
Abstract
Eukaryotic DNA is organized as a nucleoprotein polymer termed chromatin with nucleosomes serving as its repetitive architectural units. Cellular differentiation is a dynamic process driven by activation and repression of specific sets of genes, partitioning the genome into transcriptionally active and inactive chromatin domains. Chromatin architecture at individual genes/loci may remain stable through cell divisions, from a single mother cell to its progeny during mitosis, and represents an example of epigenetic phenomena. Epigenetics refers to heritable changes caused by mechanisms distinct from the primary DNA sequence. Recent studies have shown a number of links between chromatin structure, gene expression, extracellular signaling, and cellular differentiation during eye development. This review summarizes recent advances in this field, and the relationship between sequence-specific DNA-binding transcription factors and their roles in recruitment of chromatin remodeling enzymes. In addition, lens and retinal differentiation is accompanied by specific changes in the nucleolar organization, expression of non-coding RNAs, and DNA methylation. Epigenetic regulatory mechanisms in ocular tissues represent exciting areas of research that have opened new avenues for understanding normal eye development, inherited eye diseases and eye diseases related to aging and the environment.
Similar content being viewed by others


Introduction
Cellular differentiation and organogenesis require precise temporal and spatial control of gene expression. This process is confined to the nucleus, in which ∼2 m of human genomic DNA is organized as the nucleoprotein polymer structure known as chromatin. Primary sequence of our DNA stores the raw genetic information. However, the heritable content of the eukaryotic nucleus is not limited to its primary DNA sequence, but also includes DNA methylation, histone modifications, the presence of specific macromolecules including RNAs and proteins, and their distribution throughout the plasma membrane and cytoplasm. These additional sources of inherited information function through epigenetic regulatory mechanisms. Although the genetic information generally remains constant during development, epigenetic information changes during specification of individual cell lineages and during cellular differentiation in response to a variety of cues including cell-to-cell interactions and extracellular signaling. Chromatin remodeling refers to a spectrum of dynamic processes that alter local and/or global chromatin structure (see Urnov and Wolffe, 2001). These processes, mediated by chromatin remodeling enzymes, are central for understanding of development of multicellular organisms. Epigenetic regulation is a reversible process as it is possible to reach the status of pluripotency of the fertilized egg from the majority of terminally differentiated cells through the process of nuclear reprogramming.
The vertebrate eye represents an excellent system to study both genetic and epigenetic modes of gene regulation at the molecular level as regulatory genes and their hierarchies are among the best understood in mammalian systems. Interestingly, lens fiber cell differentiation culminates with the controlled self-destruction of the nuclei localized in the organelle-free zone of the lens. In contrast, terminally differentiated rod photoreceptors of nocturnal animals require an unusual inverse reorganization of their euchromatin/heterochromatin architecture to optimize the transmission of light to the photodetection apparatus. Furthermore, specific ocular cells can be transdifferentiated into other cell types such as the lens. Collectively, these findings nicely illustrate the usefulness of the eye as an advantageous system for epigenetic studies.
The regulatory mechanisms of vertebrate eye development have been previously described, from the genetic perspective, in a number of reviews focused on the early stages of eye development (see Chow and Lang, 2001; Graw, 2003), lens formation (see Grainger, 1992), function of key DNA-binding transcription factors (TFs) (see Cvekl and Piatigorsky, 1996; Ashery-Padan and Gruss, 2001; Livesey and Cepko, 2001; Hatakeyama and Kageyama, 2004; Donner et al., 2006; Medina-Martinez and Jamrich, 2007; Moore and Vetter, 2007; Mardi and Seth, 2009), and evolutionary aspects of eye formation (see Gehring and Ikeo, 1999; Jonasova and Kozmik, 2008). In contrast, epigenetic regulatory mechanisms have received far less attention (see Cvekl and Duncan, 2007). Thus, the focus of the present review is to summarize these mechanisms, mostly reflecting studies published during the last 3 years. First, we introduce principles of vertebrate eye development, formation of progenitor cell lineages, individual cell type specification, and their terminal differentiation. We also provide a list of key DNA-binding TFs that regulate lens and retinal development to understand how individual chromatin remodeling enzymes are recruited to specific regions in chromatin. Next, we summarize recent original studies of epigenetic regulatory mechanisms related to ocular development. Finally, we discuss advances in methods studying chromatin structure and their implementation for studies of eye development and disease.
Vertebrate eye development and specific DNA-binding TFs
The vertebrate eye is formed from cells of the neuronal ectoderm origin, which give rise to the retina and epithelium of iris. Surface ectoderm provides lens progenitor cells, and, subsequently, corneal epithelial cells. Neural crest-derived periocular mesenchyme is required for the formation of corneal endothelium and keratocytes, stroma of the iris, ciliary body, and trabecular meshwork cells. The key steps in the formation of ocular progenitor cells and their subsequent proliferation and differentiation are regulated by specific DNA-binding TFs that act as transcriptional activators, repressors, and in many instances as dual activators and repressors. Currently, it is estimated that the human genome contains between 1700 and 1900 genes encoding DNA-binding TFs. A manually curated database lists 1391 such genes (Vaquerizas et al., 2009) from which 349 (∼25%) show tissue-restricted expression patterns. In contrast, 123 genes (∼9%) show distinct expression in a single tissue. Interestingly, there is no truly eye-specific DNA-binding TF although genetic loss-of-function studies have identified a number of these factors with distinct panocular or more subtle eye-specific defects. A list of key specific DNA-binding TFs that regulate lens and/or retinal development is given in Table 1. A significant number of these genes, that is Pax6, pRb, Prox1, Six3, Sox2, and Sox11, control cell autonomy in both lens and retinal development. In contrast, c-Maf, Hsf4, Pitx3, and Sox1 regulate lens but not retinal development. Similarly, Crx, Mitf, Nrl, Pax2, Vax1, Vax2, and Vsx2 regulate various aspects of retinal development. At least three genes, Hes1, Lhx2, Rax, regulate cell-autonomously retinal and indirectly lens development. In contrast, AP-2α regulates cell-autonomously lens (Pontoriero et al., 2008) and indirectly general retinal development (Bassett et al., 2007).
Lens- and retinal-specific functions of these genes are thought to be controlled through temporally and spatially restricted extracellular signaling through seven canonical signal transduction pathways (FGF/MAPK/Ras, JAK/STAT, Notch, nuclear receptors, Shh, TGF-β, and Wnt), and through synergistic interactions between smaller groups of co-expressed DNA-binding TFs (for example, Pax6, c-Maf, and Sox1 in lens; and Crx, Nrl, and Nr2e3 in photoreceptors). Extracellular signaling temporally and spatially controls expression territories of these DNA-binding TFs and regulates their post-translational modifications (PTMs) such as acetylation, glycosylation, phosphorylation, sumoylation, and ubiqitination. These modifications are engaged in a variety of protein–protein interactions and modulate function of individual TFs. Signaling is also thought to regulate various activities of chromatin remodeling enzymes (see Simone, 2006). Elucidating the functional interactions between signal-regulated (Table 2) and lineage-specific (Table 1) TFs elaborates the idea of insufficiency of a single activator and reveals cooperative interactions between multiple specific DNA-binding TFs and chromatin remodeling enzymes that either promote or repress transcription (Barolo and Posakony, 2002).
A large body of data demonstrate unique roles for the TF Pax6 (Glaser et al., 1994; Quinn et al., 1996; Collinson et al., 2001; Shaham et al., 2009). Pax6 expression is regulated by FGF signaling (Faber et al., 2001) and its downstream genetic network includes a number of lineage-restricted DNA-binding factors (Lachke and Maas, 2009). An integrative model to explain multiple functions of Pax6 and other genes in eye development has been recently proposed for the Drosophila Pax6 homologue, eyeless (ey), and other members of the retinal determination network (Firth and Baker, 2009). The central premise of this model is that three distinct combinatorial codes regulate expression of individual retinal determination genes: TGF-β and Hedgehog, TGF-β and Ras, and TGF-β, Hedgehog, Ras and Notch. Thus, it would be interesting to identify upstream signals that regulate Pax6, Six3, and Sox2 expression in lens lineage (Rajagopal et al., 2009) and Rax, Pax6, Lhx2, and Hes1 in retinal progenitor cells (RPCs) and how expression of these genes regulate the signal transduction pathways introduced earlier (Tang et al., 2010).
Formation of retinal and lens progenitor cells
Vertebrate retinas originate from a single field of primitive neuroectoderm localized in the anterior part of the neural plate called the retinal field or prospective retina (see Lupo et al., 2006; Fuhrmann, 2008; Niederreither and Dolle, 2008). This field is established as a result of early neural patterning that is mediated through Wnts, FGFs, TGF-β/BMPs, retinoic acid, extensive cell migration (Rembold et al., 2006), and the coordinated expression of DNA-binding TFs such as Rax, Six3, Pax6, Six6, Hes1, and Lhx2 (Masse et al., 2007). Next, separation of the single eye field into two symmetrical retinal primordia is achieved as a combination of at least two distinct mechanisms that use sonic hedgehog (Shh) and TGF-β (nodal) signaling. Neural fold elevation and subsequent formation of the open neural tube is followed by additional patterning events in which the eye fields develop together with the forebrain and form a pair of optic vesicles (Yun et al., 2009) comprised of retinal and optic stalk progenitor cells (see Figure 1a).

A schematic diagram of mammalian eye development (a–c) and structure of the mature retina (d). See text for details, periocular mesenchyme (POM). Ganglion cells mark the ganglion cell layer (GCL) and their axons form the optic nerve, targeting synaptic connections in the optic chiasm of the brain. The inner plexiform layer (IPL) contains several substrata where ganglion cells synapse with bipolar cells of the inner nuclear layer (INL). The INL also contains amacrine and horizontal cells, which form horizontal connections. Bipolar cells are interneurons that also synapse with rod- and cone-photoreceptors of the outer nuclear layer (ONL), forming connections in the outer plexiform layer (OPL). Photoreceptor cell nuclei (mostly rods seen here) comprise the ONL, whereas their inner segments (IS) and sensory outer segments (OS) are situated adjacent to the retinal pigment epithelium (RPE). Muller glial cell processes span the neural retina from the GCL to the ONL/OS boundary. Their nuclei tend to position in the INL.
