
Abstract
Purpose: There is limited information on long-term outcomes among children with Duarte galactosemia and controversy about treatment of this potentially benign condition. This study examined developmental disabilities and issues that required special education services within a population-based sample of children with Duarte galactosemia.
Methods: Children born between 1988 and 2001 who were diagnosed with Duarte galactosemia and resided in the five-county metropolitan Atlanta area at birth and from 3 to 10 years of age were linked to the (1) Metropolitan Atlanta Developmental Disabilities Surveillance Program, an ongoing, population-based surveillance system for selected developmental disabilities and (2) Special Education Database of Metropolitan Atlanta. Special education records were reviewed for children who linked. Clinical genetics records were reviewed to assess laboratory levels at the time of diagnosis and metabolic control during treatment.
Results: Of the 59 eligible children, none were found to have intellectual disability, cerebral palsy, hearing loss, vision impairment, or an autism spectrum disorder. However, five, 8.5% of 3 to 10 years or 15.2% of eligible 8 years, were identified as having received special education services, four of whom were confirmed with a speech or language disorder, or were receiving services for speech or language or both compared with 4.5% and 5.9% of children without Duarte galactosemia, respectively.
Conclusions: Despite galactose restriction until 1 year, select developmental issues associated with special education, specifically involving speech and language, have been found among some children with Duarte galactosemia.
Similar content being viewed by others

Main
Through newborn screening for galactosemia, Duarte galactosemia has been increasingly identified in the population. The Duarte allele, with a frequency among a panethnic US population of approximately 5%, is the most common mutation affecting galactose-1-phosphate uridyl transferase (GALT).1 Duarte galactosemia is relatively common, with a prevalence of approximately 1 in 2000 births, compared with 1 in 47,000 births for classic galactosemia.1,2 Infants with this variant form of galactosemia are compound heterozygotes for the Duarte variant mutation and a classic galactosemia mutation. The presence of the Duarte variant mutation results in a GALT enzyme level approximately 25% of that of children with normal functioning GALT, compared with almost zero activity among children with classic galactosemia.
Untreated classic galactosemia can result in serious medical outcomes, including feeding problems, hepatotoxicity, renal damage, brain damage, cataracts, sepsis, and death. Treatment can prevent many of the acute complications among infants, but clinical studies report poor long-term outcomes even with excellent treatment, including neurologic dysfunction, speech disorders, growth delays, ovarian failure, and intellectual disabilities.3,4
Unlike classic galactosemia, Duarte galactosemia has been considered benign by some clinicians.5 However, there has been controversy about defining this condition as benign, as well as whether and when to implement dietary interventions.5–8 Most previous studies report no clinical abnormalities despite biochemical abnormalities but still make recommendations to initiate treatment for various elevated levels of galactose-1-phosphate (Gal-1-P).
Although data on the neurodevelopmental outcomes of children with Duarte galactosemia are limited, the few studies that have been conducted either have examined intellectual disabilities solely and not other developmental outcomes, such as learning disorders or speech and language delays, or have evaluated developmental outcomes in younger ages when standardized cognitive functioning tests are less stable than when administered to school-age children.5,6,9,10 This study examined both developmental disabilities and other developmental outcomes that require special education services in childhood among a population-based sample of children with Duarte galactosemia.
MATERIALS AND METHODS
Study population
The study population included children born during the period 1988 to 2001 who were diagnosed with Duarte galactosemia after positive identification through the Georgia Newborn Screening Program. Children were included in the study sample if they resided in the five-county metropolitan Atlanta area (Clayton, Cobb, DeKalb, Fulton, and Gwinnett counties) at birth and at any point during the period when they were 3 to 10 years. Residency at birth was based on the mother's address on the newborn screening and birth certificate records. Residency during the period when children were 3 to 10 years was determined using the Accurint® search engine based on the mother's name, address, and any other available information (such as maternal date of birth or social security number). Children were excluded if they were not followed clinically by the Division of Medical Genetics at Emory University, in Atlanta, GA.
