genome-wide evidence for the maternal ancestry is missing, and the origin of the paternal sets (PpPp; termed ‘LL’ in Stöck et al., 2010) remains unidentified. The two sexes undergo a normal tetraploid meiosis (Stöck et al., 2005), resulting in diploid sperm and ovules with one Pp and one Pm set of chromosomes each (Stöck et al., 2010). The triploid B. baturae (Stöck et al., 1999), limited to Karakoram and Hindukush and possibly the High Pamirs, has a maternal ancestor that, according to mtDNA 16S rDNA (Litvinchuk et al., 2011) is predicted to be closely related to B. shaartusiensis (Mezhzherin and Shcherbak, 1996; cf. Mezhzherin and Shcherbak, 1995; SS in Figure 2), found in southwestern Tajikistan. The ancestor of the two paternal sets (BpBp) also remains unidentified (Figure 1). Triploidy is maintained by differential male and female forms of meioses: in the male germ line, the Bm set (referred to as NOR- in Stöck et al., 2012) is eliminated, followed by a diploid meiosis of the Bp sets (referred to as NOR+ in Stöck et al., 2012) that only produces recombined haploid Bp sperm, whereas in the female germ line, the Bm set is first duplicated, followed by a tetraploid meiosis that produces diploid BmBp ovules (Stöck et al., 2012). Hence, the Bp set is sexually transmitted by both sexes, whereas the Bm set is only maternally and clonally transmitted, along with mtDNA. This unique mode of transmission (termed ‘pre-equalizing hybrid meiosis’ by Stöck et al., 2012) is expected to bear important consequences on the evolution of the Bm set, by strongly enhancing genetic drift and drastically reducing the efficiency of selection. The effective population size of the Bm set is reduced to one quarter that of the Bp sets, and the absence of recombination over large genomic regions is expected to induce strong Hill-Robertson interferences between co-segregating loci (e.g., McVean and Charlesworth, 2000): deleterious mutations can get fixed by hitchhiking with beneficial ones during selective sweeps or, conversely, beneficial alleles can be eliminated by selection against co-segregating deleterious mutations (background selection; Charlesworth, 1994). In the absence of recombination, the class of individuals with the lowest genetic load cannot be restored once eliminated by drift (Muller’s ratchet; Haigh, 1978; Gabriel et al., 1993). The small effective population size associated with strong Hill-Robertson interferences is expected to reduce the molecular adaptation of non-recombining genomic regions and lead to their rapid divergence (McVean and Charlesworth, 2000). The evolutionary forces acting on the non-recombining Bm set are similar to those of non-recombining Y or W sex chromosomes, and thus expected to induce a similar degeneration, but at the level of an entire genome.

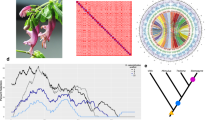
Maximum-Likelihood phylogram of the mitochondrial phylogenetic relationships of Palearctic green toads (B. viridis subgroup) based on ca. 880 bp of the mitochondrial control region (D-loop) with information on ploidy level (2n: diploid, 3n: triploid, 4n: tetraploid) and gametogenesis mode (m: meiotic, phm: pre-equalizing hybrid meiosis). Note that for some taxa ploidy and reproductive mode remain insecure (indicated by ‘?’). Numbers above branches show bootstrap support for 100 re-sampled datasets; missing numbers indicate less than 50% support. The most recent common ancestor of B. balearicus, B. turanensis, B. siculus and B. shaartusiensis was estimated to be of Pliocene age, that is, to have lived about 3.3 Mya (95% high probability density interval: 1.7–5.1 Mya) by Stöck et al. (2013).

Right: Bar plots from the Bayesian STRUCTURE clustering, hierarchically ordered from K=2 (bottom) to K=7 (top). Left: associated topology for genome phylogenies. A maximum in ΔK was found at K=4.
Our study had two main objectives. The first one was to establish a topology for the reticulate phylogeny in a species-rich group that involves several lineages, some of which very closely related. Nothing was known regarding the origins of paternal subgenomes in polyploids; hypotheses had been formulated regarding the origin of maternal subgenomes, but these were primarily based on mtDNA similarities, and needed to be tested with fast-evolving multiple nuclear markers. To achieve this first goal, we sampled adults from the two polyploid species as well as from three diploid species (turanensis, shaartusiensis and latastii) that might have contributed to their ancestry, and genotyped them with co-amplifying nuclear microsatellite markers (Supplementary Text 1 for details). We expected the patterns of amplification and allelic size range similarities to shed light on the genetic relationships among these various genomes and subgenomes.