Lens progenitor cells originate from the pre-placodal region, a band of cells that encircles the anterior neural plate facilitated by FGF signaling in synchrony with inhibition of BMP and Wnt signaling (see Streit, 2004, 2007). This inhibition is provided by the lateral mesoderm and neuroectoderm. The pre-placodal region also gives rise to the anterior pituitary, olfactory neurons, inner ear, and the trigeminal and epibranchial cranial placodes. Lineage tracing studies in chick and zebrafish have shown that anterior pituitary, olfactory, and lens progenitor cells are localized all across the pre-placodal region and migrate into their final destinations to establish the individual placodes. Lens placode formation is thus the first morphologically detectable stage of the embryonic lens. At the genetic level, lens placode formation is controlled by cell-specific expression of at least two TFs, Pax6, and Six3 (Liu et al., 2006). Lens placodes are formed precisely at the region of surface ectoderm where optic vesicles contact the surface ectoderm (Chauhan et al., 2009). Lens placodes increase their size through cell proliferation followed by reciprocal invaginations as both the lens placode and the optic vesicle buckle inward (see Figure 1b). The invagination of the lens placode produces the lens pit, a precursor stage for the formation of the lens vesicle comprised of lens precursor cells. The invagination of the optic vesicle is required for the formation of the bilayered optic cup. Its posterior layer forms the retinal pigmented epithelium (RPE) and its anterior portion forms the neuroretina. The optic cup formation is completed by a circumferential growth of both neuroretina and RPE. Defects in the formation of either the lens placode or optic cup because of mutations in TFs that control these processes (Rax, Pax6, and Lhx2) lead to anopthalmia. Lens differentiation produces only a single cell type, lens fiber cells. In contrast, optic cup is divided into three regions, neuroretina, RPE, and optic stalk (Figure 1b). Differentiation of the neuroretina generates six major cell types, that is amacrine, bipolar, horizontal, photoreceptor cells, retinal ganglion cells (RGC), and Muller radial glia cells. Amacrine, bipolar, ganglion, and photoreceptors cells are produced as many different subtypes. Recent reviews on retinal (see Harada et al., 2007; Lamb et al., 2007) and lens (see Cvekl and Duncan, 2007) development can be found elsewhere.
Lens differentiation
The lens vesicle is polarized with its anterior cells retaining proliferative capacity. In contrast, the posterior cells of the lens vesicle exit the cell cycle (see Griep, 2006) and terminally differentiate into the primary lens fibers (Figure 1c). Subsequently, the anterior cells ‘differentiate’ into the lens epithelium (see Lovicu and McAvoy, 2005). In addition to these processes, lens growth continues throughout the lifespan, as lens epithelial cells continue to proliferate moving toward the lens equator where they undergo terminal differentiation, virtually like the primary lens fiber cells, to generate the secondary lens fibers. The lens and the retina clearly influence each other's development and growth. Lens fiber cell differentiation is regulated by a combination of multiple FGFs and BMPs originating from the neuroretina (see Robinson, 2006). Lens fiber cell differentiation is marked by cellular elongation, expression of crystallins, and other lens-specific structural proteins (CP49, filensin, and MIP), and DNase IIβ, an enzyme required for denucleation of the lens fibers. c-Maf, GATA-3, Hsf4, Pitx3, Prox1, and Sox1 are essential for various aspects of lens fiber differentiation (see Table 1).
Retinal differentiation
RPCs are multipotent and capable of producing different cell types up to the final cell division. Recent data indicate intrinsic heterogeneity of RPCs (Oron-Karni et al., 2008). Neurogenesis and gliogenesis in retina are antagonistically regulated by at least eight members of the basic helix-loop-helix (bHLH) family of TFs such as Hes1, Hes5, Hesr2, Mash1, Math3, Math5, Neurod1, and Neurod4 (Table 1), acting either as activators and repressors. These TFs represent the third largest group of TFs encoded by over 200 genes (Vaquerizas et al., 2009). They can form various homo- and heterodimer complexes with other members of bHLH families and Id (HLH) proteins (without basic DNA-binding domain) and bind ‘E-box’ recognition sites. Hes1 and Hes5, downstream targets of Notch signaling (Table 2), are expressed in the progenitor cells, inhibit neuronal differentiation, and promote formation of glial cells including Muller cells. In contrast, in the absence of active Notch signaling, Atoh7/Math5, Mash1, Neurod1, and Neurod4 activators induce neuronal-specific gene expression. To explain retinal cell-type specification, a combinatorial model based on cooperative interactions between bHLH and homeodomain-containing proteins has been proposed (Harada et al., 2007). Studies of homeodomain-containing proteins showed distinct expression domains of Pax6, Six3, Prox1, Rax, Vsx2, Lim1, Crx, and Otx2 across individual retinal layers (Figure 1d, ganglion cell layer; inner nuclear layer, INL; and outer nuclear layer, ONL). Early homeodomain proteins (for example, Pax6) influence the expression of some bHLH family members, and unique combinations of bHLH activators are thought to determine cell fate within the individual retinal layers as described below.
RGCs are the first post-mitotic cells generated from the RPCs residing in the neuroretina portion of the optic cup and are followed by specifications of horizontal cells, cone photoreceptors, amacrine cells, bipolar cells, rod photoreceptors, and Muller glial cells (Harada et al., 2007). RGC cell fate is determined by Pax6 and Atoh7/Math5 (Wang et al., 2001; Matter-Sadzinski et al., 2005), with Pou4f1 and Pou4f2 used to produce diversity within the RGCs (Badea et al., 2009). Adjacent amacrine cells are marked by Pax6, Six3, Prox1 and Math3, and Neurod1. Horizontal cells express slightly different combinations of Pax6, Six3, Lim1, and Math3. Photoreceptor cells are marked by homeodomain proteins Crx and Otx2, and bHLH factors Mash1 and Neurod1. Bipolar cells are characterized by co-expression of Vsx2 and bHLH Mash1 and Math3. The RPCs that sustain Hes1, Hes5, and Hesr2 expression together with homeodomain-containing Rax will become Muller glial cells (see Harada et al., 2007). The power of this combinatorial model is that the outputs are determined by the presence of specific combinations of factors and absence of some factors that otherwise promote alternative cell fates (Riesenberg et al., 2009). However, we have very limited knowledge about downstream targets of these combined TFs and upstream signaling that regulates expression and/or activities of these lineage-specific TFs. In addition, several other TFs have been implicated in retinogenesis, for example, forkhead Foxn4 and high-mobility group proteins (HMG)-containing Sox9 are important for differentiation of amacrine and Muller cells, respectively. Dorso-ventral patterning events of the optic vesicle are also regulated by specific TFs. The posterior part of the optic cup differentiates into RPE cells (Figures 1b and c) marked early by expression of Pax2 and Pax6, and later by bHLH factor Mitf and homeodomain-containing Otx2 (Martinez-Morales et al., 2003). Optic stalk development (Figure 1b) is regulated by Pax2, Tbx2, Vax1, and Vax2 (Mui et al., 2005). A variety of extracellular signals (for example, FGFs, GDF11 from the TGF-β superfamily, BMP4, BMP7, and CNTF) are exchanged between individual ocular cells and surrounding tissues including autocrine signaling and control retinal cell proliferation, differentiation, and apoptosis as described elsewhere (see Harada et al., 2007).
Taken together, synergistic interactions between lineage-specific and signal-regulated DNA-binding TFs represent essential, though not sufficient, mechanism of specific gene regulation during individual ocular cell lineage specification and cellular differentiation. A significant number of specific DNA-binding factors recruit a plethora of chromatin remodeling complexes that alter both local and global chromatin structures, some of which can be propagated through the cell cycle.
Epigenetic regulatory mechanisms
Epigenetic regulation provides a framework to understand many general questions of gene regulation during embryonic development. First, both undifferentiated and differentiated cells possess a ‘cell memory’ of their identity. This is required for maintenance of the differentiated state of individual cell types and for pluripotency of uncommitted cells. However, some ocular cells, such as iris and RPE, can ‘transdifferentiate’ into lens cells (see Sanchez Alvarado and Tsonis, 2006). In contrast, ‘nuclear reprogramming’ reverses the differentiated state back to the state of totipotentent embryonic stem cells (Yu et al., 2007). Homeostasis of both embryonic and adult stem cells requires tight epigenetic control (Bernstein et al., 2006). Second, condensed chromatin creates an environment that severely restricts access of specific DNA-binding TFs to their binding sites. This compacted chromatin can be converted into an environment permissive for gene expression by using several distinct mechanisms collectively termed chromatin remodeling (see Urnov and Wolffe, 2001). Third, cellular differentiation is characterized by a simultaneous activation of cohorts of genes. This process could be very efficient if tightly coordinated within the nucleus (see de Laat and Grosveld, 2003; Kosak and Groudine, 2004). Recent studies of these processes provided a number of answers and testable hypotheses with epigenetic regulatory mechanisms having pivotal roles.
The primary unit of chromatin structure is a single nucleosome core particle with a molecular weight of 210 kDa. The first level of chromatin structure is the 11 nm fiber represented by a nucleosomal array, or ‘beads on a string.’ The 11 nm fiber can be coiled further like a 30-nm-thick solenoid. Condensation of individual solenoids spatially organizes the 100- and 400-nm-thick interphase chromatin fibers within the nucleus. Each nucleosome consists of a cylindrical core histone octamer with 147 bp of supercoiled DNA wrapped around its outer surface. Each nucleosome is built from a central heterotetramer of histones H3 and H4, (H3–H4)2, sandwiched between a pair of histone H2A–H2B heterodimers. Histones are small basic proteins comprised of a central globular domain flanked by N- and C-terminal peptides of variable lengths, the histone ‘tails.’ Crystallographic studies provide detailed information both related to the protein–protein interactions between individual core histones, H2A, H2B, H3, and H4, and protein–DNA interactions (Luger et al., 1997; Davey et al., 2002); however, the histone tails are not essential for the octamer formation. Biochemical assays generated large body of data demonstrating that histone tails as well as their globular domains are targets for covalent PTMs that have important functions to regulate accessibility of chromatin required to control gene expression.