Data sources
Georgia Newborn Screening Program
The Georgia Newborn Screening Program is a collaborative effort between the Division of Medical Genetics, Department of Human Genetics at Emory University School of Medicine and the Georgia Department of Human Resources, Division of Public Health. During the period 1988 to 1990, an abnormal newborn screening test result for galactosemia was defined as the absence of fluorescence on the “Beutler” assay.11 During the period 1991 to 2001, additional criteria of >10 mg/dL of total galactose on the “Hill” assay12 were added.
Emory clinical genetics records
Following an abnormal newborn screen for galactosemia, children were referred to the Division of Medical Genetics at Emory University and diagnostic testing was performed by obtaining Gal-1-P and urine galactitol levels, GALT activity, and mutation analysis. Children identified with Duarte galactosemia were treated with a galactose-restricted (primarily by lactose restriction) diet until 1 year. Typically, children were seen at 4 to 6 months of age for monitoring before beginning solids and again at 10 to 12 months of age after a milk challenge unless more frequent monitoring was indicated. If Gal-1-P levels were within normal limits on a normal diet, the child was no longer monitored by the Emory Clinical Genetics Program and was encouraged to follow-up with their pediatrician. None of the children were identified with developmental issues during the first year of life while being treated by the Emory Clinical Genetics Program.
Clinical genetics records were reviewed to collect relevant medical histories and examine laboratory results. Records were available for 88% of the study sample. Because the focus of this study was to examine long-term developmental outcomes, children whose clinical genetics records were unavailable for review were still included in this study because they were confirmed as seen by the Emory Clinical Genetics Program and met study requirements to be eligible for linkage to developmental disability and special education data. Two registered dietitians with expertise in inborn errors of metabolism reviewed laboratory results before and during treatment to assign a level of metabolic control using a four-point scale (Table 1). Two pediatric geneticists and a developmental pediatrician assessed abstracted clinical genetics records and birth certificate data to determine if there were other causes (e.g., prematurity, injury, other medical condition) that might have contributed to a developmental disability.
Developmental disabilities data
The Metropolitan Atlanta Developmental Disabilities Surveillance Program (MADDSP) is an ongoing, population-based monitoring system for select developmental disabilities (intellectual disability, cerebral palsy, hearing loss, vision impairment, and autism spectrum disorders) conducted by the Developmental Disabilities Branch at the Centers for Disease Control and Prevention. MADDSP is based on administrative record review, at multiple sources, including educational, clinical, and service records, to identify children with developmental disabilities at 8 years. Specific surveillance methods and case definitions are published elsewhere.13 This study used available data from the 1996, 2000, 2002, and 2004 surveillance years (birth cohorts 1988, 1992, 1994, and 1996). For children not born in an eligible surveillance year who were found to be receiving special education services, a special MADDSP review was conducted to determine if these children met the MADDSP case definition for a developmental disability.
The Special Education Database of Metropolitan Atlanta (SEDMA) was created by the Centers for Disease Control and Prevention Developmental Disabilities Branch by merging the electronic files from all nine school systems in metropolitan Atlanta to identify children 3 to 10 years who received special education services. SEDMA exceptionalities represent the type of special education placement (e.g., learning disorder or speech/language impairment) and not a disability specific diagnosis. Children may not have a specific exceptionality but be eligible to receive related services such as those for speech or language or both. Data were used for the 1996 to 2004 school years.
Data linkage and analysis
The study sample was linked to birth certificate records and both the MADDSP and SEDMA databases. Birth certificate data were used to assist in establishing residency during the study period and to provide additional information to corroborate clinical data on potential risk factors, other than Duarte galactosemia, that might account for a child's developmental disability or special education exceptionality. Linkage to MADDSP was used to identify defined developmental disabilities, whereas the use of SEDMA allowed examination of other developmental issues. For children whose records were linked to SEDMA, an additional review of the special education records was conducted to provide greater detail about their special education services. Using these linkages, the prevalence of having a developmental disability or receiving special education services among children with Duarte galactosemia was calculated. To obtain a comparable estimate of the birth prevalence of children receiving special education services, we used the 1998 special education December 1 count and linked these data to 1-year survivors from the 1988 to 1995 birth cohorts (3–10 years of age in 1998). The special education count for 1998 was selected because it encompassed the greatest portion of our study birth cohort for years in which complete data were available for 1-year survivors. To determine this birth prevalence, we divided the total number of children receiving special education services in these birth cohorts (children who were born in the five counties of metropolitan Atlanta) by the sum of metropolitan Atlanta 1-year survivors across all birth cohorts. Although children with Duarte galactosemia were included in the overall birth prevalence, because of its very low incidence, we do not believe this greatly affected the comparison. The birth prevalence of special education services among all 1-year survivors in the five-county metropolitan Atlanta area may be an underestimate due to out migration of children between 1 year and 3 to 10 years which are included in the denominator.
