
Abstract
An important feature of cancer is dysregulation of gene activity and gene expression, which is driven by a combination of acquired genetic and epigenetic alterations. Here, we will highlight how insights into the epigenetic processes underpinning tumor biology have led to the emerging field of cancer pharmaco-epigenomics. First, we will discuss how interference with the epigenetic machinery in cancer is leading to novel promising therapies, with several DNA methyltransferase and histone deacetylase inhibitors being approved for cancer treatment. Second, we will discuss how epigenetic markers in cancer may increasingly be used as complementary diagnostic tools, prognostic markers of disease progression, and predictive markers of treatment response. Although the anti-tumoral activities of epigenetic therapies have thus far been attributed to reactivation of silenced tumor-suppressor and/or apoptotic genes, they may also influence the tumor environment by directly affecting stromal cells. As an example, we will discuss how tumor-endothelial cells are regulated at the epigenetic level and are affected by methyltransferase and histone deacetylase inhibitors.
Similar content being viewed by others
Introduction
Cancer is a disease marked by uncontrolled cell growth. Decades of research have led to the identification of numerous genetic alterations in the DNA sequence as major drivers of carcinogenesis. Generally, these genetic alterations affect oncogenes with a dominant gain of function, and tumor-suppressor genes with a loss of function (Stratton et al., 2009). However, dysregulation of gene activity in cancer can also be achieved by mechanisms that do not involve changes in the DNA sequence and which are generally referred to as epigenetic alterations. In this review, we will first discuss epigenetic hallmarks that are altered in cancer cells, including DNA methylation changes and histone modifications. Insights into how these affect cancer and clinical therapeutic applications have led to a new research area called pharmaco-epigenomics. The first area of interest that we will discuss in this field involves the development of cancer therapies that aim to reverse epigenetic changes in cancer cells and already resulted in the approval of three drugs for the treatment of cancer patients (Jones and Baylin, 2007). Second, the recent identification of epigenetic changes as novel prognostic or predictive markers for cancer therapies will also be discussed. Finally, we will expand the scope of epigenetic changes beyond that of cancer cells, by discussing how anti-angiogenic treatment may trigger epigenetic changes in stromal cells, in particular endothelial cells of the tumor.
DNA methylation and histone modifications in cancer
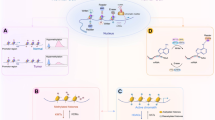
Epigenetic hallmarks commonly altered in cancer cells include changes in DNA methylation and histone modifications. Although the exact cause of these alterations is unclear, environmental cues have been shown to induce epigenetic modifications of DNA or histones (Figure 1) (Gluckman et al., 2008).

The epigenetic bridge between environment and heredity. Different environmental cues influence epigenetic modification of histones or DNA and alter access of transcription factors (TFs) to the DNA sequence, thereby affecting gene expression. Transcriptionally active chromatin is characterized by the presence of, for instance, acetyl groups (Ac) on specific lysine residues of histones in the nucleosome, which decreases their binding to DNA and results in a more open chromatin structure that permits access of transcription factors. CpG sequences in the promoter regions (P) of actively transcribed genes are generally unmethylated, allowing for the binding of TF. Transcriptionally inactive chromatin is characterized by histone deacetylation, promoter CpG methylation (as indicated by methyl groups [Me]), and decreased binding of TF. These acquired epigenetic modifications can potentially be transmitted to offspring and—in part—determine the inherited phenotype. Figure adapted from Gluckman et al. (2008).
Methylation changes often involve the hypermethylation of promoters, which leads to repression of transcription by inhibiting binding of specific transcription factors and by recruiting methyl CpG-binding proteins and their associated chromatin remodeling complexes (Figure 1) (Sasaki and Matsui, 2008). Besides hypermethylation of CpG islands, global DNA hypomethylation also occurs in cancer. Hypomethylation of the genome largely affects the intergenic regions of the DNA, particularly repeat sequences and transposable elements, and is believed to result in chromosomal instability and increased mutation events (Wilson et al., 2007). Although promoter hypermethylation is mostly associated with tumor-suppressor gene silencing, such as the retinoblastoma gene, CDKN2A, or hMLH1, global DNA hypomethylation is associated with activation of proto-oncogenes, such as c-JUN or c-MYC, and generation of genomic instability (Feinberg and Tycko, 2004; Feinberg, 2007).
A second important type of epigenetic alterations in cancer cells are histone modifications, which affect gene transcription through local relaxation of nucleosomal structure and through recruitment of nonhistone proteins (Strahl and Allis, 2000). A myriad of histone-modifying enzymes has been identified in the past 10 years. Among the most studied so far are the histone acetyltransferases and histone deacetylases (HDACs) (Kouzarides, 2007). Acetylation of histones by histone acetyltransferases promotes gene transcription by creating a more accessible chromatin structure, whereas HDAC-induced deacetylation dampens histone-DNA and histone-nonhistone protein interactions, impairing transcription (Figure 1) (Sasaki and Matsui, 2008). As such, transcriptionally silent genes are frequently associated with deacetylation of histone H3 and H4 (Ballestar et al., 2003; Jones and Baylin, 2007). Similarly to DNA methylation, histone modifications are commonly disrupted in cancer cells. For instance, global loss of monoacetylation and trimethylation of histone H4 can be considered a common hallmark of human tumor cells and altered histone modifications constitute a mechanism for inactivation of tumor-suppressor genes, as illustrated by hypermethylation of lysine 9 in histone H3 of the CDKN2A gene (Nguyen et al., 2002; Fraga et al., 2005; Seligson et al., 2005).