Regulation of DNA accessibility through a variety of mechanisms
The primary question is how are cis-binding sites of specific DNA-binding TFs (see examples in Tables 1 and 2) recognized in chromatin? The answer involves the regulation of DNA accessibility to the proteins that regulate gene expression, with many of them forming steady-state complexes that can be 10 times larger than the individual nucleosome core particle. Physicochemical studies suggest that one moderately sized protein can traverse the entire nucleus in a matter of seconds (see van Holde and Zlatanova, 2006). As there are many copies of most TFs, the bulk chromatin can be effectively searched in a relatively brief period of time. Experimental data also support the idea that chromatin is a highly dynamic structure with several parallel mechanisms assisting the DNA-binding factors to localize their target genes (see Hager et al., 2009).
Transient site exposure by dissociation and re-binding (‘breathing’ or ‘unwrapping’ of the DNA from the nucleosome octamer) near the exit of DNA from the nucleosome and adjacent to the nucleosomal dyad (see Luger, 2006) is consistent with the observations that nucleosome core particles are thermodynamically marginally stable structures (see van Holde and Zlatanova, 2006). It has been suggested that this incipient instability is an essential property of chromatin. Another way to free up DNA sequences that were previously bound by the histone octamer is to ‘slide’ the histone octamer along the DNA (see Luger, 2006). This process can occur spontaneously or can be catalyzed by ATP-dependent DNA helicases of chromatin remodelers (see below). Less is known about changes in the higher-order chromatin architecture that are thought to precede changes at the level of individual nucleosomes that fall within interacting proximal and distal regulatory regions of transcribed genes.
Alternatively, certain DNA-binding TFs (for example, FoxA1 and FoxA2) have been shown to act as ‘pioneering’ factors to ‘open’ closed chromatin structure (Cirillo et al., 2002; Sekiya et al., 2009). Spontaneous release of linker histone H1 can be followed by binding of members of HMG family (see below) near the entry/exit of DNA from the nucleosome (see van Holde and Zlatanova, 2006) and these reversible interactions can influence the process of initial recognition of regulatory site(s) on the nucleosomal DNA. These models on accessibility of DNA wrapped on the histone octamer are not mutually exclusive.
Histone code hypothesis and post-translational histone modifications
Core histone post-translational modifications (PTMs) serve as a major source of epigenetic information. Acetylation, ADP ribosylation, methylation, phosphorylation, sumoylation, and ubiquitination of specific amino acid residues were established for at least 45 residues in the H2A, H2B, H3, and H4 core histones. There are 11 known modifications of the linker histone H1. There are over 150 known histone PTMs including novel modifications outside of histone tails. Histone tail PTMs can be classified as ‘bulky’ such as sumoylation and ubiquitination. ‘Smaller’ PTMs such as mono-, di-, and tri-methyl moieties dramatically alter reactivity of the modified lysine and arginine residues. Detailed lists of these modifications are available (see Appendix 2 in Allis et al., 2007).
A variety of molecular mechanisms of histone PTMs have been proposed to explain their effects on transcription (see Margueron et al., 2005; Luger, 2006). These range from rather non-specific to highly selective mechanisms (Fischle et al., 2003). These PTMs can directly affect general DNA accessibility by altering protein–DNA interactions and influence formation of higher-order nucleosome–nucleosome interactions. The selective mechanisms are represented by the ‘histone-code’ hypothesis (see Strahl and Allis, 2000), and this concept can be extended to similar PTMs of TFs and other chromatin-associated proteins (see Sims and Reinberg, 2008; Yang and Seto, 2008). The histone code operates as a sequence of three phases: ‘writing’, ‘reading,’ and ‘erasing’ of individual histone PTMs. In the ‘writing’ phase, a set of marks is established through the action of enzymes including histone acetyltranferases (HATs), protein methyltransferases, and serine/threonine protein kinases. These enzymes contain a specific catalytical subdomain (for example, acetylase, kinase, and demethylase) associated with one or more domains that mediates contacts with DNA (for example, AT-hook, bromodomain, and SANT domain) as shown in Figure 2. In the ‘reading’ stage, a specific mark or group of marks are recognized by proteins containing specific recognition modules such as the bromo- and chromodomains that interact with acetylated and methylated histone tail residues, respectively (Table 3). Finally, these PTMs can be ‘erased’ through action of histone deacetylases (HDACs), histone demethylases, and protein phosphatases. It appears that the majority, if not all, of these modifications are reversible, although some of them are more stable, for example, H3 K27m1, and H4 K20me1, and may be propagated through the cell cycle.

Chromatin-regulatory enzymes and their modular organization. Representative members of histone acetyltransferases (EP300/p300, PCAF and TAF1), deacetylases (HDAC1), methyltransferases (MLL1), demethylases (KDM1, KDM2A, and KDM5a), and ATP-helicases (SMARCA4 and SMARCA5) are shown with their catalytical and structural domains. Additional information on recognition of core histone modifications by bromodomain and PHD finger are given in Table 3 and in recent reviews (Klose et al., 2006; Ruthenburg et al., 2007; Taverna et al., 2007). AOD, aminooxidase domain; ARID/BRIGHT, AT-rich interactive domain; Bromo, bromodomain; deacetylase, deacetylase type I; HAT, histone acetyltransferase; helicase, DEAD/H-box helicase domain (ATPase); JmjC, Jumonji C demethylase catalytical core; JmjN, Jumonji N-terminal domain associated with the JmjC; kinase, protein serine/threonine kinase domain; PHD, PHD zinc-finger; SANT, nucleosome interacting domain; SET, protein methylase domain; SWIRM, α-helical protein–protein interaction domain; Zn-finger, various Zn-finger domains.
The histone code hypothesis is supported by two key observations. Earlier studies showed that specific combinations of PTMs depend on each other, for example, methylation of H3 K4 supports a broad acetylation including both histones H3 and H4 (see Fischle et al., 2003; Margueron et al., 2005). Recent genome-wide studies of 39 histone modifications in cultured cells supported the idea about interdependence between individual modifications and indicated that multiple modifications with similar roles increase robustness of a specific chromatin state at the level of individual nucleosomes (Wang et al., 2008). From the perspective of ‘reader’ proteins, a significant fraction of chromatin remodeling enzymes and chromatin-associated proteins contain two domains that can bind modified histones (for example, HRX contains a bromodomain, and three PHD domains, see Figure 2 and Table 3; Ruthenburg et al., 2007) and/or two or more proteins with distinct ‘reader’ modules physically interact like proteins of the PTIP complex (Pax2 Transcription activation domain Interacting Protein, Patel et al., 2007). In addition to the MLL3 H3K4 methyltransferase, PTIP complex includes WDR5, a protein recognizing methylated H3 (Table 3), and other chromatin regulatory proteins ASH2L, NCOA6, RBBP5, and SMC1.
Another mechanism that links histone PTMs with transcription relates to the phosphorylation of RNA polymerase-II. Sequential phosphoryation the RNA polymerase-II CTD domain (Ser-5 then Ser-2) correlates to the distribution of H3K4 trimethylation and H3K36 methylation in active genes. Ser-5 phosphorylated Pol-II triggers release of the mediator complex and recruitment of H3K4 methyltransferase. Indeed, H3K4me3 modifications are enriched at the 5′-end of active genes (Barski et al., 2007). In yeast, transcribing Pol-II also becomes phosphorylated on Ser-2 and can then bind the H3K36 methyltransferase, Set2 (Phatnani and Greenleaf, 2006). H3K36 methylation levels are higher into the transcribed regions of many yeast genes. In the mouse neural retina, the amount of Ser-2 phosphorylated Pol-II at the midpoint of the Rhodopsin gene correlates with activation of the gene (Mali et al., 2008). This suggests that Pol-II CTD phosphorylation might have similar roles in establishing the required chromatin architecture at active genes in mature photoreceptors.
Studies of histone acetylases, deacetylases, methylases, demethylases, kinases, and phosphatases generated a large body of data, and systematic genetic approach to elucidate function of these enzymes during individual organ formation has just begun. It has been shown that two major HATs, p300 and CBP (Figure 2) are expressed in the mouse eye (Yang et al., 2007), and that nearly one-third of the TFs listed in Tables 1 and 2 (for example, c-Maf, Crx, Neurod1, Pax6, and Prox1) have been shown to form specific complexes with p300/CBP and activate gene expression (Kasper et al., 2006). In lens, p300 activated crystallin gene expression (Chen et al., 2002) and both p300 and CBP have been identified in the mouse αA-crystallin locus (Yang et al., 2007). Cited2, a CBP-interacting protein, is essential for lens morphogenesis and hyaloid vasculature development (Chen et al., 2008). In retina, a number of molecular studies focused on the Crx and Nrl, two essential genes for photoreceptor development (Table 1). It has been shown that Crx binds Rhodopsin, M-opsin, and S-opsin promoters prior to Nrl, Nr2e3, and RNA polymerase-II suggesting its role as ‘pioneering’ factor described above; Crx binding alone is insufficient to elicit gene expression. Although Nrl, Crx, and Nr2E3 mRNA concentrations are high by postnatal day 3 in the mouse retina, only Crx is detectable on the Rhodopsin promoter (Peng and Chen, 2007). Consistent with this, the transcriptionaly active form of Pol-II (Ser-2-Phosphorylated) is detected inside the active Rhodopsin gene at P25, but not at P3 (Mali et al., 2008). Acetylation of H3 in these promoters was linked to the presence of Gcn5 HAT. A model of the TF network in photoreceptor gene regulation is based on dual roles of Crx plus Nr2e3 as direct activators and repressors of rod versus cone-specific genes (Hennig et al., 2008; Haider et al., 2009). In addition, sumoylation of Crx and Nr2e3, by E3 SUMO Pias3 ligase, is required for rod photoreceptor differentiation (Onishi et al., 2009). Interestingly, inhibition of HDAC activities by trichostatin-A resulted in reduced expression of both Crx and Nrl and demonstrating unique role of HDACs in retinal development (Chen and Cepko, 2007).