RESULTS
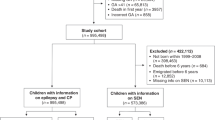
Among the 75 children with Duarte galactosemia born in the metropolitan Atlanta area, 59 (79%) continued to reside in the surveillance area during the period when they were 3 to 10 years and their records were available for linkage to MADDSP and SEDMA (Fig. 1). Of these children, none also had a developmental disability monitored by MADDSP (Fig. 1). Five (8.5%) children within the 3 to 10 year age range were identified as having received special education services (Fig. 1). Among the general population of children 3 to 10 years born and residing in metropolitan Atlanta, 4.5% (n = 13,187 of 295,939) received special education services in 1998. All five children identified as having received special education services (speech or language services or both) were 8 years at the time of their last special education evaluation. When we restricted our sample to children with Duarte galactosemia who were at 8 years and had received special education services, we found that 15.2% (5 of 33) of children with Duarte galactosemia were receiving special education services compared with 5.9% (2,264 of 38,328) of 8-year-old children in the general population (children born in 1990 and receiving special education services in 1998) (Fig. 1).

Flowchart of Duarte galactosemia cases from newborn screening identification to developmental outcome. *Birth years 1988 through 2001; therefore, 3 years of follow-up for children born in 2001 (age 3 in 2004 school year) and at least 10 years of follow-up for children born between 1988 through 1994 (at least age 10 in 1998–2004). †Birth years 1988 through 1996; therefore, all of whom had at least 8 years of follow-up in special education. This is a subset of 3 through 10 years and was restricted to establish equal follow-up time for all children.
Table 2 lists the special education exceptionalities under which these children received services. Although two children received services through a speech and language exceptionality, after record review, two additional children were confirmed to have received special education services through learning disorder exceptionality and met the eligibility criteria for related speech and language services. More specifically, Child 1 and Child 2 were served under an exceptionality of learning disorder, but also received speech and language services to address moderate to severe articulation disorder (Child 1) and a mild to moderate language disorder (Child 2). Detailed school records were not available for an additional review for Child 3 to assess the specific types of services and delays. Children 4 and 5 were served under speech and language impairment exceptionalities. Although further details were not available, it is appropriate to assume that Children 4 and 5 were receiving speech and language services.
For children identified with special education needs, newborn screening levels of galactose ranged from 13 to 30 mg/dL. Gal-1-P levels at diagnosis ranged from <1 to 23 mg/dL with four of the five children considered to have moderately high to high levels (9.2–23). Galactose restriction for these children with Duarte galactosemia was initiated at a mean age of 23 days, with a range from 16 to 34 days (Table 2).
For children with Duarte galactosemia who did not receive special education services (n = 54), newborn screening levels of Gal-1-P were available for 29 of the children with a median level of 15 mg/dL (mean = 16.4 mg/dL) and a range of 4.6 to 29 mg/dL. Figure 2 depicts Gal-1-P levels before galactose restriction (at diagnosis) and metabolic control during this restriction. Among the children who did not receive special education services, the majority (56%) had normal Gal-1-P levels by the time of diagnosis, 10% had mildly elevated levels, 15% had moderately elevated, 10% had high levels, and 8% had unknown levels. Galactose restriction for these children began at a median age of 22 days (mean = 29 days), ranging from 1 to 101 days. During dietary treatment, the majority (88%) had normal levels, 4% had mildly elevated levels, 2% had moderately elevated levels, none had high levels, and 6% had unknown levels.