Pharmaco-epigenomics: bringing epigenetics to the bedside
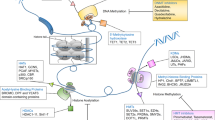
Insights into the epigenetic origin of cancer have led to the emergence of a new research field, which is called cancer pharmaco-epigenomics (Figure 2). Two main areas of interest can be defined in pharmaco-epigenomic research. The first involves the development of cancer therapies that aim to reverse epigenetic changes in cancer cells. Unlike genetic alterations, changes in histone modification and DNA methylation are reversible, and this reversible nature makes both mechanisms attractive therapeutic targets. The best-studied examples so far are agents that inhibit DNA methyltransferases (DNMT inhibitors) or histone deacetylases (HDAC inhibitors). Their relevance has been shown by the preclinical success of HDAC and DNMT inhibitors, as well as the clinical efficacy of these agents in cancer therapy. The second area of research in pharmaco-epigenomics involves the identification of epigenetic biomarkers that could be used to diagnose cancer, estimate disease progression, or predict interpersonal variations in response to therapy (Figure 2). Both areas of research as well as their clinical implications will now be assessed in detail.

Pharmaco-epigenomic treatment possibilities in cancer. The epigenetic modifications, such as DNA-methylation and histone modifications, are involved in the different preclinical and clinical steps of cancer, offering major possibilities for cancer pharmaco-epigenomics. Epigenetically modified genes can be used as diagnostic, prognostic or predictive markers of treatment response. In addition, DNA-methyltransferase (DNMT) inhibitors and histone deacethylation (HDAC) inhibitors are already used for specific clinical indications, providing proof-of-principle for future epigenetic drug development.
Epigenetic cancer therapy
Recognition of the fundamental function of epigenetic alterations in tumor biology has opened an entirely novel research avenue in cancer therapy, which is aimed at blocking or reversing the epigenetic alterations that promote malignancy and allow cancer cells to adapt to changes in the microenvironment. The two most targeted gene families in epigenetic cancer therapy are the HDAC and DNMT gene families (Herman and Baylin, 2003). The exact mechanisms underlying the anti-tumor activity of drugs that target these genes have not yet been completely elucidated. Given the vast influence of DNA methylation and histone modifications on gene expression, many cellular pathways are likely to be involved, including pathways that control cell-cycle arrest, differentiation, apoptosis, angiogenesis and metastasis (Joseph et al., 2004; Michaelis et al., 2004; Dai et al., 2005; Duan et al., 2005; Rocchi et al., 2005; Shetty et al., 2005). Recent studies also indicate that DNA demethylation treatment can rescue growth-inhibitory effects of certain microRNAs (Saito et al., 2006).
Meanwhile, it has been established that certain tumor types respond well to DNMT and HDAC inhibitor treatments, with the best clinical efficacy seen in hematologic malignancies. The DNMT inhibitor decitabine, for instance, is approved for the treatment of patients with myelodysplastic syndrome or acute myeloid leukemia (Hackanson et al., 2005; Silverman and Mufti, 2005). Decitabine has not yet been proven to be effective in solid tumors, although disease stabilization has been observed (Momparler et al., 1997; Schrump et al., 2006). The HDAC inhibitor vorinostat (suberoylanilide hydroxamic acid) has recently been approved for treatment of cutaneous T-cell lymphoma in patients with progressive, persistent, or recurrent disease (Khan and La Thangue, 2008). Other DNMT inhibitors, such as the orally active zebularine—in contrast to the intravenous administered decitabine—are under development (Cheng et al., 2003) and their potential synergy with HDAC inhibitors is being investigated as well (Cameron et al., 1999; Shaker et al., 2003).
As epigenetic therapy can induce cancer cell reprogramming, HDAC and DNMT inhibitors could act synergistically with conventional chemotherapy. This would allow chemotherapy to be applied at lower dosages, resulting in reduced toxicity, whereas the efficacy of the combined therapy would still be increased compared with monotherapy. Monotherapies often also induce genetic and epigenetic alterations that result in selection of resistant cell clones. Interestingly, tumors that have become resistant to initial treatment with chemotherapy because of epigenetic changes might become sensitive again to the drug in the presence of epigenetic therapy (Smith et al., 2007). In melanoma, for instance, one of the standard treatments is interferon, which induces tumor apoptosis, differentiation and increases the anti-tumor immune response. Silencing of genes involved in signaling downstream of interferon, such as interferon regulatory factor 8 and XIAP-associated factor 1 trigger resistance of tumor cells to interferon therapy (Reu et al., 2006; Yang et al., 2007). However, injection of decitabine in nude mice carrying melanoma xenografts led to resensitization to interferon treatment (Reu et al., 2006). A phase I clinical trial of decitabine together with interleukin-2 in melanoma patients further showed an objective response in 31% of the patients (Gollob et al., 2006). Likewise, preclinical data also suggests that epigenetic therapy can induce radiosensitization and enhance current conventional treatment regimens (Munshi et al., 2005). Molecular mechanisms underlying this radiosensitizing potential are not yet fully understood, but can be partially explained by the silencing of DNA-repair genes. The HDAC inhibitor sodium butyrate is reported to aggravate radiation-induced damage and enhance apoptosis by inactivation of repair-related genes, such as Ku70 and Ku86 (Munshi et al., 2006). Similarly, treatment with another HDAC inhibitor, Vorinostat, can prolong the appearance of repair foci identified by phosphorylated Histone 2AX (γ-H2AX), which is indicative of reduced repair efficiency and increased radiosensitivity (Munshi et al., 2006).
