
Abstract
Lysophosphatidic acid (LPA) is a bioactive lysophospholipid involved in numerous physiological responses. However, the expression of LPA receptors and the role of the Hippo signaling pathway in epithelial cells have remained elusive. In this experiment, we studied the functional expression of LPA receptors and the associated signaling pathway using reverse transcriptase–PCR, microspectrofluorimetry, western blotting and immunocytochemistry in salivary gland epithelial cells. We found that LPA receptors are functionally expressed and involved in activating the Hippo pathway mediated by YAP/TAZ through Lats/Mob1 and RhoA/ROCK. Upregulation of YAP/TAZ-dependent target genes, including CTGF, ANKRD1 and CYR61, has also been observed in LPA-treated cells. In addition, based on data suggesting that tumor necrosis factor (TNF)-α induces cell apoptosis, LPA upregulates TNF-induced caspase-3 and cleaved Poly(ADP-ribose)polymerase (PARP). However, small interfering RNA treatment to Yes-associated protein (YAP) or transcriptional co-activator with a PDZ-binding motif (TAZ) significantly decreased TNF-α- and LPA-induced apoptosis, suggesting that YAP and TAZ modulate the apoptotic pathway in salivary epithelial cells.
Similar content being viewed by others


Introduction
Lysophosphatidic acid (LPA), which is mediated by a G protein-coupled receptor (LPA1-6), regulates diverse cellular processes, including cell polarization, proliferation, differentiation, adhesion and migration.1 In particular, LPA enhances the effects of the extracellular matrix on acinus formation through a pathway that is dependent on PI3-kinase/Rho GTPases in human salivary gland cells2 and induces branching morphogenesis of embryonic mouse salivary epithelium.3 Furthermore, Sphingosine 1-phosphate (S1P), another lysophospholipid, modulates Ca2+ signaling and autoimmune disease in submandibular gland cells.4, 5 However, the functional expression and signaling pathway of LPA receptors have not been rigorously studied in secretory epithelia, including salivary glands.
The mammalian Hippo pathway is involved in cell proliferation, apoptosis, migration and differentiation,6 and this pathway contains a core kinase cascade in which the mammalian STE20-like protein kinases (MST1 and MST2) phosphorylate and activate large tumor suppressor homologs (Lats1 and Lats2). Subsequently, Lats1/2 phosphorylate their downstream targets, which include a Yes-associated protein (YAP) and a transcriptional co-activator with a PDZ-binding motif (TAZ), the homolog of YAP.6, 7 Phosphorylation of YAP/TAZ allows YAP to bind to 14-3-3 proteins in the cytoplasm;6, 8 however, dephosphorylated YAP/TAZ are translocated into the nucleus and associate with Transcriptional enhancer activator (TEA) domain family member transcription factors on DNA8 to induce the expression of their target genes, which were shown to be involved in cell proliferation and anti-apoptotic function.9 Currently, four upstream pathway are known to regulate the Hippo pathway: the Crumbs homolog complex, the MST kinase regulator, the actin cytoskeleton and adherence junctions.7, 8 In addition to these major upstream regulators, small molecules can modulate the Hippo pathway.6 A new class of Hippo pathway regulators, that is, LPA and S1P, elevate YAP/TAZ activity in human ovarian cancer and embryonic stem cells.10, 11
Recent reports suggest that defects in Hippo signaling are associated with Sjogren’s syndrome in humans because inhibition of Lats causes a reduction in E-cadherin-mediated cell adhesion, which has an important role in cell polarity.12 The mechanisms of salivary gland destruction in Sjogren’s syndrome patients remain unclear. However, it has been reported that inappropriate apoptosis causes lymphocyte infiltration with gland destruction and secretory dysfunction.13, 14 Tumor necrosis factor (TNF)-α induces apoptosis, which results in many inflammatory diseases and the loss of physiological functions.15 In this study, we report the functional expression of LPA receptors and identify the Hippo signaling pathway as a regulator of apoptosis in salivary gland epithelial cells.
Materials and methods
Materials
LPA, Carbachol (CCh), BAPTA-AM, U73211, 2-APB and Y-27632 were purchased from Sigma (St Louis, MO, USA). Fura-2 AM was obtained from Molecular Probes (Eugene, OR, USA). A Hippo signaling antibody sampler kit, β-tubulin, lamin A/C caspase-3 and PARP were purchased from Cell Signaling Technology (Danvers, MA, USA). TAZ, p-TAZ and β-actin were obtained from Santa Cruz (Dallas, TX, USA).