The second aim was to explore the potential processes of genomic reorganization that are expected to follow polyploidization. Specifically, we addressed the following questions: (i) Do markers always cluster into the same linkage groups in polyploids as in diploids, or have some translocation events occurred? (ii) Does recombination in polyploids only take place between homologous pairs, or can we find evidence for homeologous recombination? (iii) Do recombination rates increase following polyploidization events, as documented in other lineages? This second set of questions (i–iii) was addressed by rearing families from the same five species, and genotyping them for the same set of co-amplifying microsatellite markers to perform linkage analyses.
Materials and methods
Animal sampling and DNA extraction
The available pedigrees consisted of 1045 individuals from 30 families, of which 9 were of B. latastii, 3 were of B. turanensis, 4 were of B. shaartusiensis, 6 were of B. baturae and 8 were of B. pewzowi (Table 1). Of these 30 families, 27 originated from mating pairs caught in amplexus during the 2005–2010 breeding seasons from one B. pewzowi population in Kazakhstan, three B. turanensis and three B. pewzowi populations in Kyrgyzstan, one B. latastii and two B. baturae populations in Pakistan, and one B. shaartusiensis population in Tajikistan (Table 1). The three other families stemmed from laboratory crosses between two females and three males of B. baturae (families B4, B5 and B6; Table 1) caught in a reproductive state during the 2009 spring breeding period). Females from these crosses were stimulated by injection of 0.1 ml of a 0.9% NaCl-solution containing 500–1000 IU of human choriogonadotropin (Sigma-Aldrich, Inc.). Parts of the egg strings from each pair were reared, respectively, in the field or in the laboratory (Table 1) to sample whole embryos, or larvae were fed with fish food (Tetramin) until tadpoles had grown enough to allow tailtip sampling. All parents were sampled for buccal cells (Broquet et al., 2007), finger tips or blood (using sterile syringes), except for two B. shaartusiensis families (S3, S4) of which only offspring embryos were collected. Two adult specimens of B. latastii and seven of B. baturae (buccal swabs) were added to our samples (Table 1). Buccal swabs were stored dry at −20 °C; blood and tissue samples in 70% ethanol at −20 °C. DNA was extracted using the QIAGEN DNeasy Tissue Kit or the BioSprint robotic workstation (QIAGEN), both following the manufacturer’s protocols with overnight sample incubation in proteinase K at 56 °C. DNA was eluted in a 150 μl and a 50 μl volume (QIAGEN Buffer AE) and stored at −20 °C.
Microsatellite primers, amplifications and scoring
All of the 30 microsatellite markers developed by Betto-Colliard et al. (2013) were used for analyses, using the same PCR protocols (for details of their cross-amplification success, see Supplementary Text 1). Allele sizes were scored with GENEMAPPER v.4.0 (Applied Biosystems). Genotypic configurations for triploid and tetraploid individuals were easily inferred from segregation patterns among families in a majority of alleles (92.5%). In some cases (1.6%), dosage of the amplified products, based on quantitative values for microsatellite allele amplification (peak size) could be used to assign alleles in polyploid individuals (Esselink et al., 2004 and Stöck et al., 2010 for similar approaches). Whenever dosage interpretation was not trustworthy to identify allelic composition (5.9%), allele values were considered as ‘missing data’ to conserve a robust dataset. Null alleles were readily identified, given information on ploidy levels and pedigrees; for instance, in a diploid family where the father only expresses allele A and the mother allele B, the occurrence of offspring with allele A only reveals a null allele in the mother (genotype B0), while occurrence of offspring with allele B only reveals a null allele in the father (genotype A0). Deviations from Hardy-Weinberg equilibrium were tested using ARLEQUIN v. 3.5.1.3 (Excoffier and Lischer, 2010) in all subgenomes, except in the clonally and maternally transmitted Bm set.
Linkage analyses
To evaluate signs of genomic reorganization between polyploid species and their presumed diploid ancestors via homeologous recombination, changes in recombination rates or translocations events, linkage analyses were performed using CRIMAP v.5.0 (Green et al., 1990) based on all families from the resource pedigree. Analyses in the triploid B. baturae were limited to the recombining diploid Bp set, that is, excluding the clonally and maternally transmitted Bm set. In the tetraploid B. pewzowi, linkage analyses were achieved separately for the two homologous sets (PpPp and PmPm, respectively), identified from our pedigree analyses, thanks to similarity of allelic size ranges between these sets and their putative ancestors (namely, between PpPp and LL; PmPm and TT), and very low rates of male recombination (see Results). Pairwise linkage analyses, identifying linkages between any two loci, were performed using the TWOPOINT option, assuming sex-specific recombination rates. Pairwise linkage was considered significant if the logarithm of odds score exceeded 3.0 (i.e., the odds that the two loci are linked exceed 1000: 1). For linkage groups including more than two loci, we used the ALL option to select the order with the highest log likelihood. This likelihood was further compared with that of alternative orders by pairwise flipping of neighboring loci using the FLIPS option. Finally, we used the BUILD option to estimate recombination rates and Kosambi mapping distances for the sex-specific maps. Linkage maps were drawn using MAPCHART v.2.1 (Voorrips, 2002). Effects of sex, ploidy level and their interaction (explanatory variables) on the recombination rate (response variable) were tested with randomized ANOVAs over all data, in which recombination rates were randomly permuted 104 times between sexes and ploidy levels (Manly, 1997). Given the absence of ploidy effects (see Results), all genomes (LL, TT, SS, BpBp, PmPm, PpPp) were then independently tested for sex-specific differences in recombination rate, using Wilcoxon tests in R (R Development Core Team, 2009). Wilcoxon tests and randomized ANOVAs were performed on pairwise recombination rates of loci belonging to the same linkage group.