Epigenetic cancer therapy can thus be considered a promising approach because of its synergy with chemotherapy, its resensitization of chemoresistant tumors, and its increase in the efficacy of radiotherapy. Despite this great potential, the nonspecific effects of epigenetic drugs are an area of concern for clinical application in patients. For instance, given the effect of global DNA hypomethylation on genomic stability, therapy-induced hypomethylation might promote tumor formation on the long run, although this hypothesis still needs verification (Eden et al., 2003; Yang et al., 2003). Epigenetic therapy might also cause activation of imprinted or silenced genes and has indeed been shown to be mutagenic and possibly even carcinogenic (Carr et al., 1988; Jackson-Grusby et al., 1997). However, these concerns should not be exaggerated, as DNMT inhibitors only act on dividing cells, while leaving other cells unaffected. Furthermore, evidence suggests that epigenetic drugs have a tendency to activate genes that have become abnormally silenced (Karpf et al., 1999; Liang et al., 2002). Although no mechanism has been shown to explain this, it is possible that the chromatin structure of aberrantly silenced genes is more susceptible to reactivation when compared with genes silenced in normal physiological conditions. However, it should be noted that patients receiving HDAC or DNMT inhibitors in the clinic have not suffered yet from any major toxicities or unexplained long-term adverse effects (Jones and Baylin, 2002; Yoo and Jones, 2006). Although caution is warranted, current clinical evidence suggests that epigenetic therapy is reasonably safe.
In regard to potential side-effects, there is great promise for specific epigenetic therapies targeted at particular genes through the use of promoter-specific transcription factors (Moore and Ullman, 2003). This strategy has, for instance, been shown to specifically reactivate MASPIN, a tumor-suppressor gene silenced by promoter methylation in aggressive epithelial tumors (Beltran et al., 2007). Hereto, Beltran et al. constructed an artificial transcription factor consisting of six zinc-finger domains targeting unique 18-bp sequences in the MASPIN promoter, linked to an activator domain. This artificial transcription factor reactivated epigenetically silenced MASPIN, induced apoptosis of cancer cells in vitro, and suppressed tumor growth in a xenograft breast cancer model in nude mice. Hence, despite some concerns about the long-term safety, epigenetic cancer therapies clearly hold great potential and the next generation of targeted therapies could overcome possible pitfalls and improve clinical efficacy and safety of epigenetic drugs.
Epigenetic cancer management
Given the important function of epigenetic alterations in cancer, it is also likely that DNA methylation and histone modifications, similar to somatic genetic alterations, can be used for the diagnosis and molecular classification of cancer, and to predict cancer progression or response to therapy. Indeed, although the epigenetic mapping of genes in a clinical research setting is challenging due to the poor preservation of chromatin structure in clinical samples, there exists a tight correlation between methylation patterns, chromatin structure, and gene expression (Szyf, 2004). DNA methylation reflects the chromatin structure of a gene and can be considered as a stable covalent DNA mark for gene activity (Geiman and Robertson, 2002). As DNA methylation is better preserved compared with histone modification and chromatin structure, even in low-quality samples, clinical epigenetic cancer research currently relies on DNA methylation for biomarker identification. But this could change in the near future with improved sample collection, better storage methods, and novel analytical methods.
We currently distinguish three potential applications for epigenetic markers in cancer management. In particular, epigenetic markers could be used as complementary diagnostic tools, prognostic markers of disease progression, and predictive markers of treatment response.
First of all, epigenetic alterations could be used to complement existing diagnostic tools for cancer detection. Sensitive PCR-based methods have been developed to detect hypermethylated CpG islands in DNA from various sources, such as blood, urine, sputum, or tumor biopsies (Herman and Baylin, 2003). These approaches have stimulated the discovery of abnormally methylated DNA sequences as tumor markers across multiple cancer types. For instance, hypermethylation of glutathione S-transferase 1 is seen in 80–90% of prostate cancer patients, whereas benign hyperplastic prostate tissue is not hypermethylated (Esteller et al., 1998; Jeronimo et al., 2001). Glutathione S-transferase 1 methylation in prostate biopsies or urine could thus help to assist in the diagnosis of malignant prostate cancer (Cairns et al., 2001). Likewise, Melotte et al. (2009) reported a new biomarker for colorectal cancer in stool samples. The authors showed that N-Myc downstream-regulated gene 4, a tumor-suppressor candidate, is frequently silenced by promoter hypermethylation in colorectal cancer. By using a methylation-specific PCR assay for N-Myc downstream-regulated gene 4, they successfully identified 53% of colorectal cancer cases and correctly predicted which of the individuals were free of cancer. In another study, a panel detecting several hypermethylated genes in breast ductal fluids correctly identified twice as many breast cancers compared with classical cytological techniques (Fackler et al., 2006). Importantly, emerging evidence indicates that epigenetic alterations occur early in carcinogenesis before other biomarkers are detectable. For example, substantial hypermethylation of the tumor-suppressor CDKN2A can already be detected in bronchial pre-neoplastic epithelium of smokers (Belinsky et al., 1998). This makes these markers very attractive for use in diagnostic panels to detect cancer in its early stage, which is obviously important, as the odds of survival are the highest at this stage. The detection of epigenetic changes in the blood, stool, or urine even offers the additional advantage that it can be used for noninvasive diagnosis.