Metabolic control (based on galactose-1-phosphate levels) of children with Duarte galactosemia not receiving special education services.
DISCUSSION
There is a dearth of epidemiologic data on the neurodevelopmental outcomes of children with Duarte galactosemia. We are encouraged that no occurrences of intellectual disability, cerebral palsy, autism spectrum disorders, hearing loss, or vision impairment were identified among these children with Duarte galactosemia. Although based on the findings from the previous work by Van Naarden Braun et al.,14 we posited that we may identify children with Duarte galactosemia who are receiving special education services, we did not have an a priori hypothesis on the age at which these services would be received. As a result of finding that all of the five children identified as receiving special education services for speech and or language disorders were 8 years at their last evaluation, we restricted our sample to children who were 8 years and had special education evaluation(s) through the age of 8 years. As would be expected, we identified a higher percentage of the population with Duarte galactosemia receiving special education services, which was approximately two and half times that of the general population of 8-year-old children. Although this study found a higher percentage of children with Duarte galactosemia receiving special education services compared with the general population of children for both 3 to 10 years and 8 years, because of the very low prevalence of Duarte galactosemia, it was difficult to conclude using statistical testing that the prevalence of receiving special education services among children with Duarte galactosemia was higher than we would have expected by chance among the general population in the metropolitan Atlanta area.
These findings reinforced the first examination of this population conducted by Van Naarden Braun et al.,14 which similarly found a higher than anticipated number of children with Duarte galactosemia receiving special education services (n = 6, five with speech and language as exceptionality, two of whom were identified by both studies in the overlapping birth cohorts) in their examination of developmental outcomes among children identified with a metabolic disorder through newborn screening. This study expanded the previous study by also examining the control population of children with Duarte galactosemia not receiving special education services to compare laboratory findings and metabolic control during dietary treatment and confirming residency eligibility for linkages, as well as calculating the prevalence of special education services received by the general surveillance area population of 8-year-old children.
A recent study by Ficicioglu et al.,10 which examined young children (1–6 years) with Duarte galactosemia, found significant differences in adaptive scores but not language or intelligence quotient between treated and untreated children, although these outcomes did not appear to be related to biochemical markers. Because of the young age of the children in the study by Ficicioglu et al., intelligence testing may be unstable and not yet discern issues in development, a point discussed by Ficicioglu et al.9,10 This study did examine children at an older age and found several children with developmental concerns warranting placement in special education. Follow-up until at least 8 years appeared to be a crucial period for identification of developmental issues in this study.
Children with Duarte galactosemia receiving special education services had speech and language issues, with at least one child having a severe articulation disorder. Children with classic galactosemia frequently have speech and language problems, particularly verbal dyspraxia.15–18 Waisbren et al.15 administered speech evaluations to eight early treated children with galactosemia, 3.6 to 11.6 years, and found seven had at least one speech and language deficit, five of whom had deficits in articulation. Another study by Nelson et al.16 evaluated the speech of 24 patients with galactosemia and found 54% had verbal dyspraxia and 8% had an articulation disorder without verbal dyspraxia. Both these studies reported that the problems in speech and language occurred despite early galactose restriction and biochemical control. Because of this finding and the presence of elevated levels of Gal-1-P in cord blood, Waisbren et al.15 suggested the possibility of a prenatal effect. In this study, we found speech and language problems despite galactose restriction until 1 year, although the age at initiation of galactose restriction tended to be a week or two later for children with Duarte galactosemia compared with children with classic galactosemia. Although a clear correlation between days of dietary galactose ingestion and speech and language disorders among children with classic galactosemia has not been established, age at initiation of lactose-restricted diet could not be ruled out as a contributing factor in this study.3,18
One limitation of this study was that it relied on review of secondary data rather than in-person speech and language evaluations. It is possible that we missed children whose speech and language issues were not severe enough for enrollment in special education services, or who received private speech and language services and were not receiving special education services. We attempted to review the special education records to provide more details about the actual services received beyond the exceptionality listed. Unfortunately, the availability of these records was limited and not all records could be accessed (e.g., some of the records were in storage or the child had moved to a different school district). Although this study covered a 14-year period, the likelihood of being identified with a developmental disability and/or receiving special education services was greater for older children (earlier birth cohorts) than younger children. This is evidenced in our findings when we restricted our sample to children with 8 years of follow-up and suggests that including younger children may result in an underestimate of the impact of the need for special education services among children with Duarte galactosemia. In addition, although we did not have access to early intervention records, we would have only missed children whose developmental delay resolved before entry into public school and therefore further special education services were not warranted.