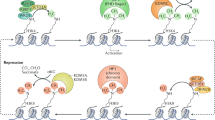
Collectively, a general model of gene activation during terminal differentiation considers both specific DNA-binding TFs and epigenetic regulation through chromatin structure (Figure 3). In the ‘closed’ 30 nm fiber conformation, the initiation TF can locate one or more binding sites through the DNA breathing and/or ‘pioneering’ activity (see above). This TF, in turn, recruits remodelers that aid the transition into the 11 nm chromatin fiber. During this process, additional cis-sites are recognized by TFs, followed by extensive histone tail PTMs that enable transcriptional machinery to initiate RNA synthesis. As these PTMs may substitute for the presence of some TFs, it is possible that their stable binding is no longer needed for gene activation. Chromatin immunoprecipitation analysis of αA-crystallin locus revealed stable binding of Pax6 with the promoter and upstream enhancer, DCR1, within a 16 kb domain of lens-specific chromatin (Yang et al., 2006). In contrast, strong recruitments of c-Maf and CREB to the promoter were found in lens chromatin compared with chromatin prepared from lens epithelial cells. However, recent conditional inactivation of Pax6 in secondary lens fibers did not find any significant changes in αA-crystallin expression though cell cycle exit and fiber cell differentiation were disrupted (Shaham et al., 2009). The simplest explanation of these finding is that Pax6 binding to its target genes in chromatin before its inactivation was sufficient to elicit epigenetic changes that were transmitted into the post-mitotic lens fibers, and Pax6 presence was no longer needed as shown in Figure 3.

A general model of gene activation of tissue-specific genes during embryonic eye development. A condensed chromatin domain, shown as 30 nm fiber, is marked by the presence of compacted nucleosomes marked by repressive PTMs (red octagons), and the presence of chromatin remodeling enzymes such as HDACs. The process of gene activation is initiated through binding of a TF (yellow) that can recognize its target sites in ‘closed’ chromatin conformation. Recruitment of distinct chromatin remodeling complexes/enzymes (for example, CR1 and CR2) through TF results in the generation of a novel set of histone PTMs, enriched for activating modifications (green octagons-methylations, blue flags-acetylations) and transition to the 11 nm chromatin conformation. Within this ‘open’ chromatin structure, TF1 (purple) and TF2 (orange), can locate their corresponding binding sites, and recruit additional nucleosome remodeling activities by reading the histone code that promotes transcription (Tx). It is possible that binding of the original TF (yellow) is no longer required at the binding site(s). See text for additional details.
ATP-dependent chromatin remodeling
At least five families of ATP-dependent chromatin remodeling machines (SWI/SNF, ISWI, INO80, SWR1, and NuRD) non-covalently alter nucleosome structure (see Saha et al., 2006). Their catalytical subunits are ATPases such as SMARCA4/BRG1, SMARCA5/SNF2H, SMARCA2/BRM, and nine variants of CHD3/4 (see Cvekl and Duncan, 2007) with DEAD/H-box helicase as their catalytical domain (Figure 2). Biochemical and single-molecule image experiments have shown that different complexes generate a variety of ‘remodeled’ substrates by processes that include promotion of histone octamer sliding (Zhang et al., 2006), histone exchange, histone eviction (see Park and Luger, 2008), conformational change of the DNA path on the nucleosomal surface, and other changes at the level of higher-order chromatin structure (see Fischle et al., 2003; Hager et al., 2009). These remodeling mechanisms promote or inhibit specific gene expression. Each remodeling machine is comprised of the core ATPase, and between 2 and 10 associated non-catalytic subunits, BAFs in case of SWI/SNF complexes. Recent studies have shown subunit exchanges in individual remodeling complexes during neural development (see Yoo and Crabtree, 2009). In retina, Baf60c is co-expressed in retinal progenitors with Brg1, but not with Brm, as the SWI/SNF catalytical subunit (Lamba et al., 2008). Baf60c is re-expressed in proliferating Muller glia. BAF60c interacts with RAR/RXR heterodimers (Table 2) through the LXXLL motif (see Cvekl and Wang, 2009), and this interaction is ligand-dependent (Flajollet et al., 2007). Smarca2/Brm regulates expression of Pou4f2 in RGC (Das et al., 2007). Thus, tissue-restricted non-catalytical subunits can have important functions in identifying specific regions of chromatin through their cognate TFs. In addition to this mechanism, TFs such as β-catenin, CREB, Hsf4, Mitf, Pax6, pRb, and Tbx2 (Tables 1 and 2) interact with SMARCA4/BRG1 as shown by protein–protein interaction assays (see Trotter and Archer, 2008). BRG1-containing complexes can also recognize acetylated histones through the bromodomain (Figure 2; Table 3) and nucleosome-binding protein, HMGA1 (see next section). Similarly, Snf2h contains two SANT domains (Figure 2), similar to the Myb DNA-binding domain, which associate with histone tails (see Cvekl and Duncan, 2007).
The zebrafish mutant young (yng) is a nonsense mutation in one of two zebrafish Brg1 genes and shows abnormal lens and retinal differentiation (Gregg et al., 2003; Kurita et al., 2003). RNA expression profiling of wild-type and yng retinas revealed that approximately one-fourth of retinal-specific genes were abnormally regulated in this system (Leung et al., 2008). Inhibition of Snf2h, a catalytical subunit of ISWI complexes, using morpholinos in Xenopus, showed that this enzyme is also required for normal lens and retinal development (Dirscherl et al., 2005). Binding of both Brg1 and Snf2h to αA-crystallin locus has been shown in lens chromatin (Yang et al., 2006). Although no similar data are available for the other ATPases, ocular coloboma is among many other developmental abnormalities caused by mutations in CHD7 helicase in humans with CHARGE syndrome (Lalani et al., 2006).
Nucleosome-binding proteins and minor core histone variants
Nucleosome-binding (or chromatin architectural) proteins are non-histone proteins that directly bind to nucleosomes and include three families of HMGs, HMGA, HMGB, and HMGN. From regulatory perspective, ‘linker histone’ H1 belongs to this group as H1 is thought to compete with HMGN proteins. Displacement of H1 favors a more open chromatin architecture. HMGA and HMGB proteins contain HMG- and AT-hook as their DNA-binding surfaces. It has been shown that HMGA1 binds to an AT-rich region of the mouse αB-crystallin promoter and recruits BRG1 (Duncan and Zhao, 2007). HMGN3a and HMGN3b have been proposed to regulate expression of glycine transporter gene 1 in the retina (West et al., 2004). Dynamic expression pattern of HMGN1/2/3 genes in the eye suggest multiple roles of these proteins in gene regulation in the cornea, lens, and retina (Lucey et al., 2008).
There are eight core histone variants (‘replacement’ histones), three for histone H3 and five for H2A, respectively (see Cvekl and Duncan, 2007). It has been shown that histone chaperone complex (HIRA) deposits H3.3 in a transcription-dependent, replication-independent manner, into multiple nucleosomes along the transcribed chromatin (Mito et al., 2005). H2A.Z histone variant is highly conserved through the evolution, is essential for early mouse embryonic development, and can be found acetylated in euchromatin and ubiquitinated in facultative heterochromatin (see Zlatanova and Thakar, 2008). In zebrafish transgenics, EGFP expression using the zebrafish histone H2A.F/Z promoter become silent in the adult zebrafish retina (Burket et al., 2008). This suggests a developmental regulation of variant histone promoters.
DNA methylation
Methylation of DNA, mostly as 5′-methylcytosine in CpG dinucleotides (see Fazzari and Greally, 2004), represents a fundamental epigenetic mechanism with a comprehensive knowledge how the DNA methylation patterns are propagated into daughter cells after cell divisions (Probst et al., 2009). Two DNA methyltransferases, DNMT3a and DNMT3b, catalyze de novo methylation reactions. In contrast, DNMT1 mediates the DNA methylation maintenance reactions. The methyl groups of methylated CpG residues reside in the major groove of the DNA helix, where a large number of sequence-specific DNA-binding proteins recognize the DNA (see Fazzari and Greally, 2004). This modification can either promote or inhibit binding of various proteins. The methyl-CpG-binding proteins (for example, MBD1 to MBD6 and MECP2) have been shown to recruit transcriptional repressor complexes to methylated promoter regions (see Klose and Bird, 2006). This process results in transcriptional silencing and is, at least partially, regulated through sumoylation of MBD1 (Lyst et al., 2006). Another global epigenetic regulatory protein, whose binding is regulated through DNA methylation, is the ubiquitously expressed zinc-finger protein CTCF, which binds to GC-rich sequences and is implicated in three-dimensional organization of chromatin (see Phillips and Corces, 2009). CTCF functions as a TF for Pax6 expression in RPCs (Li et al., 2006). However, during differentiation their expression domains segregate and Pax6 is expressed only in RGC and amacrine cells and CTCF expressed in bipolar cells, Muller cells, and outer nuclear cell layer (Canto-Soler et al., 2008).