Second, epigenetic profiles could be used as biomarkers for the prognosis of cancer patients. Given the heterogeneity of cancer and the large differences in survival observed in patients with histologically similar tumors, biomarkers that identify high-risk patients with poor survival could guide treatment selection to achieve the best possible clinical risk-benefit ratio. Current clinical practice is mainly based on immunohistological analysis, although recent progress to improve risk stratification has also been made using gene-expression (Sotiriou and Pusztai, 2009) or somatic mutation signatures (Pharoah et al., 1999; Schmidt et al., 2007). Epigenetic biomarkers could possibly complement these existing tools. For instance, hypermethylation of the APC and CDKN2A genes were shown to be associated with poor prognosis in breast and colorectal cancer, respectively (Muller et al., 2003; Wettergren et al., 2008). It has also been shown that global histone modification profiles, such as histone lysine methylation and acetylation marks, are correlated with clinical and pathological parameters of prostate cancer and can be a significant predictor of prostate cancer recurrence (Ellinger et al., 2009). Although these epigenetic profiles hold great promise, it should be mentioned that some studies used small sets of tumors, which might explain why they subsequently failed to be replicated in independent follow-up studies. Many of the markers identified in the field of epigenetics, therefore, will have to be carefully validated, preferentially in the context of randomized controlled clinical trials (Silverman et al., 2002).
Third, epigenetic alterations could also function as predictive markers to assess response to a particular cancer therapy. As it is increasingly being recognized that each tumor has its own genetic profile that needs its own specific therapy, it is expected that future cancer therapies will become tailored to the individual patient. The paradigm of personalized medicine, illustrated by the recent approval for a test that determines mutations in the KRAS gene to predict response to the EGFR inhibitor cetuximab, is an example that could apply to epigenetic markers as well (Normanno et al., 2009). It is, therefore, essential to identify epigenetic differences that explain inter-individual variation in therapy response. An excellent example is the methylation-induced silencing of the DNA-repair gene MGMT (O6-methylguanine–DNA methyltransferase) in glioma, which occurs in almost half of glioma patients (Esteller et al., 2000). MGMT is involved in the repair of DNA alkyl adducts formed by alkylating chemotherapeutic agents. Two studies have shown that silencing of MGMT through promoter hypermethylation is an independent predictive marker for response to the drugs carmustine or temozolomide (Esteller et al., 2000; Hegi et al., 2005). These findings have further been shown for cyclophosphamide (Esteller et al., 2002) and several other chemotherapies, such as cisplatin and irinotecan (Strathdee et al., 1999; Agrelo et al., 2006), illustrating the emerging theme that epigenetic inactivation of DNA-repair genes could predict response to chemotherapy. Two other DNA-repair genes that have been well studied in this respect are the so-called ‘breast cancer genes’ BRCA1 and BRCA2, which are frequently inactivated at the epigenetic level in several cancer types (Birgisdottir et al., 2006; Lee et al., 2007; Tapia et al., 2008). These genes are required for the DNA double-strand break repair processes. As a consequence, cancer cells carrying inactivated BRCA1 or BRCA2 genes are no longer capable of repairing DNA damage induced by, for instance, platinum-based compounds. Intriguingly, these cancers have recently been shown to respond well to poly(ADP-ribose) polymerase (PARP) inhibitors (Fong et al., 2009). The PARP enzyme is essential for the repair of DNA single-strand breaks. PARP inhibitors can thus enhance the cytotoxic effects of DNA damaging agents by selectively targeting cells defective in the BRCA1/2-dependent DNA-repair pathway and inhibiting their PARP-dependent repair mechanisms (Fong et al., 2009). In this context, besides mutation status, methylation changes in the BRCA1/2 promoter could also serve as an attractive biomarker to select patients eligible for PARP-targeted therapies.
Epigenetic therapy beyond the cancer cell
Although cancer research has initially focused on the growth autonomy of cancer cells, it is becoming apparent that the stroma that surrounds the cancer cells, such as fibroblasts, endothelial cells, and inflammatory cells, also has an important function in driving tumor cell proliferation (Hanahan and Weinberg, 2000). The anti-tumoral properties of novel epigenetic therapies have thus far largely been attributed to the reactivation of silenced tumor-suppressor genes in tumor cells. However, given their universal gene regulatory effects, it is likely that epigenetic therapy will also affect stromal cells.
As endothelial cells have an important function during blood vessel formation or angiogenesis, and rapidly respond to environmental changes, such as hypoxia, it is not surprising that epigenetic regulation of angiogenesis represents an important research area. Given the importance of angiogenesis in tumor progression and the clinical importance of angiogenesis inhibitors, we will here discuss the effects of epigenetic therapy on angiogenesis in greater detail.