Clustering analyses
To investigate the genetic relationships between lineages, we performed a Bayesian clustering analyses using STRUCTURE v.2.3.4 (Pritchard et al., 2000) based on the genotypes of 69 adults, comprising the 60 parents (those of families S3 and S4 were inferred from offspring genotypes) and the nine supplementary specimens (two B. latastii and seven B. baturae individuals; Table 1), where the subgenomes of polyploid species (Bm, BpBp, PmPm, PpPp) were considered as distinct entities. Each run, replicated 10 times, comprised a ‘burn-in’ period of 104 and 105 iterations. The number of clusters (K) was varied from 2 to 7 (this maximal value corresponding to the expected number of distinct subgenomes: LL, TT, SS, Bm, BpBp, PmPm, PpPp), to establish a hierarchical clustering and provide insights on the depth of splitting among the genomes. The most likely number of clusters was inferred from the ad hoc ΔK statistic (Evanno et al., 2005). Topology was also inferred from a Neighbor Net Tree, built with the program SPLITSTREE (v.4.13.1; Huson and Bryant, 2006) based on the FST-matrix of pairwise distances between genomes (computed with ARLEQUIN v.3.5.1.3; Excoffier and Lischer, 2010). A principal component analysis was conducted to visualize the differentiation between genomes, using PCAGEN (Goudet, 1999) with 104 randomizations of genotypes to test for significance of axes.
Results
Assignment of maternal and paternal sets of chromosomes
In the allotriploid B. baturae, the single maternal set Bm was easily identified from our pedigree analyses because of its purely clonal and maternal transmission; it furthermore displayed the same patterns of amplification and range of allelic sizes as the putative maternal ancestor B. shaartusiensis (Supplementary file S1). In the allotetraploid B. pewzowi, the maternal Pm set also often displayed the same pattern of amplification and range of allelic sizes as in its putative maternal ancestor B. turanensis, while the Pp set displayed similarity with its putative paternal ancestor B. latastii (Supplementary file S1). In some ambiguous cases, identification was possible by the patterns of linkage with other already assigned markers, thanks to the extremely low levels of recombination in males (see below). In a few doubtful cases, markers were excluded; altogether, 4 out of 30 markers (BaC111, BaC212, BaD5, BlD114) could not be assigned with certainty and were excluded from analyses. BaD103 was also excluded from linkage analyses, being non-informative (monomorphic throughout).
Linkage groups
The patterns of marker amplification, polymorphism and assignments to different linkage groups based on the program CRIMAP are provided in Table 2. Depending on species and subgenomes, three to seven linkage groups could be formed (Supplementary Text 1). Some markers remained unlinked in several genomes (Table 2) because of a lack of polymorphism and statistical power. The linkage groups that could be established were largely consistent within and between species and subgenomes, pointing to a strong synteny across species and ploidy levels with no signs of intergenomic recombination. Loci orders, however, were not always conserved (Supplementary Figure S1a–c), possibly suggesting some inversions. More importantly, a few cases of potential translocations could be established (Table 2). Interestingly, these translocations affected only the paternal sets of the allotriploid baturae (Bp) and the allotetraploid pewzowi (Pp) genomes, and consisted only of marker movement out of LG1, which had previously been identified as the sex chromosome in several diploid species of the B. viridis radiation (Stöck et al., 2011, 2013). Another striking outcome was the heterogeneity found among the paternal sets of B. baturae families: two markers (BaD11 and BlB223) mapped to LG1 in four families (families B1, B2, B3, B5; group 1, termed Bp1) but to different linkage groups (respectively, LG6 and LG2) in families B4 and B6 (group 2, termed Bp2).
Recombination rates
In all species, average recombination rates were much lower in males (0.02–0.16) than in females (0.34–0.46), a highly significant difference (Wilcoxon tests, P<0.001 for all comparisons). These sex differences occurred on all linkage groups (Supplementary Table S1 and Supplementary Text 1). Accordingly, maps were always much shorter in males than in females (Supplementary Figure S1a–c). In contrast, recombination did not vary with ploidy level; there was no tendency for a larger recombination rate in polyploid species (Supplementary Table S1). The interaction between sex and ploidy level on recombination rate was not significant (Supplementary Table S1).