Although studies of DNA methylation often serve as a gateway to more complex epigenetic studies, the currently available data on lens and retina are limited to a small number of genes. In lens, methylation studies of several crystallin gene promoters were consistent with the standard model in which methylated DNA inhibits gene expression (Sullivan et al., 1991; Klok et al., 1998). In retina, site-specific hypomethylation of interphotoreceptor retinoid-binding protein regulates its expression in photoreceptors as the promoter is hypermethylated in other retinal cells (Boatright et al., 2000). DNA methyltransferase-1 (DNMT1) is required for terminal differentiation of retina, intestine, and exocrine pancreas whereas liver and endocrine differentiation were normal in zebrafish (Rai et al., 2006). Interestingly, a recent study of DNA methyltransferase-2 (DNMT2) in zebrafish revealed its essential role as cytoplasmic enzyme methylating tRNAs. Morpholino knockdown of Dnmt2 induced differentiation defects in the eye, brain, and liver (Rai et al., 2007). Finally, similar studies of zebrafish Dnmt3, an orthologue of mammalian DNMT3B, showed its essential role in both lens fiber cell differentiation and for differentiation of RPCs into precursors of bipolar, horizontal, and photoreceptor cells. In contrast, formation of RGC appeared normal (Rai et al., 2010).
Nuclear organization and compartmentalization of transcription
Cellular differentiation is also characterized by spatial organization of chromatin harboring active and inactive genes within the cell nucleus. A special feature of transcriptional networks is the establishment of organized centers, or hubs, that contain multiple co-regulated genes connected together through long-range interactions between genes on the same or different chromosome (see de Laat and Grosveld, 2003; Kosak and Groudine, 2004).
Lens fiber cell differentiation involves a tightly regulated process of denucleation (see Bassnett, 2009) leading to the destruction of genetic information in terminally differentiated lens fibers. This process is distinct from apoptosis (see Bassnett, 2009) and karyolysis of mammalian erythrocytes through nuclei engulfment by macrophages (Yoshida et al., 2005). In mouse, the initial DNA breakpoints are observed at E15.5 of embryonic development and these changes are followed by the formation of ‘pyknotic’ nuclei. The final process of denucleation of the primary lens fibers occurs between E16 and E18. Thus, perturbed nuclear structure coincidences with peak expression levels of several lens crystallins. It would be interesting to determine the nuclear structure and long-range chromatin interactions of lens fiber cells nuclei between E14.5 and E15.5, and whether crystallin genes are organized as transcriptional hubs similar to those found for α- and β-globin genes in red blood cells (Ragoczy et al., 2006).
The conventional nuclear architecture, with transcriptionally active euchromatin located mostly to the nuclear center and densely compacted heterochromatin found at the nuclear periphery (see Kosak and Groudine, 2004), was found to be reversed in the rod photoreceptors in nocturnal mammals (Solovei et al., 2009). During early neonatal stages, rod progenitor cells exhibit a normal nuclear architecture and become post-mitotic by postnatal day 5. The nuclear reorganization is initiated around the time of eye opening and takes additional 2 weeks to be completed. The inverted structure is represented by a central region of heterochromatin. This region is surrounded by a thick layer of non-centromeric heterochromatin and the outer shell is comprised of euchromatin. This inverted nuclear pattern affects optical properties of rod photoreceptors in the ONL of the retina (Figure 1d). It reduces light scattering compared with the nuclei with ‘normal’ organization and generates higher, yet uniform, refractive index at the central region of the nucleus (Solovei et al., 2009). Interestingly, the Rhodopsin gene, common housekeeping genes, and inactive genes all reside in the euchromatin zone in the nuclear perifery. Thus, epigenetic regulatory mechanisms such as core histone modifications specific for heterochromatin (H3 K9me3 and H3 K20me3), and for euchromatin (H3 K4me3), are not only used for gene regulation but also for compaction of nuclear architecture as an adaptation to improve night vision. This nocturnal rod nuclear architecture is lost in NRL−/− mice and rescued by ectopic expression of NRL, driven from the S-opsin gene promoter (Oh et al., 2007). NRL is also expressed in rod photoreceptors of diurnal species, therefore, other factors must contribute to this morphological difference. The follow-up studies should probe the molecular mechanisms underlying this process and details of the organization of individual chromosomes and rod-specific genes within the compacted nuclei.
Non-coding RNAs
Non-protein-coding RNAs are powerful regulatory molecules of chromatin structure and transcriptional elongation. Much of the genome is actually transcribed into non-protein-coding RNAs with as yet unknown functions. Distinct spatiotemporal localization of 13 representative microRNAs (miRNAs) in the mouse eye identified several candidate molecules for functional studies of lens and retinal development (Karali et al., 2007). Several miRNAs have been shown to participate in retinal development (Huang et al., 2008). Correct eye development in Xenopus requires microRNA-24a, as it represses apoptosis through inhibition of pro-apoptotic factors caspase 9 and Apaf1 (Walker and Harland, 2009). Dicer, an endoribonuclease in the RNase III family, cleaves double-stranded and pre-miRNAs into short double-stranded small interfering RNAs. Inactivation of dicer in retinal cells caused morphological and physiological defects through reduced levels of expression of several miRNAs (Damiani et al., 2008). Similarly, inactivation of this enzyme in lens and corneal progenitor cells showed that dicer is required for terminal differentiation of lens fiber cells and protection of these cells from apoptosis (Li and Piatigorsky, 2009). Altered retinal miRNA expression profiles were identified in a mouse model of retinitis pigmentosa (Loscher et al., 2008). Additional studies are required to elucidate the roles of at least 21 potentially retinal-specific miRNAs identified earlier (Xu et al., 2007).
Non-coding RNAs directly participate in chromatin condensation as shown in Drosophila and other model organisms. Formation of silent heterochromatin requires Dicer, followed by incorporation of small interfering RNAs into the RITS complex (RNA-induced Initiation of Transcriptional gene Silencing). A part of the molecular mechanism is a self-reinforcing cycle using KMT1A/SUV39H1 protein methyltranferase, HDACs, HP1, and possibly other proteins (see Allis et al., 2007). It would be interesting to determine if these small interfering RNA-dependent mechanisms are used in heterochromatin formation in rod photoreceptor nuclei described above.
Propagation of epigenetic information through the cell cycle
Transcription also faces another challenge. During cellular division, the cell must decide either to maintain the ‘cellular’ memory of its transcriptional status or to reset its transcriptional programme by using epigenetic mechanisms. During mitosis, most but not all TFs dissociate from DNA. In contrast, the stable protein–DNA complexes in mitosis might help the rapid reactivation of transcription upon exit from mitosis as indicated by nuclease-hypersensitive sites that persist during mitosis (Martinez-Balbas et al., 1995; Christova and Oelgeschlager, 2002). It is thought, that ‘chromatin’ memory is important for lineage specification and commitment during development of multicellular organisms. Thus, it would be important to determine interactions of lineage-specific TFs such as Crx, Math5, Neurod1, Pax6, and Rax, with DNA through the cell cycle. Mitotic retention of lineage-specific TF Runx2 in osteogenic cells (Young et al., 2007) supports experimental testing of this model in ocular cells.
Transdifferentiation and nuclear reprogramming
One of the hallmark experiments of tissue regeneration in vertebrates is transdifferentiation of lens from dorsal iris through dedifferentiation of pigment epithelium in a few species of salamander (see Sanchez Alvarado and Tsonis, 2006). It is reasonable to hypothesize that the dedifferentiation process shares similar features with nuclear reprogramming, a process used to generate totipotent cells from mature cell types (see Jaenisch and Young, 2008). Epigenetic changes are likely to have key roles in the process of lens regeneration. To test this hypothesis, Tsonis and co-workers have recently initiated identification of proteins including stem cell pluripotency factors during lens and limb regeneration (Roddy et al., 2008; Maki et al., 2009) that could provide novel information about epigenetic changes in transdifferentiation. This information has enormous value for understanding the regenerative potential of human ocular tissues and the physiology of adult retinal stem cells (see Lamba et al., 2009).
Epigenetic mechanisms and eye diseases
A number of recent studies of aging and cancer showed that a plethora of epigenetic regulatory mechanisms are important in human disease mechanisms that opened novel opportunities for ocular drug research. Several eye diseases were recently linked to enzymes that catalyze histone PTMs. Spinocerebellar ataxia type 7 (SCA7) is a neurodegenerative disorder cause by expansion of polyglutamine tract in the Ataxin-7 protein. Although Ataxin-7 expression is not cell-type specific, the most affected genes are targets of CRX. Ataxin-7 is an integral component of STAGA (SPT3-TAF9-ADA-GCN5 acetyltransferase) complex, and mediates a direct interaction of STAGA with CRX. Normal Ataxin-7/CRX interaction is perturbed in polyglutamine expanded Ataxin-7. Chromatin immunoprecipitation assays demonstrated retinal-specific association of CRX, GCN5, and acetylated histone H3 with CRX target genes (Palhan et al., 2005). Interestingly, recent studies showed that STAGA complex contain a histone H2A and H2B deubiquitinase activity (Zhao et al., 2008). Sirtuins (SIRT1–7) are deacetylases implicated in a range of diseases and longevity (see Dali-Youcef et al., 2007). Reduced expression of Sirt1was found in the mouse rd10 retinal degeneration model, which might reduce the protective functions of Sirt1 in double DNA stranded DNA-break repair mechanisms and in maintaining energy homeostasis in photoreceptor cells (Jaliffa et al., 2009).
The potential to pharmacologically modify gene expression through the manipulation of histone acetylation or DNA methylation is an area of intense interest (see Acharya et al., 2005). Furthermore, investigation of some currently prescribed compounds reveals potential off target effects on these epigenetic modifications. Two potent HDAC inhibitors, valproic acid and trichostatin A, increase expression and secretion of Clusterin/APO-J in a human RPE cell line, ARPE-19 (Suuronen et al., 2007). Clusterin/APO-J is one component of macular drusen, a protein/lipid deposition that forms under the RPE basement membrane in eyes affected by age-related macular degeneration. These basement membrane changes are thought to disrupt the RPE's communication with the underlying choroidal blood supply, causing hypoxia, neovascular response, and loss of the RPE. A recent report showed that HDAC4 regulates the survival of retinal neurons by preventing apoptosis of rod photoreceptors and bipolar cells through stabilization of TFs HIFα and possibly other mechanisms (Chen and Cepko, 2009).