Epigenetic therapy targeting tumor angiogenesis
Angiogenesis is a remarkably dynamic process that is tightly controlled by a balance of stimulatory and inhibitory angiogenic signals. Consequently, an imbalance in these signals will result either in shortage or excess of blood vessels, which will contribute to ischemic or malignant disorders, respectively (Carmeliet, 2005). Since 2004, the first angiogenesis inhibitors are widely being used in first-line treatments of various solid tumors in combination with chemotherapy (Kerbel, 2008). Tumor angiogenesis by itself does not initiate malignancy, but has a critical function in cancer by promoting tumor progression and metastasis (Carmeliet, 2005). Activation of the so-called angiogenic switch is considered as one of the hallmarks of cancer that promotes tumor growth and metastasis (Hanahan and Weinberg, 2000). Emerging evidence indicates that epigenetic alterations of genes involved in angiogenesis are involved in this switch, and may cause tumors to recruit new blood vessels and sustain their growth (Buysschaert et al., 2008). Glioblastomas, for instance, are typically characterized by excessive blood vessel development and frequently display epigenetic inactivation of the anti-angiogenic thrombospondin-1 (THBS-1) gene (Li et al., 1999). THBS-1 is also suppressed early in breast carcinogenesis by histone modifications (Hinshelwood et al., 2007) and THBS-1 silencing through methylation is observed in a significant portion of primary colorectal adenomas (Rojas et al., 2008). Interestingly, oxygen-glucose deprivation, which frequently occurs in tumors, was shown to increase THBS-1 promoter methylation and subsequent silencing. This transcriptional inactivation could be reversed by reoxygenation (Hu et al., 2006). Interfering with the epigenetic machinery could thus be used to reactivate silenced anti-angiogenic factors and inhibit new blood vessel growth or restore the normal function of the chaotic, disorganized microvascular network.
Several molecular mechanisms underlying the anti-angiogenic activities of epigenetic therapies have also been elucidated. For instance, the HDAC inhibitor TSA impairs blood vessel formation in vitro and in vivo by downregulating pro-angiogenic signaling factors, such as the vascular endothelial growth factor (VEGF) and by upregulating angiostatic factors, such as ADAMTS-1 (Deroanne et al., 2002; Rossig et al., 2002; Chou and Chen, 2008). Furthermore, TSA induces the expression of tumor-suppressors p53 and VHL and downregulates HIF-1α, a transcription factor that activates hypoxia-induced angiogenic signaling pathways (Kim et al., 2001). However, when interpreting the effects of HDAC inhibitors, and in particular their anti-angiogenic mode of action, a major challenge lies in determining whether they act directly on blood vessels or indirectly through tumor cells. Hellebrekers et al. (2007) recently identified several downregulated genes in tumor-conditioned endothelial cells versus normal endothelial cells, which included the anti-angiogenic genes clusterin, fibrillin 1, and quiescin Q6. They showed that expression of these genes could be reactivated by treatment with the HDAC inhibitor TSA. These findings show that the anti-angiogenic effects of HDAC inhibitors can at least in part be explained by their direct influence on endothelial gene expression. Another recently identified mechanism of action of HDAC inhibitors seems to be the impairment of endothelial progenitor cell function (Rossig et al., 2005). Adult progenitor cells possess stem cell-like properties and are able to differentiate into endothelial cells that assist in the growth of new blood vessels (Rossig et al., 2005; Young et al., 2007). HDAC inhibitors can block their differentiation into endothelial cells through repression of the transcription factor HoxA9, a master regulator of expression for endothelial-committed genes, such as eNOS, VEGFR-2, and VE-cadherin (Rossig et al., 2005).
Similar to HDAC inhibitors, DNMT inhibitors can also reactivate epigenetically silenced genes in tumors and decrease tumor growth in vitro and in vivo (Suzuki et al., 2002; Baylin, 2004). Again, these results cannot be interpreted without considering the effect of DNMT inhibitors on blood vessels. Indeed, the specific inhibitors decitabine and zebularine can decrease vessel formation and inhibit proliferation of tumor-conditioned endothelial cells by reactivation of growth-inhibiting genes, such as THBS-1, JUNB, and IGFBP3, known to be silenced in tumor-conditioned endothelial cells (Hellebrekers et al., 2006b). Furthermore, these compounds can restore expression of the epigenetically silenced intercellular adhesion molecule-1 on tumor-conditioned endothelial cells in vitro and in vivo by reversal of histone modifications in the intercellular adhesion molecule-1 promoter. This results in restored leukocyte-endothelial cell adhesion and enhanced leukocyte infiltration (Hellebrekers et al., 2006a).
Taken together, HDAC and DNMT inhibitors act on multiple cell types, most notably on tumor and endothelial cells, hereby affecting tumor angiogenesis and cancer cell survival.
Epigenetic biomarkers for anti-angiogenic therapy
The epigenetic status of a particular angiogenic growth factor itself might be a potential predictor of response to treatment. As such, a study by Rini et al. (2006) investigated the predictive effect of genetic and epigenetic inactivation of the VHL gene, a negative regulator of VEGF, in renal cell carcinoma patients treated with the monoclonal anti-VEGF antibody bevacizumab. Patients with VHL inactivation had an improved response to therapy and displayed a strong trend toward prolonged time to disease progression. Although still speculative, it is possible that VHL inactivation in tumors renders these patients more dependent on VEGF for initiating and sustaining angiogenesis and, therefore, makes them more susceptible to VEGF inhibition. Although these results need further validation, they clearly show that epigenetic biomarkers could become a valuable predictive tool for anti-VEGF therapy.