The systematic location of data on metabolic treatment and control was also a challenge. Because of the small case size and limited availability of laboratory data, statistical testing was not conducted to compare children receiving and not receiving special education services. In general, there did not appear to be major differences in Gal-1-P levels among the two groups. Despite galactose restriction, many children were considered to have higher than normal Gal-1-P levels. Although this could indicate that the child was not given the recommended diet, more often it was due to the time it took to lower a high initial Gal-1-P level, due to either later age of initiation of galactose restriction or higher than average level at birth. Furthermore, because all children with Duarte galactosemia in this study were treated until at least 1 year, it was not possible to evaluate developmental trajectory without galactose restriction.
However, despite these limitations, the study had important strengths: in particular, the study used a population-based approach to examine both defined developmental disabilities and other developmental issues among children with Duarte galactosemia. By establishing residency from birth to 3 to 10 years, we were able to calculate the prevalence of children with Duarte galactosemia, receiving special education services and compare it with that among the general population of children born in metropolitan Atlanta for both the 3 to 10 year-old cohorts as well as those aged 8 years. The prevalence of receiving special education services among 1-year survivors may be an underestimate due to out migration between 3 and 10 years. Although both estimates are potential undercounts, our calculations use comparable cohorts of 1-year survivors on which to base the comparison of the prevalence of children receiving special education services with and without Duarte galactosemia.
Although Duarte galactosemia is considered by some to be a benign condition, these data raise questions as to whether this is a valid conclusion and suggest that further investigation is needed. The population-based approach to conducting surveillance of developmental disabilities used in this study currently is being replicated at 10 US sites as a part of the Autism and Developmental Disabilities Monitoring Network.19 Because newborn screening programs are operational in all states across the United States, with additional resources, this study might be replicated across the Autism and Developmental Disabilities Monitoring Network and data could be pooled. With a larger sample size, more conclusive evidence might be obtained. The importance of long-term, follow-up studies of Duarte galactosemia cohorts within public health programs has been underscored by a recent statement of the US Secretary of Health and Human Services' Advisory Committee on Heritable Disorders and Genetic Diseases in Newborns and Children stating that the principle goal of long-term follow-up is to “assure the best possible outcomes for individuals with disorders identified through newborn screening.”20
This study evaluated long-term developmental outcomes among a population-based sample of children with Duarte galactosemia. Despite galactose restriction until 1 year, select developmental issues associated with special education services specifically involving speech and language issues have been found among some children with Duarte galactosemia. The initial Gal-1-P level and duration of elevation before galactose restriction might play a role in developmental outcomes. However, there may be other biochemical indices that are better markers for long-term outcomes. Further large-scale, epidemiologic studies are needed to evaluate these findings and explore the effect of Duarte galactosemia on long-term outcomes.
REFERENCES
Suzuki M, West C, Beutler E . Large-scale molecular screening for galactosemia alleles in a pan-ethnic population. Hum Genet 2001; 109: 210–215.
Elsas L, Dembure P, Langley S, Paulk E, Hjelm L, Fridovich-Keil J . A common mutation associated with the Duarte galactosemia allele. Am J Hum Genet 1994; 54: 1030–1036.
Waggoner D, Buist N, Donnell G . Long-term prognosis in galactosemia: results of a survey of 350 cases. J Inherit Metab Dis 1990; 13: 802–808.
Schweitzer S, Shin Y, Jakobs C, Brodehl J . Long-term outcome in 134 patients with galactosemia. Eur J Pediatr 1993; 152: 36–43.
Levy HL, Spe SJ, Walton DS, et al. Galactose-1-phosphate uridyl transferase deficiency due to Duarte/galactosemia combined variation: clinical and biochemical studies. J Pediatr 1978; 92: 390–393.