There are enormous implications of DNA methylation to understand eye disease. Apart from perturbed global methylation in cancer cells, DNA methylation patterns can change as a result of aging (see Calvanese et al., 2009) and diet (see Waterland, 2009). As the majority of eye diseases such as age-related macular degeneration, cataract, and glaucoma have age as the major risk factor, it is reasonable to hypothesize that environmental factors in combination with aging can influence DNA methylation of critical genes required for normal function of the visual system.
Histone H2AX is a variant histone involved in DNA damage response and repair. Studies of H2afx−/− mice have shown an inhibition of the neovascular response to hypoxia, without any apparent affects on normal vascular development in the retina (Economopoulou et al., 2009). This suggests that the regulation of the chromatin architecture is integral to the retinal neovascular response, which is a detrimental process involved in many retinal diseases including diabetic retinopathy, retinopathy of prematurity, and AMD.
External ocular tissues are directly exposed to the outside environment and may be particularly sensitive to these effects. A good clinical example is a condition of the external eye surface known as pterygia, which involves abnormal proliferation of the conjunctiva around and into the edge of the clear cornea. Both hypermethylation of the p16 tumor suppressor gene promoter and elevated expression of DNMT3b are correlated to diminished expression of p16 in human pterygia tissue samples (Chen et al., 2006).
Concluding remarks
In this review, we summarized current knowledge about epigenetic regulatory mechanisms in vertebrate eye with the primary focus on lens and retina. Similar to other cells and organs, chromatin dynamics in ocular cells is tightly regulated by a coordinated action of ATP-dependent chromatin remodelers, histone-chaperones, histone-modifying enzymes, and nucleosome-binding proteins in conjunction with the ‘breathing’ of nucleosomal DNA. A number of examples of epigenetic eye-specific processes discussed earlier should stimulate additional studies to identify novel eye-specific roles of chromatin remodeling enzymes, miRNAs, and specific PTM of nucleosomes associated with expression of eye-specific genes. Comprehensive studies of model genes such as myogenin, βA-globin, and interferon β, revealed a diverse order in recruitment of individual chromatin remodeling complexes and specific TFs. In contrast, it is reasonable to hypothesize that expression of genes that are regulated by shared specific DNA-binding TFs, for example, lens crystallins (through Pax6, Maf, and Sox proteins), and individual photoreceptor genes (through Crx, Nr2e3, and Nrl), would use similar molecular mechanisms of their activation in lens and cone photoreceptor terminal differentiation, respectively.
Although a number of TFs discussed here were shown to recruit Brg1 (SWI/SNF) and p300/CBP chromatin remodeling enzymes/complexes (Table 1), significantly less is known about recruitment of methyltransferases, demethylases, and deacetylases to specific regions of chromatin. The notable exception is the retinoblastoma protein, pRB, as it has an ability to associate with at least six distinct chromatin remodeling systems (Table 1) and is essential for both lens fiber (Morgenbesser et al., 1994) and retinal (Xu et al., 2009) differentiation. Purification of proteins that interact with key lens and retinal lineage TFs is likely to identify proteins that function in chromatin remodeling.
To determine maps of the chromatin landscape in differentiated ocular cells, implementation of the rationale and procedures used by The Encyclopedia of DNA Elements (ENCODE) Project will be needed mostly through the use of chromatin immunoprecipitation and massively parallel DNA sequencing, chromatin immunoprecipitation-seq (see Mendenhall and Bernstein, 2008), and other high-throughput procedures to map DNA methylation and transcriptional units of protein coding and non-coding genes in the individual cell epigenomes. Although the appropriate chromatin mapping technologies are improving and their costs have dropped, a specific problem for developmental biologists is the limited amounts of chromatin that can be prepared from individual cell types. Some of the technical limitations can be resolved by using FACS sorted cells, use of tagged core histones expressed using tissue-specific promoters, and use of cells generated by lineage-specific differentiation of human and mouse embryonic stem cells. Cell isolation might affect the epigenetic regulation of some genes, so the development of methods toward analysis of single cells in their tissue context would be ideal. In the mean time, studies of cell-specific genes can be targets of exploration in the lens, neural retina, and the RPE during ocular development. Importantly, epigenetic studies of chromatin remodeling enzymes and nucleosome proteins in the eye are aided by an increasing repertoire of cre-lines established for conditional gene targeting in different ocular cell lines, and at different stages of eye development. In addition, although not discussed here, Drosophila is a powerful model system to identify various chromatin remodeling enzymes and the functions of Polycomb (PcG) and Trithorax (Tx), using compound eye ommatidia as the primary tissue of interest (Carrera et al., 2008). When these limiting issues are resolved, we will be rewarded with system-level insights about gene regulation in normal eye development, in adult ocular cells and tissues, and in normal and disease states.
References
Acharya MR, Sparreboom A, Venitz J, Figg WD (2005). Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol 68: 917–932.
Allis CD, Jenuwein T, Reinberg D (2007). Overview and concepts. In: Allis CD, Jenuwein T, Reinberg D (eds) Epigenetics. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, pp 23–61.
Ashery-Padan R, Gruss P (2001). Pax6 lights-up the way for eye development. Curr Opin Cell Biol 13: 706–714.
Badea TC, Cahill H, Ecker J, Hattar S, Nathans J (2009). Distinct roles of transcription factors brn3a and brn3b in controlling the development, morphology, and function of retinal ganglion cells. Neuron 61: 852–864.
Barolo S, Posakony JW (2002). Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev 16: 1167–1181.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837.
Bassett EA, Pontoriero GF, Feng W, Marquardt T, Fini ME, Williams T et al. (2007). Conditional deletion of activating protein 2α (AP-2α) in the developing retina demonstrates non-cell-autonomous roles for AP-2α in optic cup development. Mol Cell Biol 27: 7497–7510.
Bassnett S (2009). On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res 88: 133–139.
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326.
Boatright JH, Nickerson JM, Borst DE (2000). Site-specific DNA hypomethylation permits expression of the IRBP gene. Brain Res 887: 211–221.
Burket CT, Montgomery JE, Thummel R, Kassen SC, LaFave MC, Langenau DM et al. (2008). Generation and characterization of transgenic zebrafish lines using different ubiquitous promoters. Transgenic Res 17: 265–279.
Calvanese V, Lara E, Kahn A, Fraga MF (2009). The role of epigenetics in aging and age-related diseases. Ageing Res Rev 8: 268–276.
Canto-Soler MV, Huang H, Romero MS, Adler R (2008). Transcription factors CTCF and Pax6 are segregated to different cell types during retinal cell differentiation. Dev Dyn 237: 758–767.
Carrera I, Zavadil J, Treisman JE (2008). Two subunits specific to the PBAP chromatin remodeling complex have distinct and redundant functions during drosophila development. Mol Cell Biol 28: 5238–5250.
Chauhan BK, Disanza A, Choi SY, Faber SC, Lou M, Beggs HE et al. (2009). Cdc42- and IRSp53-dependent contractile filopodia tether presumptive lens and retina to coordinate epithelial invagination. Development 136: 3657–3667.
Chen B, Cepko CL (2007). Requirement of histone deacetylase activity for the expression of critical photoreceptor genes. BMC Dev Biol 7: 78.
Chen B, Cepko CL (2009). HDAC4 regulates neuronal survival in normal and diseased retinas. Science 323: 256–259.
Chen PL, Cheng YW, Chiang CC, Tseng SH, Chau PS, Tsai YY (2006). Hypermethylation of the p16 gene promoter in pterygia and its association with the expression of DNA methyltransferase 3b. Mol Vis 12: 1411–1416.
Chen Q, Dowhan DH, Liang D, Moore DD, Overbeek PA (2002). CREB-binding protein/p300 co-activation of crystallin gene expression. J Biol Chem 277: 24081–24089.
Chen Y, Doughman YQ, Gu S, Jarrell A, Aota S, Cvekl A et al. (2008). Cited2 is required for the proper formation of the hyaloid vasculature and for lens morphogenesis. Development 135: 2939–2948.
Chow RL, Lang RA (2001). Early eye development in vertebrates. Annu Rev Cell Dev Biol 17: 255–296.
Christova R, Oelgeschlager T (2002). Association of human TFIID-promoter complexes with silenced mitotic chromatin in vivo. Nat Cell Biol 4: 79–82.
Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9: 279–289.
Collinson JM, Quinn JC, Buchanan MA, Kaufman MH, Wedden SE, West JD et al. (2001). Primary defects in the lens underlie complex anterior segment abnormalities of the Pax6 heterozygous eye. Proc Natl Acad Sci USA 98: 9688–9693.
Cvekl A, Duncan MK (2007). Genetic and epigenetic mechanisms of gene regulation during lens development. Prog Retin Eye Res 26: 555–597.
Cvekl A, Piatigorsky J (1996). Lens development and crystallin gene expression: many roles for Pax-6. Bioessays 18: 621–630.
Cvekl A, Wang WL (2009). Retinoic acid signaling in mammalian eye development. Exp Eye Res 89: 280–291.
Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J (2007). Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med 39: 335–345.
Damiani D, Alexander JJ, O’Rourke JR, McManus M, Jadhav AP, Cepko CL et al. (2008). Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci 28: 4878–4887.
Das AV, James J, Bhattacharya S, Imbalzano AN, Antony ML, Hegde G et al. (2007). SWI/SNF chromatin remodeling ATPase Brm regulates the differentiation of early retinal stem cells/progenitors by influencing Brn3b expression and Notch signaling. J Biol Chem 282: 35187–35201.
Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ (2002). Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 A resolution. J Mol Biol 319: 1097–1113.
de Laat W, Grosveld F (2003). Spatial organization of gene expression: the active chromatin hub. Chromosome Res 11: 447–459.