Another anti-angiogenic strategy that is currently evaluated in clinical studies is anti-integrin therapy. Integrins are cell surface receptors involved in angiogenesis and are, therefore, an attractive target to inhibit blood vessel growth in tumors. A potential biomarker for anti-integrin therapy might be the ADAM23 gene, which acts as a negative regulator of the integrinαVβIII receptor. ADAM23 is frequently silenced by promoter hypermethylation and as its silencing correlates with tumor progression, it might be associated with the acquisition of an angiogenic and metastatic phenotype (Verbisck et al., 2009). Tumors with ADAM23 hypermethylation might, therefore, depend more on αV integrin signaling for their vessel growth, which would render them more eligible for anti-integrin therapy.
Future perspectives
It is expected that in the next few years, information from DNA methylation, histone modification, and gene-expression arrays will lead to the identification of epigenetic cancer drivers, which could subsequently be targeted through epigenetic therapy or serve as biomarkers for diagnosis, prognosis or prediction of therapy response. Moreover, combining current chemo- or radiotherapy with epigenetic therapy could result in increased efficacy, whereas allowing lower doses and reducing toxicity. This approach might also delay the emergence of resistance caused by high-dose chemotherapeutic treatments. Although current epigenetic drugs are nonspecific, the prospect that promoters of particular important genes could selectively become demethylated also holds great promise. As such, pharmaco-epigenomics could overcome some of the limitations of traditional genetic-focused medicine, making it possible that epigenomic cancer management and therapy will become an essential part of clinical practice in the future. Ultimately, clinicians might be able to quickly identify the most relevant epigenetic lesions for every patient and translate them into effective personalized epigenomic medicine.
Another promising future application of pharmaco-epigenomics lies in epigenetic alterations as diagnostic, prognostic, or predictive markers in cancer. In particular, the evaluation of genome-wide epigenetic profiles to identify the most common and relevant epigenetically altered genes in a certain cancer type is expected to dramatically increase our knowledge in this field. Large-scale efforts to completely characterize the (epi)genomic origin of cancer are underway. For instance, The Cancer Genome Atlas project has already identified methylation profiles with strong prognostic value in ovarian cancer, and many other cancer types are currently being analyzed. Eventually, these novel epigenetic signatures, regardless of their diagnostic, prognostic, or predictive nature, will have to be validated on large sets of tumors, preferentially collected in the context of controlled clinical studies.
References
Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF et al. (2006). Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA 103: 8822–8827.
Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J et al. (2003). Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J 22: 6335–6345.
Baylin SB (2004). Reversal of gene silencing as a therapeutic target for cancer--roles for DNA methylation and its interdigitation with chromatin. Novartis Found Symp 259: 226–233;discussion 234–237, 285–288.
Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E et al. (1998). Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci USA 95: 11891–11896.
Beltran A, Parikh S, Liu Y, Cuevas BD, Johnson GL, Futscher BW et al. (2007). Re-activation of a dormant tumor suppressor gene maspin by designed transcription factors. Oncogene 26: 2791–2798.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE (2006). Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res 8: R38.
Buysschaert I, Schmidt T, Roncal C, Carmeliet P, Lambrechts D (2008). Genetics, epigenetics and pharmaco-(epi)genomics in angiogenesis. J Cell Mol Med 12 (6B): 2533–2551.
Cairns P, Esteller M, Herman JG, Schoenberg M, Jeronimo C, Sanchez-Cespedes M et al. (2001). Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin Cancer Res 7: 2727–2730.
Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB (1999). Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 21: 103–107.
Carmeliet P (2005). Angiogenesis in life, disease and medicine. Nature 438: 932–936.
Carr BI, Rahbar S, Asmeron Y, Riggs A, Winberg CD (1988). Carcinogenicity and haemoglobin synthesis induction by cytidine analogues. Br J Cancer 57: 395–402.
Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE et al. (2003). Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst 95: 399–409.
Chou CW, Chen CC (2008). HDAC inhibition upregulates the expression of angiostatic ADAMTS1. FEBS Lett 582: 4059–4065.
Dai Y, Rahmani M, Dent P, Grant S (2005). Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol 25: 5429–5444.
Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N et al. (2002). Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21: 427–436.
Duan H, Heckman CA, Boxer LM (2005). Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol 25: 1608–1619.
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003). Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300: 455.
Ellinger J, Kahl P, von der Gathen J, Rogenhofer S, Heukamp LC, Gutgemann I et al. (2009). Global levels of histone modifications predict prostate cancer recurrence. Prostate 70: 61–69.
Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG (1998). Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res 58: 4515–4518.
Esteller M, Gaidano G, Goodman SN, Zagonel V, Capello D, Botto B et al. (2002). Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. J Natl Cancer Inst 94: 26–32.
Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V et al. (2000). Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343: 1350–1354.
Fackler MJ, Malone K, Zhang Z, Schilling E, Garrett-Mayer E, Swift-Scanlan T et al. (2006). Quantitative multiplex methylation-specific PCR analysis doubles detection of tumor cells in breast ductal fluid. Clin Cancer Res 12 (11 Part 1): 3306–3310.
Feinberg AP (2007). Phenotypic plasticity and the epigenetics of human disease. Nature 447: 433–440.
Feinberg AP, Tycko B (2004). The history of cancer epigenetics. Nat Rev Cancer 4: 143–153.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M et al. (2009). Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G et al. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400.
