
Abstract
Purpose:
Autoimmune lymphoproliferative syndrome is a disorder of lymphocyte apoptosis. Although FAS molecules bearing mutations in the signal-transducing intracellular death domain exhibit dominant-negative interference with FAS-mediated apoptosis, mechanisms for pathology of non-death domain FAS mutations causing autoimmune lymphoproliferative syndrome are poorly defined.
Methods:
RNA stability, protein expression, ligand binding, and ability to transmit apoptosis signals by anti-FAS antibody or FAS ligand were determined for a cohort of 39 patients with non-death domain autoimmune lymphoproliferative syndrome. Correlations between mutation type and disease penetrance were established in mutation-positive family members.
Results:
Frameshifts or transcriptional stop mutations before exon 7 resulted in messenger RNA haploinsufficiency, whereas an amino-terminal signal sequence mutation and certain intracellular truncations prevented cell surface localization of FAS. All resulted in decreased FAS localization, inability to bind FAS ligand, and reduced FAS ligand-induced apoptosis. Extracellular missense mutations and in-frame deletions expressed defective FAS protein, failed to bind FAS ligand, and exhibited dominant-negative interference with FAS-mediated apoptosis. Mutation-positive relatives with haploinsufficient or extracellular mutations had lower penetrance of autoimmune lymphoproliferative syndrome clinical phenotypes than did relatives with death domain mutations.
Conclusion:
We have defined molecular mechanisms by which non-death domain FAS mutations result in reduced lymphocyte apoptosis, established a hierarchy of genotype-phenotype correlation among mutation-positive relatives of patients with autoimmune lymphoproliferative syndrome, and demonstrated that FAS haploinsufficiency can lead to autoimmune lymphoproliferative syndrome.
Genet Med 2012:14(1):81–89
Similar content being viewed by others


Introduction
Autoimmune lymphoproliferative syndrome (ALPS; OMIM # 601859) a rare, heritable disorder of immune homeostasis is characterized by chronic lymphadenopathy and/or splenomegaly and expansion of a normally rare population of mature TCRαβ+CD3+CD4−CD8− T (double-negative T; DNT) cells.
The majority of patients with ALPS have germline or somatic point mutations in the tumor necrosis family receptor superfamily 6 gene FAS, also known as TNFRSF6, APO1, APT1, and CD95.1,2 These patients have recently been reclassified as ALPS-FAS to distinguish them from patients with ALPS with other molecular diagnoses.3 FAS, a transmembrane signaling protein for a critical immune homeostasis apoptosis pathway, regulates immune responses by removing autoreactive lymphocytes.4,5,6,7,8 A surface homotrimer of FAS molecules interacts with homotrimerized FAS ligand (FAS-L) and initiates a cascade of caspase cleavages resulting in programmed cell death. Defects in the FAS gene result in impaired death signaling through the FAS/FAS-L pathway. Mutations disrupting the highly conserved FAS death domain (DD) have been well characterized, and the presence of one or two mutant chains in the homotrimer is sufficient to disrupt the functioning of the assembled receptor2 Thus, the majority of FAS mutations are dominant negative and heterozygous, resulting in a mutant protein that interferes with the function of the normal protein encoded on the wild-type allele.
Additional mechanisms may be present in some patients with ALPS, such as haploinsufficiency, in which a reduced amount of normal protein is insufficient for full physiological function. Haploinsufficiency has been associated with inadequate protein leading to disruption of homeostasis, such as in type I hereditary angioedema,9 Carney complex,10 or hereditary hypercholesterolemia.11 Haploinsufficiency can also lead to diminished transmission of growth signals, as in sonic hedgehog mutations in holoprosencephaly.12 A single case report by Roesler et al.13 described a patient with a mutation causing in-frame loss of exon 6 and overproduction of soluble FAS. They concluded that the clinical phenotype was not due to increased soluble FAS, but rather due to haploinsufficiency, as increasing the surface expression of FAS reversed the in vitro apoptosis defect, whereas it was not corrected in the presence of excess soluble FAS.
We report comprehensive evidence of FAS haploinsufficiency causing nonmalignant lymphoproliferation and autoimmunity in human disease. Approximately half of the FAS mutations of patients with ALPS are outside the exon 9 DD region. We determined functional consequences of these mutations and found that most mutations causing frameshifts or stops 5′ to exon 7 resulted in haploinsufficiency, whereas extracellular missense mutations and in-frame deletions exhibited dominant-negative interference. A signal sequence mutation and certain splicing mutations caused protein-positive functional haploinsufficiency rather than dominant-negative interference. Mutation-positive relatives of probands with haploinsufficient or extracellular mutations had a lower penetrance of ALPS clinical phenotypes than did relatives with DD mutations.