Dirscherl SS, Henry JJ, Krebs JE (2005). Neural and eye-specific defects associated with loss of the imitation switch (ISWI) chromatin remodeler in Xenopus laevis. Mech Dev 122: 1157–1170.
Donner AL, Lachke SA, Maas RL (2006). Lens induction in vertebrates: variations on a conserved theme of signaling events. Semin Cell Dev Biol 17: 676–685.
Duncan B, Zhao K (2007). HMGA1 mediates the activation of the CRYAB promoter by BRG1. DNA Cell Biol 26: 745–752.
Economopoulou M, Langer HF, Celeste A, Orlova VV, Choi EY, Ma M et al. (2009). Histone H2AX is integral to hypoxia-driven neovascularization. Nat Med 15: 553–558.
Faber SC, Dimanlig P, Makarenkova HP, Shirke S, Ko K, Lang RA (2001). Fgf receptor signaling plays a role in lens induction. Development 128: 4425–4438.
Fazzari MJ, Greally JM (2004). Epigenomics: beyond CpG islands. Nat Rev Genet 5: 446–455.
Firth LC, Baker NE (2009). Retinal determination genes as targets and possible effectors of extracellular signals. Dev Biol 327: 366–375.
Fischle W, Wang Y, Allis CD (2003). Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172–183.
Flajollet S, Lefebvre B, Cudejko C, Staels B, Lefebvre P (2007). The core component of the mammalian SWI/SNF complex SMARCD3/BAF60c is a coactivator for the nuclear retinoic acid receptor. Mol Cell Endocrinol 270: 23–32.
Fuhrmann S (2008). Wnt signaling in eye organogenesis. Organogenesis 4: 60–67.
Gehring WJ, Ikeo K (1999). Pax 6: mastering eye morphogenesis and eye evolution. Trends Genet 15: 371–377.
Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL (1994). PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet 7: 463–471.
Grainger RM (1992). Embryonic lens induction: shedding light on vertebrate tissue determination. Trends Genet 8: 349–355.
Graw J (2003). The genetic and molecular basis of congenital eye defects. Nat Rev Genet 4: 876–888.
Gregg RG, Willer GB, Fadool JM, Dowling JE, Link BA (2003). Positional cloning of the young mutation identifies an essential role for the Brahma chromatin remodeling complex in mediating retinal cell differentiation. Proc Natl Acad Sci USA 100: 6535–6540.
Griep AE (2006). Cell cycle regulation in the developing lens. Semin Cell Dev Biol 17: 686–697.
Hager GL, McNally JG, Misteli T (2009). Transcription dynamics. Mol Cell 35: 741–753.
Haider NB, Mollema N, Gaule M, Yuan Y, Sachs AJ, Nystuen AM et al. (2009). Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction. Exp Eye Res 89: 365–372.
Harada T, Harada C, Parada LF (2007). Molecular regulation of visual system development: more than meets the eye. Genes Dev 21: 367–378.
Hatakeyama J, Kageyama R (2004). Retinal cell fate determination and bHLH factors. Semin Cell Dev Biol 15: 83–89.
Hennig AK, Peng GH, Chen S (2008). Regulation of photoreceptor gene expression by Crx-associated transcription factor network. Brain Res 1192: 114–133.
Huang KM, Dentchev T, Stambolian D (2008). MiRNA expression in the eye. Mamm Genome 19: 510–516.
Jaenisch R, Young R (2008). Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132: 567–582.
Jaliffa C, Ameqrane I, Dansault A, Leemput J, Vieira V, Lacassagne E et al. (2009). Sirt1 involvement in rd10 mouse retinal degeneration. Invest Ophthalmol Vis Sci 50: 3562–3572.
Jonasova K, Kozmik Z (2008). Eye evolution: lens and cornea as an upgrade of animal visual system. Semin Cell Dev Biol 19: 71–81.
Karali M, Peluso I, Marigo V, Banfi S (2007). Identification and characterization of microRNAs expressed in the mouse eye. Invest Ophthalmol Vis Sci 48: 509–515.
Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A et al. (2006). Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol 26: 789–809.
Klok EJ, van Genesen ST, Civil A, Schoenmakers JG, Lubsen NH (1998). Regulation of expression within a gene family. The case of the rat γB- and γD-crystallin promoters. J Biol Chem 273: 17206–17215.
Klose RJ, Bird AP (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 31: 89–97.
Klose RJ, Kallin EM, Zhang Y (2006). JmjC-domain-containing proteins and histone demethylation. Nature Rev Genet 7: 715–727.
Kosak ST, Groudine M (2004). Form follows function: The genomic organization of cellular differentiation. Genes Dev 18: 1371–1384.
Kurita R, Sagara H, Aoki Y, Link BA, Arai K, Watanabe S (2003). Suppression of lens growth by alphaA-crystallin promoter-driven expression of diphtheria toxin results in disruption of retinal cell organization in zebrafish. Dev Biol 255: 113–127.
Lachke SA, Maas RL (2009). Building the developmental oculome: systems biology in vertebrate eye development and disease. Wiley Interdiscip Rev Syst Biol Med (http://dx|.doi.org/10.1002/wsbm.59).
Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE et al. (2006). Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet 78: 303–314.
Lamb TD, Collin SP, Pugh Jr EN (2007). Evolution of the vertebrate eye: opsins, photoreceptors, retina and eye cup. Nat Rev Neurosci 8: 960–976.
Lamba DA, Hayes S, Karl MO, Reh T (2008). Baf60c is a component of the neural progenitor-specific BAF complex in developing retina. Dev Dyn 237: 3016–3023.
Lamba DA, Karl MO, Reh TA (2009). Strategies for retinal repair: cell replacement and regeneration. Prog Brain Res 175: 23–31.
Leung YF, Ma P, Link BA, Dowling JE (2008). Factorial microarray analysis of zebrafish retinal development. Proc Natl Acad Sci USA 105: 12909–12914.
Li T, Lu Z, Lu L (2006). Pax6 regulation in retinal cells by CCCTC binding factor. Invest Ophthalmol Vis Sci 47: 5218–5226.
Li Y, Piatigorsky J (2009). Targeted deletion of Dicer disrupts lens morphogenesis, corneal epithelium stratification, and whole eye development. Dev Dyn 238: 2388–2400.
Liu W, Lagutin OV, Mende M, Streit A, Oliver G (2006). Six3 activation of Pax6 expression is essential for mammalian lens induction and specification. EMBO J 25: 5383–5395.
Livesey R, Cepko C (2001). Neurobiology. Developing order. Nature 413: 471–473.
Loscher CJ, Hokamp K, Wilson JH, Li T, Humphries P, Farrar GJ et al. (2008). A common microRNA signature in mouse models of retinal degeneration. Exp Eye Res 87: 529–534.
Lovicu FJ, McAvoy JW (2005). Growth factor regulation of lens development. Dev Biol 280: 1–14.
Lucey MM, Wang Y, Bustin M, Duncan MK (2008). Differential expression of the HMGN family of chromatin proteins during ocular development. Gene Expr Patterns 8: 433–437.
Luger K (2006). Dynamic nucleosomes. Chromosome Res 14: 5–16.
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251–260.
Lupo G, Harris WA, Lewis KE (2006). Mechanisms of ventral patterning in the vertebrate nervous system. Nat Rev Neurosci 7: 103–114.
Lyst MJ, Nan X, Stancheva I (2006). Regulation of MBD1-mediated transcriptional repression by SUMO and PIAS proteins. EMBO J 25: 5317–5328.
Maki N, Suetsugu-Maki R, Tarui H, Agata K, Del Rio-Tsonis K, Tsonis PA (2009). Expression of stem cell pluripotency factors during regeneration in newts. Dev Dyn 238: 1613–1616.
Mali RS, Peng GH, Zhang X, Dang L, Chen S, Mitton KP (2008). FIZ1 is part of the regulatory protein complex on active photoreceptor-specific gene promoters in vivo. BMC Mol Biol 9: 87.
Mardi SB, Seth B (2009). Vertebrate retina and hypothalamus development. Wiley Interdiscip Rev Syst Biol Med 1: 380–389.
Margueron R, Trojer P, Reinberg D (2005). The key to development: interpreting the histone code? Curr Opin Genet Dev 15: 163–176.
Martinez-Balbas MA, Dey A, Rabindran SK, Ozato K, Wu C (1995). Displacement of sequence-specific transcription factors from mitotic chromatin. Cell 83: 29–38.
Martinez-Morales JR, Dolez V, Rodrigo I, Zaccarini R, Leconte L, Bovolenta P et al. (2003). OTX2 activates the molecular network underlying retina pigment epithelium differentiation. J Biol Chem 278: 21721–21731.
Masse K, Bhamra S, Eason R, Dale N, Jones EA (2007). Purine-mediated signalling triggers eye development. Nature 449: 1058–1062.
Matter-Sadzinski L, Puzianowska-Kuznicka M, Hernandez J, Ballivet M, Matter JM (2005). A bHLH transcriptional network regulating the specification of retinal ganglion cells. Development 132: 3907–3921.
Medina-Martinez O, Jamrich M (2007). Foxe view of lens development and disease. Development 134: 1455–1463.
Mendenhall EM, Bernstein BE (2008). Chromatin state maps: new technologies, new insights. Curr Opin Genet Dev 18: 109–115.
Mito Y, Henikoff JG, Henikoff S (2005). Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 37: 1090–1097.
Moore KB, Vetter ML (2007). Retinal development. In: Moody SA (ed). Principles of Developmental Genetics. Publisher: Academic Press: San Diego, CA, pp 548–573.
Morgenbesser SD, Williams BO, Jacks T, DePinho RA (1994). p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature 371: 72–74.
Mui SH, Kim JW, Lemke G, Bertuzzi S (2005). Vax genes ventralize the embryonic eye. Genes Dev 19: 1249–1259.