Geiman TM, Robertson KD (2002). Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem 87: 117–125.
Gluckman PD, Hanson MA, Cooper C, Thornburg KL (2008). Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73.
Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M, Moran K et al. (2006). Phase I trial of sequential low-dose 5-aza-2′-deoxycytidine plus high-dose intravenous bolus interleukin-2 in patients with melanoma or renal cell carcinoma. Clin Cancer Res 12: 4619–4627.
Hackanson B, Robbel C, Wijermans P, Lubbert M (2005). In vivo effects of decitabine in myelodysplasia and acute myeloid leukemia: review of cytogenetic and molecular studies. Ann Hematol 84 (Suppl 1): 32–38.
Hanahan D, Weinberg RA (2000). The hallmarks of cancer. Cell 100: 57–70.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352: 997–1003.
Hellebrekers DM, Castermans K, Vire E, Dings RP, Hoebers NT, Mayo KH et al. (2006a). Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res 66: 10770–10777.
Hellebrekers DM, Jair KW, Vire E, Eguchi S, Hoebers NT, Fraga MF et al. (2006b). Angiostatic activity of DNA methyltransferase inhibitors. Mol Cancer Ther 5: 467–475.
Hellebrekers DM, Melotte V, Vire E, Langenkamp E, Molema G, Fuks F et al. (2007). Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res 67: 4138–4148.
Herman JG, Baylin SB (2003). Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349: 2042–2054.
Hinshelwood RA, Huschtscha LI, Melki J, Stirzaker C, Abdipranoto A, Vissel B et al. (2007). Concordant epigenetic silencing of transforming growth factor-beta signaling pathway genes occurs early in breast carcinogenesis. Cancer Res 67: 11517–11527.
Hu CJ, Chen SD, Yang DI, Lin TN, Chen CM, Huang TH et al. (2006). Promoter region methylation and reduced expression of thrombospondin-1 after oxygen-glucose deprivation in murine cerebral endothelial cells. J Cereb Blood Flow Metab 26: 1519–1526.
Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R (1997). Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc Natl Acad Sci USA 94: 4681–4685.
Jeronimo C, Usadel H, Henrique R, Oliveira J, Lopes C, Nelson WG et al. (2001). Quantitation of GSTP1 methylation in non-neoplastic prostatic tissue and organ-confined prostate adenocarcinoma. J Natl Cancer Inst 93: 1747–1752.
Jones PA, Baylin SB (2002). The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428.
Jones PA, Baylin SB (2007). The epigenomics of cancer. Cell 128: 683–692.
Joseph J, Mudduluru G, Antony S, Vashistha S, Ajitkumar P, Somasundaram K (2004). Expression profiling of sodium butyrate (NaB)-treated cells: identification of regulation of genes related to cytokine signaling and cancer metastasis by NaB. Oncogene 23: 6304–6315.
Karpf AR, Peterson PW, Rawlins JT, Dalley BK, Yang Q, Albertsen H et al. (1999). Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci USA 96: 14007–14012.
Kerbel RS (2008). Tumor angiogenesis. N Engl J Med 358: 2039–2049.
Khan O, La Thangue NB (2008). Drug Insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat Clin Pract Oncol 5: 714–726.
Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW et al. (2001). Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med 7: 437–443.
Kouzarides T (2007). Chromatin modifications and their function. Cell 128: 693–705.
Lee MN, Tseng RC, Hsu HS, Chen JY, Tzao C, Ho WL et al. (2007). Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin Cancer Res 13: 832–838.
Li Q, Ahuja N, Burger PC, Issa JP (1999). Methylation and silencing of the thrombospondin-1 promoter in human cancer. Oncogene 18: 3284–3289.
Liang G, Gonzales FA, Jones PA, Orntoft TF, Thykjaer T (2002). Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Res 62: 961–966.
Melotte V, Lentjes MH, van den Bosch SM, Hellebrekers DM, de Hoon JP, Wouters KA et al. (2009). N-Myc downstream-regulated gene 4 (NDRG4): a candidate tumor suppressor gene and potential biomarker for colorectal cancer. J Natl Cancer Inst 101: 916–927.
Michaelis M, Michaelis UR, Fleming I, Suhan T, Cinatl J, Blaheta RA et al. (2004). Valproic acid inhibits angiogenesis in vitro and in vivo. Mol Pharmacol 65: 520–527.
Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J (1997). Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs 8: 358–368.
Moore M, Ullman C (2003). Recent developments in the engineering of zinc finger proteins. Brief Funct Genomic Proteomic 1: 342–355.
Muller HM, Widschwendter A, Fiegl H, Ivarsson L, Goebel G, Perkmann E et al. (2003). DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res 63: 7641–7645.
Munshi A, Kurland JF, Nishikawa T, Tanaka T, Hobbs ML, Tucker SL et al. (2005). Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res 11: 4912–4922.
Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE (2006). Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther 5: 1967–1974.
Nguyen CT, Weisenberger DJ, Velicescu M, Gonzales FA, Lin JC, Liang G et al. (2002). Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res 62: 6456–6461.
Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F (2009). Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol 6: 519–527.
Pharoah PD, Day NE, Caldas C (1999). Somatic mutations in the p53 gene and prognosis in breast cancer: a meta-analysis. Br J Cancer 80: 1968–1973.