Materials and Methods
Patients
Patient samples and data were obtained from previously unreported, unrelated patients with ALPS (N = 39) and their family members (N = 160) with informed consent under a protocol approved by the institutional review board of National Human Genome Research Institute and National Institute of Allergy and Infectious Diseases (protocol no. 95-I-0066). All probands met criteria for ALPS in the most severe phenotype class 1 (PC1) established by Vacek et al.14 Mutation-positive relatives exhibited a range of penetrance of ALPS features and were scored accordingly.2,14 Ficoll-separated lymphocytes were cultured in complete RPMI medium for 3 days with 1 µg/mL phytohemagglutinin then expanded with 50 U/mL interleukin-2. DNA was extracted from whole blood (Puregene DNA extraction kit, Qiagen, Valencia, CA).
FAS sequencing
The nine exons and associated splice sites of the FAS locus were polymerase chain reaction (PCR) amplified, sequenced as described,15 and the resulting sequence was compared with the genomic reference NM_000043.3.
Flow cytometry
T lymphocytes were stained for surface FAS as described1 or were incubated with cross-linked rhFAS-L or anti-6X-His and stained with FITC goat antimouse secondary (Invitrogen, Carlsbad, CA) for FAS-L binding. Fluorescence index (FI) was calculated as the ratio of the geometric mean channel of CD95-stained cells compared with isotype-stained cells or the ratio of geometric mean channel of cells stained with cross-linked FAS-L compared with crosslinker alone.
Cloning and transfection
Full-length FAS complementary DNA (cDNA) was isolated as described1 and cloned into pcDNA4-topo (Invitrogen). DNA minipreps from 20 to 40 independent colonies and the original reverse transcriptase (RT)-PCR product were sequenced for the presence of each patient’s specific mutation.
Wild-type (WT) and mutant FAS cDNA expression constructs in pCI expression vector (Promega, San Luis Obispo, CA) were sequence verified. Transfection was performed as described1; apoptosis was induced with either anti-CD95 CH11 antibody (Millipore, Billerica, MA) or cross-linked FAS-L (R&D Systems, Minneapolis, MN), and after 18 hours, propidium iodide live dead analysis was performed counting by constant time1 on a FACSCalibur (BD, San Jose, CA).
Penetrance analysis
Individuals were divided into four groups according to their FAS mutation type. Penetrance was scored in mutation-positive relatives, a family score generated for each family, and Fisher exact test was used to test for a significant association between the mutation group and a low, nonpenetrant, family score (see Methods, Supplementary Data online).
Results
Non-DD FAS mutations in ALPS
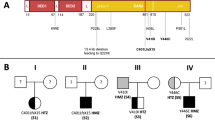
Genomic sequencing of FAS identified 18 distinct variants in the extracellular region of FAS in 22 of 39 unrelated probands ( Table 1 ). These mutations, only one of which has been reported previously, are shown in Figure 1 and included four each missense, nonsense, splice site, simple insertion/deletion, and two complex insertion/deletion mutations. The missense mutation in exon 3, c.332 A>G causing H111R, was seen in four unrelated individuals. Of the 17 remaining patients, 16 different mutations affecting the transmembrane and non-DD intracellular regions were identified, including two nonsense, 10 splice site, two deletions, one insertion, and one complex insertion/deletion.

Newly identified non-death domain FAS mutations. Diagram of the FAS gene identifying intron/exon structure and protein domains. Mutations are indicated at their approximate location and identified by symbols corresponding to mutation type.
Analysis of RNA stability in Non-DD mutations in FAS
We undertook analysis of RNA stability to determine whether haploinsufficiency is a pathologic mechanism in ALPS. Nonsense-mediated decay (NMD) is often observed in cases of premature termination codons occurring more than 54 bp before the terminal splice event.17 The predicted occurrence of NMD based on that generalization ( Table 1 ) required verification of the cDNA species present in patient’s primary T cells. As some mutations resulted in exon skipping or reduced, but not absent, levels of mutant RNA, neither real time nor allele-specific PCR was sufficient to determine the effect of a mutation at the RNA level. We, therefore, isolated FAS RNA from each patient’s activated T-cell cultures and sequenced a panel of independent full-length cDNA clones to measure the relative expression of the mutant versus wild-type allele.10 The patients were separated into two groups based on prediction of transcript stability; the mutant versus wild-type allele frequency was plotted for each group ( Figure 2 ). The 11 mutations predicted to cause NMD had a mutant allele frequency (MAF) of 0.18, significantly lower than the expected wild-type MAF of 0.5 (one sample t test, P = 0.0002); in contrast, the 14 mutations predicted to result in stable transcripts had a mean MAF of 0.52.

Mutant allele frequency of patients with FAS mutations. For each patient, the allelic expression ratio of clones from the mutation-bearing allele to the total clones isolated is shown. The predicted ratio for a healthy control is 0.5, whereas that corresponding to complete absence of mutant allele expression is 0.0. Patients were grouped by prediction of the effect of their mutations ( Table 1 ). ***P = 0.0002. NMD, nonsense-mediated decay.