Niederreither K, Dolle P (2008). Retinoic acid in development: towards an integrated view. Nat Rev Genet 9: 541–553.
Oh EC, Khan N, Novelli E, Khanna H, Strettoi E, Swaroop A. (2007). Transformation of cone precursors to functional rod photoreceptors by bZIP transcription factor NRL. Proc Natl Acad Sci USA 104: 1679–1684.
Onishi A, Peng GH, Hsu C, Alexis U, Chen S, Blackshaw S (2009). Pias3-dependent SUMOylation directs rod photoreceptor development. Neuron 61: 234–246.
Oron-Karni V, Farhy C, Elgart M, Marquardt T, Remizova L, Yaron O et al. (2008). Dual requirement for Pax6 in retinal progenitor cells. Development 135: 4037–4047.
Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y et al. (2005). Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA 102: 8472–8477.
Park YJ, Luger K (2008). Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol 18: 282–289.
Patel SR, Kim D, Levitan I, Dressler GR (2007). The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell 13: 580–592.
Peng GH, Chen S (2007). Crx activates opsin transcription by recruiting HAT-containing co-activators and promoting histone acetylation. Hum Mol Genet 16: 2433–2452.
Phatnani HP, Greenleaf AL (2006). Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 20: 2922–2936.
Phillips JE, Corces VG (2009). CTCF: master weaver of the genome. Cell 137: 1194–1211.
Pontoriero GF, Deschamps P, Ashery-Padan R, Wong R, Yang Y, Zavadil J et al. (2008). Cell autonomous roles for AP-2α in lens vesicle separation and maintenance of the lens epithelial cell phenotype. Dev Dyn 237: 602–617.
Probst AV, Dunleavy E, Almouzni G (2009). Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol 10: 192–206.
Quinn JC, West JD, Hill RE (1996). Multiple functions for Pax6 in mouse eye and nasal development. Genes Dev 10: 435–446.
Ragoczy T, Bender MA, Telling A, Byron R, Groudine M (2006). The locus control region is required for association of the murine beta-globin locus with engaged transcription factories during erythroid maturation. Genes Dev 20: 1447–1457.
Rai K, Chidester S, Zavala CV, Manos EJ, James SR, Karpf AR et al. (2007). Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev 21: 261–266.
Rai K, Jafri IF, Chidester S, James SR, Karpf AR, Cairns BR et al. (2010). Dnmt3 and G9a cooperate for tissue-specific development in zebrafish. J Biol Chem 285: 4110–4121.
Rai K, Nadauld LD, Chidester S, Manos EJ, James SR, Karpf AR et al. (2006). Zebrafish Dnmt1 and Suv39h1 regulate organ-specific terminal differentiation during development. Mol Cell Biol 26: 7077–7085.
Rajagopal R, Huang J, Dattilo LK, Kaartinen V, Mishina Y, Deng CX et al. (2009). The type I BMP receptors, Bmpr1a and Acvr1, activate multiple signaling pathways to regulate lens formation. Dev Biol 335: 305–316.
Rembold M, Loosli F, Adams RJ, Wittbrodt J (2006). Individual cell migration serves as the driving force for optic vesicle evagination. Science 313: 1130–1134.
Riesenberg AN, Liu Z, Kopan R, Brown NL (2009). Rbpj cell autonomous regulation of retinal ganglion cell and cone photoreceptor fates in the mouse retina. J Neurosci 29: 12865–12877.
Robinson ML (2006). An essential role for FGF receptor signaling in lens development. Semin Cell Dev Biol 17: 726–740.
Roddy M, Fox TP, McFadden JP, Nakamura K, Del Rio-Tsonis K, Tsonis PA (2008). A comparative proteomic analysis during urodele lens regeneration. Biochem Biophys Res Commun 377: 275–279.
Ruthenburg AJ, Li H, Patel DJ, Allis CD (2007). Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8: 983–994.
Saha A, Wittmeyer J, Cairns BR (2006). Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol 7: 437–447.
Sanchez Alvarado A, Tsonis PA (2006). Bridging the regeneration gap: genetic insights from diverse animal models. Nat Rev Genet 7: 873–884.
Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS (2009). Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev 23: 804–809.
Shaham O, Smith AN, Robinson ML, Taketo MM, Lang RA, Ashery-Padan R (2009). Pax6 is essential for lens fiber cell differentiation. Development 136: 2567–2578.
Simone C (2006). SWI/SNF: the crossroads where extracellular signaling pathways meet chromatin. J Cell Physiol 207: 309–314.
Sims 3rd RJ, Reinberg D (2008). Is there a code embedded in proteins that is based on post-translational modifications? Nat Rev Mol Cell Biol 9: 815–820.
Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T et al. (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137: 356–368.
Strahl BD, Allis CD (2000). The language of covalent histone modifications. Nature 403: 41–45.
Streit A (2004). Early development of the cranial sensory nervous system: from a common field to individual placodes. Dev Biol 276: 1–15.
Streit A (2007). The preplacodal region: an ectodermal domain with multipotential progenitors that contribute to sense organs and cranial sensory ganglia. Int J Dev Biol 51: 447–461.
Sullivan CH, O’Farrell S, Grainger RM (1991). Delta-crystallin gene expression and patterns of hypomethylation demonstrate two levels of regulation for the delta-crystallin genes in embryonic chick tissues. Dev Biol 145: 40–50.
Suuronen T, Nuutinen T, Ryhanen T, Kaarniranta K, Salminen A (2007). Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem Biophys Res Commun 357: 397–401.
Tang K, Xie X, Park J-I, Jamrich M, Tsai S, Tsai M-J (2010). COUP-TFs regulate eye development by controlling factors essential for optic vesicle morphogenesis. Development 137: 725–734.
Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ (2007). How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 14: 1025–1040.
Trotter KW, Archer TK (2008). The BRG1 transcriptional coregulator. Nucl Recept Signal 6: e004.
Urnov FD, Wolffe AP (2001). Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene 20: 2991–3006.
van Holde K, Zlatanova J (2006). Scanning chromatin: a new paradigm? J Biol Chem 281: 12197–12200.
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009). A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10: 252–263.
Walker JC, Harland RM (2009). microRNA-24a is required to repress apoptosis in the developing neural retina. Genes Dev 23: 1046–1051.
Wang NP, Chen PL, Huang S, Donoso LA, Lee WH, Lee EY (1990). DNA-binding activity of retinoblastoma protein is intrinsic to its carboxyl-terminal region. Cell Growth Differ 1: 233–239.
Wang SW, Kim BS, Ding K, Wang H, Sun D, Johnson RL et al. (2001). Requirement for math5 in the development of retinal ganglion cells. Genes Dev 15: 24–29.
Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S et al. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897–903.
Waterland RA (2009). Is epigenetics an important link between early life events and adult disease? Horm Res 71 (Suppl 1): 13–16.
West KL, Castellini MA, Duncan MK, Bustin M (2004). Chromosomal proteins HMGN3a and HMGN3b regulate the expression of glycine transporter 1. Mol Cell Biol 24: 3747–3756.
Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D (2007). MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem 282: 25053–25066.
Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D et al. (2009). Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 137: 1018–1031.
Yang XJ, Seto E (2008). Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 31: 449–461.
Yang Y, Stopka T, Golestaneh N, Wang Y, Wu K, Li A et al. (2006). Regulation of αA-crystallin via Pax6, c-Maf, CREB and a broad domain of lens-specific chromatin. EMBO J 25: 2107–2118.
Yang Y, Wolf LV, Cvekl A (2007). Distinct embryonic expression and localization of CBP and p300 histone acetyltransferases at the mouse αA-crystallin locus in lens. J Mol Biol 369: 917–926.
Yoo AS, Crabtree GR (2009). ATP-dependent chromatin remodeling in neural development. Curr Opin Neurobiol 19: 120–126.
Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S (2005). Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437: 754–758.
Young DW, Hassan MQ, Yang XQ, Galindo M, Javed A, Zaidi SK et al. (2007). Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proc Natl Acad Sci USA 104: 3189–3194.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920.
Yun S, Saijoh Y, Hirokawa KE, Kopinke D, Murtaugh LC, Monuki ES et al. (2009). Lhx2 links the intrinsic and extrinsic factors that control optic cup formation. Development 136: 3895–3906.
Zhang Y, Smith CL, Saha A, Grill SW, Mihardja S, Smith SB et al. (2006). DNA translocation and loop formation mechanism of chromatin remodeling by SWI/SNF and RSC. Mol Cell 24: 559–568.
Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S et al. (2008). A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol Cell 29: 92–101.
Zlatanova J, Thakar A (2008). H2A Z: view from the top. Structure 16: 166–179.
Acknowledgements
We thank Drs John Greally, Deyou Zheng, and Neena Haider for critical reading of this manuscript and Tatyana Harris for preparation of figures. Owing to spaces limitations, we apologize for a selective list of references. This work was supported by NIH Grants R01 EY012200 (AC), EY014237 (AC), EY014626 (KPM), and EY014626 (Oakland ERI), OU-William Beaumont Multidisciplinary Award (KPM). AC is a recipient of the Irma T Hirschl Career Scientist Award.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Cvekl, A., Mitton, K. Epigenetic regulatory mechanisms in vertebrate eye development and disease. Heredity 105, 135–151 (2010). https://doi.org/10.1038/hdy.2010.16
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2010.16
Keywords
This article is cited by
-
A multi-omics longitudinal study of the murine retinal response to chronic low-dose irradiation and simulated microgravity
Scientific Reports (2022)
-
Chromatin remodeling protein HELLS is critical for retinoblastoma tumor initiation and progression
Oncogenesis (2020)
-
Prospects and modalities for the treatment of genetic ocular anomalies
Human Genetics (2019)
-
DNA Methylomes Reveal Biological Networks Involved in Human Eye Development, Functions and Associated Disorders
Scientific Reports (2017)
-
CpG site methylation in CRYAA promoter affect transcription factor Sp1 binding in human lens epithelial cells
BMC Ophthalmology (2016)