Reu FJ, Bae SI, Cherkassky L, Leaman DW, Lindner D, Beaulieu N et al. (2006). Overcoming resistance to interferon-induced apoptosis of renal carcinoma and melanoma cells by DNA demethylation. J Clin Oncol 24: 3771–3779.
Rini BI, Jaeger E, Weinberg V, Sein N, Chew K, Fong K et al. (2006). Clinical response to therapy targeted at vascular endothelial growth factor in metastatic renal cell carcinoma: impact of patient characteristics and Von Hippel-Lindau gene status. BJU Int 98: 756–762.
Rocchi P, Tonelli R, Camerin C, Purgato S, Fronza R, Bianucci F et al. (2005). p21Waf1/Cip1 is a common target induced by short-chain fatty acid HDAC inhibitors (valproic acid, tributyrin and sodium butyrate) in neuroblastoma cells. Oncol Rep 13: 1139–1144.
Rojas A, Meherem S, Kim YH, Washington MK, Willis JE, Markowitz SD et al. (2008). The aberrant methylation of TSP1 suppresses TGF-beta1 activation in colorectal cancer. Int J Cancer 123: 14–21.
Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Forstermann U et al. (2002). Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res 91: 837–844.
Rossig L, Urbich C, Bruhl T, Dernbach E, Heeschen C, Chavakis E et al. (2005). Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J Exp Med 201: 1825–1835.
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA et al. (2006). Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9: 435–443.
Sasaki H, Matsui Y (2008). Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet 9: 129–140.
Schmidt MK, Tollenaar RA, de Kemp SR, Broeks A, Cornelisse CJ, Smit VT et al. (2007). Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J Clin Oncol 25: 64–69.
Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF et al. (2006). Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res 12: 5777–5785.
Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M et al. (2005). Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435: 1262–1266.
Shaker S, Bernstein M, Momparler LF, Momparler RL (2003). Preclinical evaluation of antineoplastic activity of inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone deacetylation (trichostatin A, depsipeptide) in combination against myeloid leukemic cells. Leuk Res 27: 437–444.
Shetty S, Graham BA, Brown JG, Hu X, Vegh-Yarema N, Harding G et al. (2005). Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol Cell Biol 25: 5404–5416.
Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R et al. (2002). Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20: 2429–2440.
Silverman LR, Mufti GJ (2005). Methylation inhibitor therapy in the treatment of myelodysplastic syndrome. Nat Clin Pract Oncol 2 (Suppl 1): S12–S23.
Smith LT, Otterson GA, Plass C (2007). Unraveling the epigenetic code of cancer for therapy. Trends Genet 23: 449–456.
Sotiriou C, Pusztai L (2009). Gene-expression signatures in breast cancer. N Engl J Med 360: 790–800.
Strahl BD, Allis CD (2000). The language of covalent histone modifications. Nature 403: 41–45.
Strathdee G, MacKean MJ, Illand M, Brown R (1999). A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene 18: 2335–2341.
Stratton MR, Campbell PJ, Futreal PA (2009). The cancer genome. Nature 458: 719–724.
Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP et al. (2002). A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 31: 141–149.
Szyf M (2004). Toward a discipline of pharmacoepigenomics. Curr Pharmacogenomics 2: 357–377.
Tapia T, Smalley SV, Kohen P, Munoz A, Solis LM, Corvalan A et al. (2008). Promoter hypermethylation of BRCA1 correlates with absence of expression in hereditary breast cancer tumors. Epigenetics 3: 157–163.
Verbisck NV, Costa ET, Costa FF, Cavalher FP, Costa MD, Muras A et al. (2009). ADAM23 negatively modulates alpha(v)beta(3) integrin activation during metastasis. Cancer Res 69: 5546–5552.
Wettergren Y, Odin E, Nilsson S, Carlsson G, Gustavsson B (2008). p16INK4a gene promoter hypermethylation in mucosa as a prognostic factor for patients with colorectal cancer. Mol Med 14: 412–421.
Wilson AS, Power BE, Molloy PL (2007). DNA hypomethylation and human diseases. Biochim Biophys Acta 1775: 138–162.
Yang AS, Estecio MR, Garcia-Manero G, Kantarjian HM, Issa JP (2003). Comment on ‘Chromosomal instability and tumors promoted by DNA hypomethylation’ and ‘Induction of tumors in nice by genomic hypomethylation’. Science 302: 1153; author reply 1153.
Yang D, Thangaraju M, Greeneltch K, Browning DD, Schoenlein PV, Tamura T et al. (2007). Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res 67: 3301–3309.
Yoo CB, Jones PA (2006). Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 5: 37–50.
Young PP, Vaughan DE, Hatzopoulos AK (2007). Biologic properties of endothelial progenitor cells and their potential for cell therapy. Prog Cardiovasc Dis 49: 421–429.
Acknowledgements
BC is supported by the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT) and DL by the Fund for Scientific Research in Flanders (FWO), Belgium.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Claes, B., Buysschaert, I. & Lambrechts, D. Pharmaco-epigenomics: discovering therapeutic approaches and biomarkers for cancer therapy. Heredity 105, 152–160 (2010). https://doi.org/10.1038/hdy.2010.42
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.42
Keywords
This article is cited by
-
Signification of Hypermethylated in Cancer 1 (HIC1) as Tumor Suppressor Gene in Tumor Progression
Cancer Microenvironment (2012)