Patterns of genome differentiation
Several microsatellite markers in the paternal subgenome Pp but none or only few markers in the other subgenomes (L, T, S, Bp and Pm) failed to meet Hardy-Weinberg expectations (Supplementary Text 1). Results from STRUCTURE analyses served to infer the relationships of genomes (Figure 2), together with a tree that summarizes the differential clustering obtained from our hierarchical approach for K, varying from 2 to 7. The first divergence (K=2) occurred between the paternal (L, Bp and Pp) and the maternal (Bm, S, Pm and T) genomes or subgenomes. K=3 separated L from Bp and Pp within the paternal branch. K=4 (corresponding to a maximum in ΔK) separated the baturae (S and Bm) from the pewzowi lineages (T and Pm) within the maternal branch. K=5 separated the baturae maternal lineage (Bm) from its presumptive ancestor (S), whereas K=6 separated the two paternal lineages of pewzowi and baturae. Finally, fixing K to 7 generated a split among several clonal lineages of baturae maternal subgenome (Bm). It is worth noting that the pewzowi maternal lineages always clustered together with turanensis, and that one copy of baturae’s maternal subgenome always clustered with shaartusiensis.
The neighbor net tree (Supplementary Figure S2) provided a very similar topology, with the deepest split between the paternal (L, Pp, Bp) and maternal (T, Pm, S, Bm) lineages, and the shallowest one between the pewzowi maternal subgenome and the turanensis genome. The only deviation from the STRUCTURE clustering corresponded to an earlier branching of Bm from the other maternal lineages in the FST-based tree, possibly caused by strong drift and Hill-Robertson interferences affecting this clonal lineage.
These patterns were also highly consistent with the outcomes of the principal component analyses (Figure 3). The seven genomes were best discriminated along the first and fourth axes; the first axis (accounting for 13.67% of the variance) separated the paternal (left) from the maternal (right) genomes, while the fourth axis (accounting for 4.63% of the variance) separated the maternal pewzowi (Pm, T; top) from the maternal baturae (Bm, S; bottom) lineages.

Principal component analysis based on adult and parental samples; results plotted along the first (horizontal) and fourth (vertical) axes.
Discussion
Allopolyploid origins and genome topologies
Clustering analyses based on our multi-locus genotype data, the vast majority of which represent newly established, cross-amplifying microsatellite loci, showed that the maternal ancestor of B. pewzowi (Pm) was closely related to B. turanensis, in accordance with previous suggestions that had been based on limited mtDNA and nuDNA sequence evidence (Stöck et al., 2006, 2010). The strong genetic similarity between these two genomes pointed to a very recent hybridization event: for all K values investigated in the STRUCTURE analyses, the Pm and T genomes clustered together (Figure 2). We also confirmed with nuclear markers that the maternal ancestor (Bm) of B. baturae was related to B. shaartusiensis. These new data represent the first nuclear and multilocus evidence for the maternal ancestry of B. baturae, which is one of the few gonochoristic all-triploid vertebrates (Stöck et al., 2012), for which previous hints on this hybrid species’ maternal ancestor came only from mtDNA (Litvinchuk et al., 2011). The close relationship between B. baturae and B. shaartusiensis, that had been inferred from identical mtDNA sequences (Litvinchuk et al., 2011; Figure 1), contrasts with the deeper divergence that we found between Bm and S based on nuclear data (Figure 2 and Supplementary Figure S2). This latter pattern might result from the haploid and clonal transmission of the Bm subgenome in B. baturae: the small effective population size and strong Hill-Robertson interferences affecting this non-recombining subgenome is bound to induce large genetic drift and rapid divergence. This affects not only the differentiation from other lineages, but also among different clonal Bm lineages, as revealed by several sub-clusters of Bm occurring for STRUCTURE setting K=7 (Figure 2). Interestingly, one of the Bm lineages of B. baturae always clustered with S in STRUCTURE analyses (Figure 2), for any given K value, indeed supporting the very recent hybridization history suggested by the strong mtDNA similarity (Litvinchuk et al., 2011).
Our data also provided a completely new line of evidence regarding the paternal ancestries of both allotriploid B. baturae and allotetraploid B. pewzowi. In contrast to the maternal origins, for which mtDNA had provided hints, nothing was known about the paternal origins of these hybrid polyploids. Our data suggest deeper divergence between the paternal subgenomes (Bp and Pp) and the lineage represented by the latastii genome (Figure 2 and Supplementary Figure S2). Given the strong similarities of the maternal lines Bm and Pm with the S and T genomes, respectively, we assume that the paternal genomes Bp and Pp stem from two unsampled (and/or possibly extinct) species that had already largely diverged from B. latastii when hybridization events occurred. At this time, these two paternal species had also likely diverged from each other: the origin of pewzowi cannot predate the Pm-T divergence, which our results suggested to be very recent (Figure 2), in line with previous mtDNA evidence (Stöck et al., 2006, 2010).