All four missense mutations in exons 1–5 (p.L7P, p.D108G, p.H111R, and p.C135Y) had equivalent numbers of mutation-bearing and wild-type clones, indicating stable expression of full-length mutant messenger RNAs (mRNAs). Nonsense mutations, c.189T>A, p.C63X, in exon 2 and, c.273C>A, p.Y91X, in exon 3 were not represented in any cDNA clones nor was the point mutation seen in the pooled RT-PCR product. In the latter case, the c.273C>A mutant allele generated several clones skipping exons 3 and 4 to produce a frame-shifted transcript resulting in premature termination. Two of the extracellular mutations with exon 2 premature stop codons (c.53T>G, p.L18X, and c.133G>T, p.E45X) had mutant cDNA present, possibly due to the proximity of the mutation to the start codon.18 In both cases, the translation of the mRNA would be predicted to result in a short, secreted peptide.
Insertion/deletion and splice mutations resulted in a variety of changes at the mRNA level. The three-nucleotide deletion, c.324_326delTGA, causing deletion of D108, gave rise to stable mRNA. In contrast, splice mutant c.30 + 1G>A and complex mutant c.397_398delTTinsA had no cDNA clones represented, indicating NMD causing mRNA instability. The c.46_47delGC frameshift mutation was represented by fewer than 15% of cDNA clones isolated, also consistent with NMD. Intron 2 splice mutation, c.197–1G>A was predicted to cause the inframe, 46 amino acid loss of exon 3. We found reduced numbers of mutant clones, 8 of 23 (MAF: 0.26), half of which deleted exon 3. Two of the remaining clones deleted exons 3 and 4, whereas two deleted exons 3, 4, and 6, both resulting in frameshifts. Similarly, the intron 4 splice mutation, c.443 + 5G>A, yielded 6 of 20 clones from the mutant allele (MAF: 0.30), four deleting exons 3 and 4, one deleting exons 3, 4, and 6, and one deleting exons 4 and 6, all predicted to produce frameshifts. Exon 5 complex mutation, c.475_489del15insA, had multiple RNA species from the mutant allele: although five clones contained the c.475_489del15insA frameshift mutation alone, six clones contained the mutation and various splice products deleting exon 4 and restoring the correct translation frame, encoding a protein with an extracellular deletion.
One mutation, c.341A>T, predicted to cause p. E98V, was shown by cDNA clone analysis to alter normal splicing. In mRNA from T cells bearing this mutation, 18 of 20 clones were from the wild-type allele, whereas the two clones from the mutant allele had frameshift deletions either of exon 4 alone or exons 3 and 4 together. Analysis of the wild-type and mutated sequences by human splicing factor19 predicted the c.341A>T mutation to destroy an exonic splice enhancer, possibly accounting for the observed exon deletions.
Mutations identified in exon 6, encoding the transmembrane domain, and exons 7–9, encoding the intracellular region of FAS, had different effects on mRNA processing and stability compared with the mutations in extracellular domain encoding exons 1–5. Three mutations, c.506–6C>G, c.506–16A>G, and c.568G>A, led to in-frame skipping of exon 6. Translation of this mRNA species would result in a protein lacking the transmembrane domain. The resulting soluble protein has been detected in supernatants of cultured cells using FAS-specific antibodies.13,20 Transcripts deleting exon 6 from each of these mutations accounted for the majority of the clones analyzed (MAF = 1.00, 0.92, and 0.79, respectively). The high rate of detection of exon 6 deletion transcripts may be due to the fact that this is a naturally occurring alternative splice form of FAS. As the base change resulting in the splice mutation was not present in the resulting transcript, it was not possible to distinguish between the mutant and wild-type alleles.
To ascertain whether the sequenced clones accurately reflected the cDNA species amplified by RT-PCR, the total RT-PCR product was sequenced as well. The sequence was carefully examined for the presence of the patient mutation or splice products seen in the clones. In the cases of missense mutations such as c.20T>C, the product resembled the heterozygous sequence seen in the genomic sequencing of the patient, reflecting equal presence of both alleles. For splice mutant c.30 + 1G>A and complex mutant c.397_398delTTinsA, there was no visible peak of the mutant alleles in the PCR product. There was, however, a visible mutant tracing present in the sequence from c.46_47delGC, although it was much smaller than the wild-type sequence. The transcripts deleting exon 6 in the patients with exon 6 splice mutations were the predominant species seen in the sequence of the RT-PCR product. Estimating allele frequencies of pooled DNA by relative peak heights in fluorescent sequence has been demonstrated to be accurate and reliable.21
CD95 expression and FAS-L binding
As many of the mutations identified in Table 1 were predicted to decrease mRNA stability or prevent protein localization at the cell membrane, we examined cell surface expression of FAS/CD95 on activated patient T cells. Cells were stained with a FAS-specific antibody or isotype control, and the FI was calculated. The difference in individual patient’s cell surface expression of FAS is apparent when examining histograms ( Figure 3a ). Wild-type control cells had an FI of 7.72, and the extracellular missense mutations with stable mRNA p.D108G and p.H111R had nearly equal FIs of 6.89 and 7.03, respectively. Cells from three patients with demonstrated mutant allele mRNA instability (p.Y91X, p.F133IfsX52, and p.W176X shown in Figure 3a , left panel) were analyzed at the same time, and all had FIs below 4, approximately half the staining intensity of the wild-type control and the cells with extracellular missense FAS defects.