Schwarz HP, Zuppinger KA, Zimmerman A, Dauwalder H, Scherz R, Bier DM . Galactose intolerance in individuals with double heterozygosity for the Duarte variant and galactosemia. J Pediatr 1982; 100: 704–749.
Kelly S . Significance of the Duarte/classical galactosemia genetic compound. J Pediatr 1979; 94: 937–940.
Gitzelmann R, Bosshard NU . Partial deficiency of galactose-1-phosphate uridyl transferase. Eur J Pediatr 1995; 154: S40–S44.
Kamphaus R . Clinical assessment of child and adolescent intelligence, 2nd ed. Boston: Allyn & Bacon, 2001; 64–70.
Ficicioglu C, Thomas N, Yager C, et al. Duarte (DG) galactosemia: a pilot study of biochemical and neurodevelopmental assessment in children detected by newborn screening. Mol Genet Metab 2008; 95: 206–212.
Beutler E, Baluda MC . A simple spot screening test for galactosemia. J Lab Clin Med 1966; 68: 137–141.
Hochella NJ, Hill JB . Fluorometric screening for galactosemia utilizing the autoanalyzer. Clin Chem 1969; 10: 949–960.
Bhasin T, Brocksen S, Avchen R, Van Naarden Braun K . Prevalence of four developmental disabilities among children aged 8 years—Metropolitan Atlanta Developmental Disabilities Surveillance Program, 1996 and 2000. MMWR Surveill Summ 2006; 55: 1–9.
Van Naarden Braun K, Yeargin-Allsopp M, Schendel D, Fernhoff P . Long-term developmental outcomes of children identified through a newborn screening program with a metabolic or endocrine disorder: a population-based approach. J Pediatr 2003; 143: 236–242.
Waisbren SE, Norman TR, Schnell RR, Levy HL . Speech and language deficits in early treated children with galactosemia. J Pediatr 1983; 102: 75–77.
Nelson CD, Waggoner DD, Donnell GN, Tuerck JM, Buist NR . Verbal dyspraxia in treated galactosemia. Pediatrics 1991; 88: 346–350.
Robertson A, Singh RH, Guerrero NV, Hundley M, Elsas LJ . Outcomes analysis of verbal dyspraxia in classic galactosemia. Genet Med 2000; 2: 142–148.
Ridel KR, Leslie ND, Gilbert DL . An updated review of the long-term neurological effects of galactosemia. Pediatr Neurol 2005; 33: 153–161.
Rice CE, Baio J, Van Naarden Braun K, Doernberg N, Meaney FJ, Kirby RS, ADDM Network. A public health collaboration for the surveillance of autism spectrum disorders. Paediatr Perinat Epidemiol 2007; 21: 179–190.
Kemper AR, Boyle CA, Aceves J, et al. Long-term follow-up after diagnosis resulting from newborn screening: statement of the US Secretary of Health and Human Services' Advisory Committee on Heritable Disorders and Genetic Diseases in Newborns and Children. Genet Med 2008; 10: 259–261.
Acknowledgements
We acknowledge Ms. Tricia Page, Newborn Screening Manager at Emory University, for her assistance in obtaining newborn screening data and facilitating clinical record review.
Author information
Authors and Affiliations
Corresponding author
Additional information
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Powell, K., Van Naarden Braun, K., Singh, R. et al. Long-term speech and language developmental issues among children with Duarte galactosemia. Genet Med 11, 874–879 (2009). https://doi.org/10.1097/GIM.0b013e3181c0c38d
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181c0c38d
Keywords
This article is cited by
-
Developmental outcomes of children with Duarte galactosemia: exploring the bases of an apparent contradiction in the literature
Genetics in Medicine (2019)
-
Appropriateness of newborn screening for classic galactosaemia: a systematic review
Journal of Inherited Metabolic Disease (2016)
-
Newborn screening for galactosemia: a 30-year single center experience
World Journal of Pediatrics (2015)
-
A brief overview of galactosemia newborn screening in the United States
Journal of Inherited Metabolic Disease (2014)