The clonal transmission of the maternal subgenome Bm combined with the departure from Hardy-Weinberg equilibrium in several markers from the paternal subgenome Pp could have affected the topology arising from our STRUCTURE analyses. In the future, gene sequencing should provide alternative and powerful tools to investigate our working hypotheses, and confirm or falsify the genome relationships proposed here (Figures 2 and 3). In particular, comparative investigations of series of genes from the maternal lineages (Bm, S, Pm and T) would offer a fascinating opportunity to compare the evolution of a given genome put in distinct evolutionary settings: namely, in either a recombining diploid state (S and T), a recombining polyploid state (Pm) or a non-recombining polyploid state (Bm). Such comparisons should allow disentangling the consequences of polyploidization from those of Hill-Robertson interferences on the evolutionary dynamics of genomes.
Sex, but not ploidy level, affects recombination rates
Our data also provided indications that sex had a major impact on recombination rates: all linkage groups and species investigated displayed strongly female-biased recombination. Heterochiasmy (i.e., sex differences in recombination rates) seems widespread throughout animals, with a trend for strongly reduced male recombination across amphibians (e.g., Sumida and Nishioka, 2000; Berset-Brändli et al., 2008; Rodrigues et al., 2013) and fishes (Moens et al., 2004). Heterochiasmy might have evolved as a pleiotropic consequence of selection on sex chromosomes (Haldane, 1922; Huxley, 1928; Nei, 1969): the easiest or quickest way to reduce sex-chromosome recombination in the heterogametic sex might be to reduce recombination over the whole genome in this sex. Accordingly, several diploid species of the B. viridis radiation have been shown to be male heterogametic (Stöck et al., 2011, 2013). If this correlation also holds for polyploids as our data support, the strongly female-biased recombination documented here for B. baturae and B. pewzowi suggests maintenance of the initial male heterogamety during polyploidization events. This point obviously deserves further investigation, given that sex chromosomes and sex-determination systems have been seen as major obstacles to polyploidization (Muller, 1925; Orr, 1990).
In contrast, recombination rates did not vary with ploidy level. These patterns differ from those recently documented from plants (Arabidopsis and Brassica), where auto- and allopolyploids were shown to display higher recombination rates than their diploid ancestors (Leflon et al., 2010, Pecincka et al., 2011, Grandont et al., 2013). However, those polyploid plant lineages resulted from recent laboratory crosses, and thus presumably did not have time to evolve advanced states of re-diploidization of genomes, a process that follows polyploidization (e.g., Soltis and Soltis, 1999; Wendel, 2000; Tayalé and Parisod, 2013). Despite their relatively young evolutionary ages, polyploids from the B. viridis subgroup already show some signs of re-diploidization, as illustrated by the physical deletion of entire rDNA clusters (NORs; Stöck et al., 2002, 2005). Thus, the increased recombination rates documented from laboratory-produced plant polyploids might only constitute a transient state, with a return towards lower rates during the process of re-diploidization.
Genome synteny and reorganization
Our linkage group maps (Supplementary Figure S1a–c) pointed to a generally strong synteny among the genomes investigated, with however some evidence for a few reorganization events. A relocation of BaC101 from LG1 to LG2 was found both in pewzowi (Pp) and in the two baturae groups (Bp1 and Bp2), and thus likely occurred early in the evolution of these subgenomes, before their divergence. The relocation of BlB223 and BaD11 from this same LG1 to LG2 and LG6, respectively, was only observed in the Bp2 group, and thus likely occurred much more recently. Although still anecdotal, these events point to patterns that might be more than coincidental: all three translocations occurred in the paternal ancestors’ subgenomes of polyploid species, and all affected the same linkage group (LG1), identified in the studies by Stöck et al. (2011, 2013) as the sex chromosome in several diploid species from the B. viridis radiation.
Extensive chromosomal rearrangements are expected following polyploidization, as repeatedly documented in polyploid plant species (Soltis and Soltis, 1999; Wendel, 2000; Bento et al., 2011; Tayalé and Parisod, 2013). Accordingly, the translocation events documented here in polyploid green toads contrast with the strong synteny that otherwise characterizes diploid species from this group, and amphibians in general (e.g., Miura, 1995; Sumida and Nishioka, 2000; Brelsford et al., 2013). It might also be meaningful that such translocations were limited not only to the paternal subgenomes, but also to the supposed sex-chromosome pairs. Assuming parental species to be male heterogametic (as are other diploid species from this radiation; Stöck et al., 2011, 2013), one would expect sex in polyploid green toads to be determined by paternal subgenomes (Bp and Pp, respectively). Similarly, assuming sex-determination systems to be conserved during polyploidization, then sex-determining genes with functional X and Y alleles should localize on the paternal LG1 in both B. baturae and B. pewzowi. Given that sex-determination systems have been proposed to constitute major barriers to polyploidization in animals (Muller, 1925; Otto and Whitton, 2000), the reorganization of sex chromosomes that may follow polyploidization is an important issue, which deserves further investigation by sequence analyses of paternal subgenomes in polyploid species from the B. viridis radiation. Very strong mtDNA similarity in the D-loop (Stöck et al., 2006 and our still unpublished data) suggests a single maternal origin of all-triploid B. baturae sampled here in two isolated high mountain valleys in the Karakoram. However, the observed LG1 differences interpreted here as translocations, could alternatively stem from, so far undocumented, multiple paternal contributions to the allotriploid genome.