FAS expression and FAS-L binding in T cells of patient with ALPS. (a) FAS surface expression in phytohemagglutin-inactivated, IL-2-expanded T cells from a control subject (top) and patients with ALPS with heterozygous FAS defects, clustered by mutation type (wild type, missense, and nonsense) and ordered 5′–3′. Results shown are representative of five experiments. (b) Ratio of intensity of cell surface expression of FAS in patient versus control T cells, measured as the fluorescence index (FI) of anti-FAS-stained cells from each patient versus simultaneously analyzed control. Data are grouped by mutation group ( Table 1 , right column). WT, ratios of FAS fluorescence intensity of T cells from one control relative to another simultaneously assayed control. **P = 0.0025, ***P ≤ 0.0001. (c) FAS-L binding, in phytohemagglutinin-activated, IL-2 expanded T cells from a control subject (top) and patients with ALPS with heterozygous FAS defects, measured in T cells from a wild-type control (same controls as in panel a) and three patients with ALPS, two with FAS mutations affecting the signal sequence and one with early frameshift mutation. Numbers in panels a and c are FI values (see “Methods” for details). Results shown are representative of five experiments.
We saw an identical pattern of reduced surface FAS expression in two unrelated patients with the signal sequence mutation L7P ( Figure 3a , right panel). Although cells from these subjects had a cDNA MAF of 0.40, they lacked cell surface expression of the mutant protein. Similarly, cells with a heterozygous c.341A>T, p.G112DfsX37 mutation showed surface expression of CD95 diminished to a haploinsufficient level ( Figure 3a , right panel), consistent with the corresponding cDNA analysis revealing a frameshifting splice deletion of exon 4 and loss of mRNA stability.
Although cell surface FAS FI was somewhat variable between control cell cultures ( Figure 3b , left), the ratio of the patient versus control fluorescence was consistently lower for patients with mutations causing absent surface expression of FAS ( Figure 3a,b ). Mutations predicted to result in haploinsufficiency ( Figure 3b , center) had significantly reduced levels of cell surface FAS (two-tailed t test, P < 0.0001) compared with control cells. Cells with mutations predicted to permit expression of a mutant protein ( Figure 3b , right) had lower FI than wild type (two-tailed t test, P = 0.0025) but significantly higher than haploinsufficient mutations (two-tailed t test, P = 0.0001).
We examined the ability of FAS-mutant T lymphocytes from patients with ALPS to bind FAS-L ( Figure 3c ). Control cells bound FAS-L with an FI of 4.17, whereas cells from patients with mutations in the ligand-binding domain (p.D108G and p.H111R) showed reduced bound FAS-L FI values of 2.26 and 1.46, respectively. Cells with haploinsufficient mutations (p.Y91X, pF133IfsX52, and p.W176X, Figure 3c , left panel; p.G112DfsX37, Figure 3c , right panel) were only minimally able to bind FAS-L, with FI values ranging from 1.19 to 1.48. Similarly, the patients with a mutated signal sequence, p.L7P, not only failed to have normal levels of cell surface FAS expression but were also unable to bind FAS-L ( Figure 3c , right panel), suggesting a threshold surface FAS expression requirement for effective FAS–FAS-L engagement.
Apoptosis of transfected BW cells
Although mutations causing mRNA instability led to loss of cell surface staining and FAS-L binding, there were several mutations predicted to result in production of an altered protein. To determine apoptotic potential and dominant-negative inhibition by these altered proteins, as demonstrated for DD mutations of FAS,1,2 pCI expression constructs of eight mutations that generated stable mRNA transcripts were tested. Transfection into FAS-nonexpressing cell line BW5147.1 was performed with equal amounts of plasmids encoding mutant FAS and either green fluorescent protein (GFP) or wild-type FAS, and apoptosis was induced by incubation with CH11 or multimeric FAS-L.
One example of cells transfected with GFP plus the mutant construct is Figure 4a . Cells transfected with the pCI-GFP plus empty pCI vector show a population of GFP-positive cells, indicated by the box in the upper left scatter plot. Treatment with CH11 and FAS-L did not induce apoptosis, as evidenced by the persistence of the GFP-positive cells in the middle and right upper scatter plots. In the second row, after transfection with GFP plus wild-type FAS, the GFP-positive population was largely eliminated after CH11 or FAS-L exposure, reflecting apoptosis induced by cross-linking of wild-type FAS at the cell surface. The third row shows cells transfected with a construct containing the DD mutation, p.I262T, known not to induce apoptosis when transfected alone, as shown here by the persistence of GFP-positive cells after CH11 or FAS-L treatment, and moreover to impair apoptosis by WT FAS in a dominant manner.1 As expected, the p.L7P construct, with a defective signal sequence preventing surface protein expression, is unable to permit either CH11 or FAS-L-induced apoptosis in BW5147.1 cells. In contrast, the p.D108G missense mutation construct in the bottom row is expressed along with GFP and induces apoptosis after CH11 treatment. When treated with FAS-L, however, many GFP-positive cells fail to undergo apoptosis. As CH11 crosslinks the FAS molecule at surface residues 126–135,22 this site is intact in the p.D108G mutant, whereas the binding of cross-linked FAS-L is reduced, presumably because the p.D108G mutation disrupts the FAS-L binding site.23,24

Flow cytometric analysis of cell death in transfected BW5147.1 cells. (a) Forward scatter versus GFP fluorescence in cells transfected with pCI-GFP and pCI containing the indicated FAS cDNA constructs. Loss of FAS/GFP-positive cells (black box, upper left plot) is attributed to apoptosis induced by either CH11 anti-CD95 antibody (middle column) or FAS-L (right column). (b) Percent cell death normalized to wild-type FAS control. Cells cotransfected with equal amounts of plasmids containing FAS cDNA with the specified mutation and either GFP or wild-type FAS were treated with either CH11 or FAS-L. Data are representative of five experiments. *Indicates >50% reduction from control.
Further experiments were performed to assess whether extracellular FAS mutations could act in a dominant-negative manner to inhibit apoptosis by wild-type FAS, as do the well-studied mutations in the intracellular DD. As shown in Figure 4b , mutant constructs cotransfected with either GFP or wild-type FAS permitted determination of their intrinsic ability to induce apoptosis and their dominant-negative effects. The apoptosis of WT FAS after exposure to either CH11 antibody (black bars) or FAS-L (white bars), designated 100% ( Figure 4b , left), was nearly completely inhibited, as expected, by the previously known FAS intracellular DD mutant p.I262T (0% cell loss with CH11 and 10% with FAS-L, Figure 4b , right). The p.L7P mutant, which alone was unable to induce apoptosis ( Figure 4b , p.L7P + GFP), when expressed together with wild-type FAS had no significant inhibitory effect on WT apoptosis ( Figure 4b , p.L7P + WT), consistent with failure to be delivered to the cell surface. The next FAS mutant, an in-frame deletion of 46 amino acids, including part of the FAS-L binding domain, did not induce apoptosis alone and reduced CH11-induced apoptosis to about 45% of WT. FAS-L-induced killing of WT by this construct was inhibited to an even greater degree, suggesting that not only does p.P65_G110del fail to bind FAS-L on its own but also the mutant molecule when incorporated into FAS trimers prevents FAS-L binding. The p.D108G construct was able to induce apoptosis on its own when stimulated with CH11 and also in conjunction with wild-type FAS. When stimulated by cross-linked FAS-L, however, the p.D108G FAS mutant allowed only 30% killing compared with the wild type. When cotransfected with wild type, the p.D108G reduced but did not prevent apoptosis. In contrast, the p.H111R construct did not conduct an apoptosis signal from either CH11 or FAS-L and exhibited strong dominant inhibition of apoptosis by WT FAS to a degree similar to that of the DD mutant.
Those constructs with mutations resulting in the loss of the DD (p.Q196TfsX16 and p.E218MfsX4) were similarly not able to induce apoptosis on their own, but when cotransfected with wild-type FAS, p. P201fs inhibited 40% of the apoptosis that occurred with WT FAS alone, whereas p.Q196TfsX16 did not have significant inhibitory activity.
Penetrance of ALPS is lower with Non-DD than with DD FAS mutations
Using the rating scale for severity of the ALPS phenotype established by Vacek et al.,14 we analyzed 160 mutation-positive relatives of ALPS probands with all types of mutations. Descriptive statistics are provided for previously defined phenotype severity class 1 (PC1) by mutation group.