Conclusions and perspectives
The multiple and independent events of hybrid-origin polyploidization that characterize Palearctic green toads (B. viridis subgroup) offer remarkable opportunities to investigate the rise and consequences of polyploidization and thus polyploid hybrid speciation in animals. Our sibship analyses of microsatellite markers allowed us to generate working hypotheses regarding ancestry relationships and genome topologies, to identify translocation events presumably associated with the reorganization of sex chromosomes, and to suggest Hill-Robertson effects on clonally transmitted subgenomes. Sequence analyses of autosomal and sex-linked genes will allow further tests of the manifold consequences of polyploidization and clonal transmission on the reshaping of genomes on general, and that of sex chromosomes in particular.
Data archiving
Genotype data available from the Dryad Digital Repository: doi:10.5061/dryad.1jc02.
References
Adams KL . (2007). Evolution of duplicate gene expression in polyploid and hybrid plants. J Hered 98: 136–141.
Ainouche ML, Jenczewski E . (2010). Focus on polyploidy. New Phytol 186: 1–4.
Bento M, Perry Gustafson J, Viegas W, Silva M . (2011). Size matters in Triticeae polyploids: larger genome have higher remodeling. Genome 54: 175–183.
Berset-Brändli L, Jacquiéry J, Broquet T, Ulrich Y, Perrin N . (2008). Extreme heterochiasmy and nascent sex chromosomes in European tree frogs. Proc R Soc Lond B 275: 1577–1585.
Betto-Colliard C, Sermier R, Perrin N, Stöck M . (2013). Development and cross-amplification of thirty microsatellite loci in five diploid and polyploid Central Asian species of Palearctic green toads (Bufo viridis subgroup). Conserv Genet Resour 5: 243–249.
Bi K, Bogart JP . (2006). Identification of intergenomic recombinations in unisexual salamanders of the genus Ambystoma by genomic in situ hybridization (GISH). Cytogenet Genome Res 112: 307–312.
Bi K, Bogart JP . (2010). Probing the meiotic mechanism of intergenomic exchanges by genomic in situ hybridization on lampbrush chromosomes of unisexual Ambystoma (Amphibia: Caudata). Chromosome Res 18: 371–382.
Bogart JP . (1980). Evolutionary implications of polyploidy in amphibians and reptiles. In: Lewis WH ed Polyploidy: Biological relevance. Plenum Press: New York. pp 341–378.
Braasch I, Postlethwait JH . (2012). Polyploidy in fish and the teleost genome duplication. In: Soltis PS, Soltis DE (eds). Polyploidy and genome evolution. Springer: Berlin Heidelberg. pp 341–383.
Brelsford A, Stöck M, Betto-Colliard C, Dubey S, Dufresnes C, Jourdan-Pineau H et al . (2013). Homologous sex chromosomes in three deeply divergent anuran species. Evolution 67: 2434–2440.
Broquet T, Berset-Braendli L, Emaresi G, Fumagalli L . (2007). Buccal swabs allow efficient and reliable microsatellite genotyping in amphibians. Conserv Genet 8: 509–511.
Buerkle CA, Morris RJ, Asmussen MA, Rieseberg LH . (2000). The likelihood of homoploid hybrid speciation. Heredity 84: 441–451.
Buggs RJA, Zhang L, Miles N, Tate JA, Gao L, Schnable PS et al . (2011). Genomic and transcriptomic shock generate evolutionary novelty in a newly formed natural allopolyploid plant. Curr Biol 21: 1–6.
Chapman MA, Burke JM . (2007). Genetic divergence and hybrid speciation. Evolution 61: 1773–1780.
Charlesworth B . (1994). The effect of background selection against deleterious mutations on weakly selected, linked variants. Genet Res 63: 213–227.
Colliard C, Sicilia A, Turrisi GF, Arculeo M, Perrin N, Stöck M . (2010). Strong reproductive barriers in a narrow hybrid zone of West-Mediterranean green toads (Bufo viridis subgroup) with Plio-Pleistocene divergence. BMC Evol Biol 10: 232.