The 160 relatives were from 54 families, ranging from 1 to 10 family members with an average of three individuals per family, whereas 18 families had just one member. There was appreciable within-family correlation, with the intraclass correlation coefficient of 0.52 for PC1. The distribution of family PC1 scores in each mutation group (1, haploinsufficient; 2, extracellular; 3, transmembrane and intracellular non-DD; and 4, DD mutations) showed a wide range of PC1 scores; however, there were more families with low PC1 scores with the haploinsufficient and extracellular mutations (mutation classes 1 and 2, 67% and 80%, respectively) than with intracellular or DD mutations (mutation classes 3 and 4, 33%, and 27%, respectively) (Figure, Supplementary Data online). Corresponding to previous studies demonstrating high penetrance with DD mutations,2,25,26 only 27% of individuals in families with DD mutations failed to meet the PC1 threshold of 1.5 indicating most mutation-positive family members had a penetrant phenotype. The median family PC1 score was 1.3 (interquartile range: 1.0 –1.6) for the haploinsufficient mutations; 1.3 (interquartile range: 1.2–1.3) for extracellular mutations; 2.1 (interquartile range: 1.5–2.9) for intracellular mutations, and 2.0 (interquartile range: 1.5–2.5) for DD mutations. There was a significant association between low family PC1 score and mutation group (P = 0.022).
Discussion
We identified and characterized 35 FAS non-DD mutations in previously unreported patients with ALPS. The effects of several of the mutations on mRNA and protein were not always predictable by genomic sequence analysis alone. Non-DD mutations were evenly distributed between extracellular and transmembrane/intracellular regions and included a majority (11/18) that were premature terminations or frameshifts. A recurrent mutation of the N-terminal signal sequence and three splicing mutations that resulted in deletion of the transmembrane domain caused failure of the protein to be transported to or anchored at the cell surface, resulting in functional haploinsufficiency. The extracellular missense mutations were found to have mRNA stability equal to wild-type FAS and to encode proteins with normal surface expression.
Currently, FAS mutations reported in ALPS, both germline1,2,25 and somatic27,28 have suggested dominant-negative interference by the mutant protein when assembled into the cell membrane spanning FAS homotrimer, and this has been invoked as the general pathophysiologic mechanism for the clinical phenotype of heterozygous ALPS. Mutations leading to predicted premature terminations and frameshifts outside the intracellular DD of FAS have been found, but other than a single report by Roesler et al.,13 their biological effect has not been previously elucidated. Hence, our analysis documents the extent of haploinsufficiency as an alternate to dominant interference for explaining the mechanism of a nonmalignant immunodysregulatory condition such as ALPS.
We have delineated three functional groups of FAS mutations in addition to the DD mutations that cause dominant interference. First, functional haploinsufficiency mutations include those that cause mRNA instability or prevent the surface expression of a FAS allele, thus resulting in a marked decrease in the quantity of FAS available for homotrimerization and FAS-L engagement. The abnormal FAS protein molecules in this group do not cause dominant-negative interference with the wild-type molecule, as evidenced by our cotransfection analysis. Considering that cell surface FAS molecules congregate into lipid rafts after engagement with FAS-L, and the increased density of FAS chains allows formation of the death-inducing signaling complex necessary for transducing the apoptotic signal, 29 haploinsufficiency may reduce surface expression of FAS below a threshold level essential for death-inducing signaling complex formation. This is supported by one previous report13 describing an ALPS-associated FAS splice mutation deleting the transmembrane region for which the apoptotic defect was reversed by increasing surface expression of FAS by transfection of a wild-type FAS expression plasmid. Furthermore, our data show that T cells with reduced surface FAS expression are unable to bind FAS-L in vitro, accounting for their diminished ability to undergo apoptosis.
Mutations before exon 7 causing frameshifts or terminations resulted in haploinsufficiency, shown by reduction of mutant cDNA clones recovered from patient mRNA, reduced T-cell surface expression of FAS, and inability of these T cells to bind FAS-L.
A second group of mutations comprises extracellular FAS missense mutations and in-frame deletions, which retain expression of FAS mutant protein at the cell surface, but fail to bind FAS-L, presumably due to their structural alterations. On exposure to FAS-L, FAS proteins with extracellular defects coexpressed with wild-type FAS demonstrate variable amounts of dominant-negative interference.
The third group of mutations we have described are intracellular mutations that lie outside the FAS DD. Both of the juxtamembrane intracellular truncation mutations of FAS we analyzed, p.Q196TfsX16 and p.E218MfsX4, failed to cause apoptosis in singly transfected cells; however, they had mild effects when cotransfected with wild-type FAS. These results are similar to the previously published p.T241K mutation, which fails to transduce a death signal alone but is a weak dominant-negative inhibitor when cotransfected with wild-type FAS.2 Siegel et al.30 demonstrated the p.T241K mutation is unable to assemble into signaling protein oligomerization transduction structures. Further, this region is associated with the formation of lipid rafts and the palmitoylation of the FAS molecule,31,32 both possible mechanisms for the defect caused by these proteins. Two additional mutations we identified, p.R203X and p.L229X, also lie in this region of lipid raft assembly. The final intracellular, non-DD mutation, c.985_1008* 4del28, p.N329KfsX21, occurs after the DD region. There is a similar mutation, c.987ins20, causing a frameshift at the same amino acid that has been identified in a patient with homozygous inheritance,33 which had undetectable surface expression of FAS. By analogy, our patient may have a functional haploinsufficiency as a result of the mutation or there may be some other mechanism; unfortunately, patient cells were not available for this study.