Comai L . (2005). The advantages and disadvantages of being polyploidy. Nat Rev Genet 6: 836–846.
Coyne JA, Orr HA . (2004). Speciation. Sinauer Associates: Sunderland, MA.
Dufresnes C, Bonato L, Novarini N, Betto-Colliard C, Perrin N, Stöck M . (2014). Inferring the degree of incipient speciation in secondary contact zones of closely related lineages of Palearctic green toads (Bufo viridis subgroup). Heredity 113: 9–20.
Esselink G, Nybom H, Vosman B . (2004). Assignment of allelic configuration in polyploids using the MAC-PR (microsatellite DNA allele counting-peak ratios). Theor Appl Gen 109: 402–408.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Evans BJ, Alexander Pyron R, Wiens JJ . (2012). Polyploidization and sex chromosome evolution in Amphibians. In: Soltis PS, Soltis DE eds. Polyploidy and genome evolution. Springer: Berlin Heidelberg. pp 385–410.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5.1.3: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564–567.
Ferris SD, Whitt GS . (1977). Loss of duplicate gene expression after polyploidization. Nature 265: 258–260.
Gabriel W, Lynch M, Bürger R . (1993). Muller’s ratchet and mutational meltdowns. Evolution 47: 1744–1757.
Gaeta RT, Pires JC, Iniguez-Luy F, Leon E, Osborn TC . (2007). Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19: 3403–3417.
Goudet J . (1999). PCAGen: http://www2unilch/popgen/softwares/pcagenhtm.
Grandont L, Jenczewski E, Lloyd A . (2013). Meiosis and its deviations in polyploid plants. Cytogenet Genome Res 140: 171–180.
Green P, Falls K, Crooks C . (1990) Documentation for CRIMAP version 24 Washington University School of Medicine St Louis MO.
Gregory TR, Mable BK . (2005). Polyploidy in plants. In: Gregory TR (ed). The evolution of the genome. Elsevier: New York. pp 427–517.
Haigh J . (1978). The accumulation of deleterious genes in a population. Theor Pop Biol 14: 251–267.
Haldane JBS . (1922). Sex ratio and unisexual sterility in hybrid animals. J Genet 12: 101–109.
Huson DH, Bryant D . (2006). Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267.
Huxley JS . (1928). Sexual differences of linkage in Gammarus chevreuxi. J Genet 20: 145–156.
Kawamura T . (1984). Polyploidy in amphibians. Zool Sci 1: 1–15.
Le Comber SC, Smith C . (2004). Polyploidy in fishes: patterns and processes. Biol J Linn Soc 82: 431–442.
Leflon M, Grandont L, Eber F, Huteau V, Coriton O, Chelysheva L et al . (2010). Crossovers get a boost in Brassica allotriploid and allotetraploid hybrids. Plant Cell 22: 2253–2264.
Litvinchuk SN, Mazepa GO, Pasynkova RA, Saidov A, Satorov T, Chikin YA et al. Influence of environmental conditions on the distribution of Central Asian green toads with three ploidy levels. J Zool Sys Evol Res,. (2011) 9: 233–239
Mable BK, Alexandrou MA, Taylor MI . (2011). Genome duplication in amphibians and fish: an extended synthesis. J Zool 284: 151–182.
Mallet J . (2007). Hybrid speciation. Nature 446: 279–283.
McVean GA, Charlesworth B . (2000). The effects of Hill-Robertson interference between weakly-selected mutations on patterns of molecular evolution and variation. Genetics 155: 929–944.
Mezhzherin SV, Pisanets EM . (1995). Genetic structure and origin of the tetraploid toad Bufo danatensis Pisanetz, 1978 (Amphibia, Bufonidae) from Central Asia: differentiation of geographic forms and genetic relationships between diploid and tetraploid species. Russ J Genet 31: 342–352.
Miura I . (1995). The late replication banding patterns of chromosomes are highly conserved in the genera Rana Hyla and Bufo (Amphibia: Anura). Chromosoma 103: 567–574.
Moens T, Høyheim B, Munck H, Gomez-Raya L . (2004). A linkage map of Atlantic salmon (Salmo salar) reveals an uncommonly large difference in recombination rate between the sexes. Anim Genet 35: 81–92.
Muller HJ . (1925). Why polyploidy is rarer in animals than in plants. Amer Nat 59: 346–353.
Nei M . (1969). Linkage modification and sex difference in recombination. Genetics 63: 681–699.
Ohno S . (1970). Evolution by Gene Duplication. Springer: New York.
Orr HA . (1990). “Why polyploidy is rarer in animals than in plants” revisited. Amer Nat 136: 759–770.
Otto SP, Whitton J . (2000). Polyploid incidence and evolution. Annu Rev Genet 34: 401–437.