When functional FAS pathways were evaluated in patients’ expanded T cells, decreased surface expression and/or altered FAS-L binding was demonstrated. Within these populations of cells studied, there were no detectable second mutations suggesting the primary mutation is sufficient to impair FAS-driven apoptosis. Second hit somatic mutations were recently described in hematopoietic cells of patients with ALPS,34 thought in some cases to contribute to disease progression severity. Longitudinal evaluation of our patients with germ line extracellular or haploinsufficient mutations, with periodic screening for somatic mutations during clinical exacerbations, may reveal whether somatic mutations contribute to manifestations developing over time.
We have now accumulated sufficient patients and mutation-bearing relatives with all four groups of mutations to refine the analysis first performed by Jackson et al.2 As demonstrated in analysis of mutation-positive relatives, the DD mutations and other intracellular and transmembrane mutations are considerably more penetrant than mutations that are functionally haploinsufficient or that affect the extracellular domain. These results correlate with the degree of impairment of apoptosis observed in vitro. Our data have clinical significance; individuals with intracellular and DD FAS mutations may be at higher risk of complications, and this genotype should be factored into genetic counseling and medical management decisions, such as whether to undertake periodic lymphoma surveillance and consider allogeneic bone marrow transplantation for severely affected phenotypes.35,36
Herein, we provide a framework for analyzing the molecular consequences of FAS mutations. Once a nucleotide change is identified, mRNA stability and protein production can be predicted by analogy to the mutations reported here. Reduced cell surface staining, performed with proper control cells, may be used to demonstrate haploinsufficiency, whereas reduced or absent FAS-L binding confirms the predicted functional effect of extracellular missense mutations. The mutations published herein, along with those previously published or catalogued at the ALPS Leiden Open Variation Database website (http://www.ncbi.nlm.nih.gov/lovd/home.php) form a comparison set for newly identified sequence variants.
Disclosure
The authors declare no conflict of interest.
References
Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995;81:935–946.
Jackson CE, Fischer RE, Hsu AP, et al. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet 1999;64:1002–1014.
Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood 2010;116:35–40.
Ballmaier M, Schulze H, Cremer M, Folman CC, Strauss G, Welte K . Defective c-Mpl signaling in the syndrome of thrombocytopenia with absent radii. Stem Cells 1998;16(suppl 2):177–184.
Mountz JD, Zhou T, Wu J, Wang W, Su X, Cheng J . Regulation of apoptosis in immune cells. J Clin Immunol 1995;15:1–16.
Peters AM, Kohfink B, Martin H, et al. Defective apoptosis due to a point mutation in the death domain of CD95 associated with autoimmune lymphoproliferative syndrome, T-cell lymphoma, and Hodgkin’s disease. Exp Hematol 1999;27:868–874.
Russell JH . Activation-induced death of mature T cells in the regulation of immune responses. Curr Opin Immunol 1995;7:382–388.
Strasser A . Life and death during lymphocyte development and function: evidence for two distinct killing mechanisms. Curr Opin Immunol 1995;7:228–234.
Zuraw BL . Clinical practice. Hereditary angioedema. N Engl J Med 2008;359:1027–1036.
Stratakis CA . Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit (PRKAR1A) in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas” (Carney complex). Ann N Y Acad Sci 2002;968:3–21.
Hobbs HH, Leitersdorf E, Leffert CC, Cryer DR, Brown MS, Goldstein JL . Evidence for a dominant gene that suppresses hypercholesterolemia in a family with defective low density lipoprotein receptors. J Clin Invest 1989;84:656–664.
Roessler E, El-Jaick KB, Dubourg C, et al. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Hum Mutat 2009;30:E921–E935.
Roesler J, Izquierdo JM, Ryser M, et al. Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95{Delta}TM), is the basis for ALPS Ia in a family with duplicated 3’ splice site AG in CD95 intron 5 on one allele. Blood 2005;106:1652–1659.
Vacek MM, Schäffer AA, Davis J, et al. HLA B44 is associated with decreased severity of autoimmune lymphoproliferative syndrome in patients with CD95 defects (ALPS type Ia). Clin Immunol 2006;118:59–65.
Niemela JE, Hsu AP, Fleisher TA, Puck JM . Single nucleotide polymorphisms in the apoptosis receptor gene TNFRSF6. Mol Cell Probes 2006;20:21–26.
Clementi R, Dagna L, Dianzani U, et al. Inherited perforin and Fas mutations in a patient with autoimmune lymphoproliferative syndrome and lymphoma. N Engl J Med 2004;351:1419–1424.
Zhang J, Sun X, Qian Y, Maquat LE . Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA 1998;4:801–815.
Inácio A, Silva AL, Pinto J, et al. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem 2004;279:32170–32180.
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C . Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67.
Papoff G, Cascino I, Eramo A, Starace G, Lynch DH, Ruberti G . An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J Immun 2006;156:4622–4630.
Cao P, Wang QJ, Zhu XT, Zhou H, Li R, Wang WP . Quantitative determination of allele frequency in pooled DNA by using sequencing method. J Chromatogr B Analyt Technol Biomed Life Sci 2011;879:527–532.
Fadeel B, Thorpe J, Chiodi F . Mapping of the linear site on the Fas/APO-1 molecule targeted by the prototypic anti-Fas mAb. Int Immunol 1995;7:1967–1975.
Banner DW, D’Arcy A, Janes W, et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell 1993;73:431–445.
Bajorath J . Identification of the ligand binding site in Fas (CD95) and analysis of Fas-ligand interactions. Proteins 1999;35:475–482.
Rieux-Laucat F, Blachère S, Danielan S, et al. Lymphoproliferative syndrome with autoimmunity: A possible genetic basis for dominant expression of the clinical manifestations. Blood 1999;94:2575–2582.
Vaishnaw AK, Toubi E, Ohsako S, et al. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum 1999;42:1833–1842.
Dowdell KC, Niemela JE, Price S, et al. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood 2010;115:5164–5169.
Holzelova E, Vonarbourg C, Stolzenberg MC, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med 2004;351:1409–1418.
Eramo A, Sargiacomo M, Ricci-Vitiani L, et al. CD95 death-inducing signaling complex formation and internalization occur in lipid rafts of type I and type II cells. Eur J Immunol 2004;34:1930–1940.
Siegel RM, Muppidi JR, Sarker M, et al. SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol 2004;167:735–744.
Chakrabandhu K, Hérincs Z, Huault S, et al. Palmitoylation is required for efficient Fas cell death signaling. EMBO J 2007;26:209–220.
Rossin A, Kral R, Lounnas N, et al. Identification of a lysine-rich region of Fas as a raft nanodomain targeting signal necessary for Fas-mediated cell death. Exp Cell Res 2010;316:1513–1522.
van der Burg M, de Groot R, Comans-Bitter WM, et al. Autoimmune lymphoproliferative syndrome (ALPS) in a child from consanguineous parents: a dominant or recessive disease? Pediatr Res 2000;47:336–343.
Magerus-Chatinet A, Neven B, Stolzenberg MC, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest 2011;121:106–112.
Benkerrou M, Le Deist F, de Villartay JP, et al. Correction of Fas (CD95) deficiency by haploidentical bone marrow transplantation. Eur J Immunol 1997;27:2043–2047.
Dimopoulou MN, Gandhi S, Ghevaert C, et al. Successful treatment of autoimmune lymphoproliferative syndrome and refractory autoimmune thrombocytopenic purpura with a reduced intensity conditioning stem cell transplantation followed by donor lymphocyte infusion. Bone Marrow Transplant 2007;40:605–606.
Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health, National Human Genome Research Institute, and National Institute of Allergy and Infectious Disease, Bethesda, MD 20892. The authors thank the patients and their families, Roxanne E. Fischer for her valuable contributions related to sample processing, Tom Fleisher and Janet Dale for phenotypic delineation, and Mike Lenardo and the late Stephen Straus for their helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article
Cite this article
Hsu, A., Dowdell, K., Davis, J. et al. Autoimmune lymphoproliferative syndrome due to FAS mutations outside the signal-transducing death domain: molecular mechanisms and clinical penetrance. Genet Med 14, 81–89 (2012). https://doi.org/10.1038/gim.0b013e3182310b7d
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.0b013e3182310b7d
Keywords
This article is cited by
-
Whole Exome Sequencing Reveals Pathogenic Variants in ADA2 and FAS Causing DADA2 and ALPS
Journal of Clinical Immunology (2023)
-
ALPS, FAS, and beyond: from inborn errors of immunity to acquired immunodeficiencies
Annals of Hematology (2022)
-
Differential cytokine withdrawal-induced death sensitivity of effector T cells derived from distinct human CD8+ memory subsets
Cell Death Discovery (2017)
-
FAS Haploinsufficiency Caused by Extracellular Missense Mutations Underlying Autoimmune Lymphoproliferative Syndrome
Journal of Clinical Immunology (2015)
-
A Rapid Ex Vivo Clinical Diagnostic Assay for Fas Receptor-Induced T Lymphocyte Apoptosis
Journal of Clinical Immunology (2013)