Pecinka A, Fang W, Rehmsmeier M, Levy AA, Mittelsten Scheid O . (2011). Polyploidization increases meiotic recombination frequency in Arabidopsis. BMC Biol 9: 24.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
R Development Core Team. (2009). R: a language and environment for statistical computing. R Foundation for Statistical Computing: Vienna.
Rodrigues N, Betto-Colliard C, Jourdan-Pineau H, Perrin N . (2013). Within-population polymorphism of sex-determination systems in the common frog (Rana temporaria). J Evol Biol 26: 1569–1577.
Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G et al . (2012). The fate of duplicated genes in a polyploid plant genome. Plant J 73: 143–153.
Schmid M . (1980). Chromosome evolution in Amphibia. In: Müller H (ed). Cytogenetics of vertebrates. Birkhäuser, Basel: Boston, Stuttgart. pp 4–27.
Schultz RJ . (1980). Role of polyploidy in the evolution of fishes. In: Lewis WH (ed). Polyploidy: Biological relevance. Plenum Press: New York. pp 313–340.
Soltis DE, Soltis PS . (1999). Polyploidy: recurrent formation and genome evolution. Trends Ecol Evol 14: 348–352.
Stöck M, Schmid M, Steinlein C, Grosse WR . (1999). Mosaicism in somatic triploid specimens of the Bufo viridis complex in the Karakoram with examination of calls morphology and taxonomic conclusions. Ital J Zool 66: 215–232.
Stöck M, Lamatsch DK, Steinlein C, Epplen JT, Grosse WR, Hock R et al . (2002). A bisexually reproducing all-triploid vertebrate. Nat Genet 30: 325–328.
Stöck M, Steinlein C, Lamatsch DK, Schartl M, Schmid M . (2005). Multiple origins of tetraploid taxa in the Eurasian Bufo viridis subgroup. Genetica 124: 255–272.
Stöck M, Moritz C, Hickerson M, Frynta D, Dujsebayeva T, Eremchenko V et al . (2006). Evolution of mitochondrial relationships and biogeography of Palearctic green toads (Bufo viridis subgroup) with insights in their genomic plasticity. Mol Phylogenet Evol 41: 663–689.
Stöck M, Ustinova J, Lamatsch DK, Schartl M, Perrin N, Moritz C . (2010). A vertebrate reproductive system involving three ploidy levels: hybrid origin of triploids in a contact zone of diploid and tetraploid paleartic green toads (Bufo viridis subgroup). Evolution 64: 944–959.
Stöck M, Croll D, Dumas Z, Biollay S, Wang J, Perrin N . (2011). A cryptic heterogametic transition revealed by sex-linked DNA markers in Palearctic green toads. J Evol Biol 24: 1064–1070.
Stöck M, Ustinova J, Betto-Colliard C, Schartl M, Moritz C, Perrin N . (2012). Simultaneous Mendelian and clonal genome transmission in a sexually reproducing all-triploid vertebrate. Proc R Soc Lond B 279: 1293–1299.
Stöck M, Savary R, Betto-Colliard C, Biollay S, Jourdan-Pineau H, Perrin N . (2013). Low rates of X-Y recombination not turnovers account for homomorphic sex chromosomes in several diploid species of Palearctic green toads (Bufo viridis subgroup). J Evol Biol 26: 674–682.
Sumida M, Nishioka M . (2000). Sex-linked genes and linkage maps in amphibians. Comp Biochem Physiol Part B: Biochem Mol Biol 126: 257–270.
Tayalé A, Parisod C : Natural pathways to polyploidy in plants and consequences for genome reorganization. In: Stöck M, Lamatsch D (eds): Trends in polyploidy research in animals and plants;. Cytogenet Genome Res,. (2013) 140: 79–96
Voorrips RE . (2002). MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J Hered 93: 77–78.
Wendel JF . (2000). Genome evolution in polyploids. Plant Mol Biol 42: 225–249.
Acknowledgements
We thank L. Grandont, E. Jenczewski and A. Lloyd for access to a manuscript in press. This work was supported by a research grant (STO 493/3-1) and a Heisenberg-fellowship (STO 493/2-1) from the German Research Foundation (DFG) to MS, and a grant from the Swiss National Science Foundation (31003A- 129894) to NP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article

Cite this article
Betto-Colliard, C., Sermier, R., Litvinchuk, S. et al. Origin and genome evolution of polyploid green toads in Central Asia: evidence from microsatellite markers. Heredity 114, 300–308 (2015). https://doi.org/10.1038/hdy.2014.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.100
This article is cited by
-
Low neutral and immunogenetic diversity in northern fringe populations of the green toad Bufotes viridis: implications for conservation
Conservation Genetics (2022)
-
Genotypic similarities among the parthenogenetic Darevskia rock lizards with different hybrid origins
BMC Evolutionary Biology (2020)
-
Natural and domestic introgressions in the marble trout population of Soča River (Slovenia)
Hydrobiologia (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}