Cell culture
The HSG cells were a generous gift from Professor Kazuo Hosoi at the Tokushima University in Japan. The cells were grown in suspension in 10-ml tissue culture plates at 37 °C in 95% air–5% CO2 and were maintained in a minimum essential medium supplemented with 10% fetal bovine serum. Each plate was replated twice a week.
Measurement of the cytoplasmic-free calcium concentration ([Ca2+]i)
The intracellular Ca2+ concentration ([Ca2+]i) in HSG cells loaded with 2 μM Fura-2 in 2 ml of culture medium for 30 min at 37 °C was measured. The cells were gently washed once by replacing the medium and then incubated in normal solution for 10 min prior to the experiment to allow for maximal de-esterification of the dye. The MetaFluor version 6.1 imaging system (Universal Imaging, West Chester, PA, USA) was used for recording and analysis. Fura-2 fluorescence was recorded at 37 °C at an excitation wavelength of 510 nm, and the results are presented as the ratio of fluorescence measured at 340 nm to that measured at 380 nm (Ca2+ fluorescence ratio, F340/F380).
Nuclear-cytoplasmic fractionation and western blot analysis
The cells were homogenized in lysis buffer (50 mM Tris, pH 7.5, 1% Triton X-100, 100 mM NaCl, 10 mM tetrasodium pyrophosphate, 10 mM NaF, 1 mM EDTA, 1 mM NaV, 1 mM EGTA, 1 mM phenylmethylsulphonyl fluoride and 1 μg ml−1 aprotinin, leupeptin and pepstatin) followed by gentle sonication on ice. For nuclear-cytoplasmic fractionation, intracellular proteins were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer’s protocol.
Following protein concentration determination, the proteins were separated on an SDS-polyacrylamide gel and transferred electrophoretically to a polyvinylidene fluoride (PVDF) membrane, which was then incubated with primary antibody.
Immunocytochemistry
The cells were grown in Dulbecco’s modified Eagle’s medium medium and allowed to adhere to coverslips for 2 h at 37 °C in 5% CO2. Treated (LPA) and untreated cells were fixed with 4% paraformaldehyde for 10 min at room temperature. Fixed cells were washed in phosphate-buffered saline 3X for 5 min and then placed in blocking solution (2% bovine serum albumin, 5% fetal bovine serum (FBS) and 0.1% Triton X-100). The cells were then incubated overnight at 4 °C with primary antibody, and after being washed, the cells were incubated at room temperature for 1 h with secondary antibody.
Reverse transcriptase–PCR analysis
The amplification conditions included 60 s at 94 °C, 30 s at 55 °C and 45 s at 72 °C for 35 cycles, and a final 5 min at 72 °C. The primers that were used (forward, reverse and product size (bp)) were as follows: LPA1, 5′-ATCGGGATACCATGATGAGT-3′ and 5′-TCCGTTCTAAACCACAGAGTG-3′, 342; LPA2, 5′-GTCCTCATTACCCAGTCATACCG-3′ and 5′-CTGATGGACTCCACCCTTTAGCT-3′, 426; LPA3, 5′-TGTCAACCGCTGGCTTCT-3′ and 5′-CAGTCATCACCGTCTCATTAG-3′, 437; LPA4, 5′-GTGGCGGTATTTCAGCCTCT-3′ and 5′-GAGTTGCAAGGCACAAGGTG-3′, 402; LPA5, 5′-GATTCCGCCCTCTGAACACA-3′ and 5′-AACCTGGTGCTCTTCAGCTC-3′, 409; LPA6, 5′-TGGGTTGGACTCGTTGACTG-3′ and 5′-TTTCGGACTTTGAGGACGCA-3′, 458; and β-Actin, 5′-TCCCTGGAGAAGAGCTACGA-3′ and 5′-ATCTGCTGGAAGGTGGACAG-3′, 362.
Real-time PCR
Total RNA was isolated from cells using TRIzol (Gibo-BRL, Invitrogen) and then used in reverse transcription reactions for cDNA synthesis (Invitrogen, Carlsbad, CA, USA). Real-time PCRs were performed with SYBR green premix buffer and an ABI Prism 7300 sequence detector (Perkin-Elmer Applied Biosystems, Foster City, CA, USA), and relative expression levels were determined after normalization to the threshold cycle (CT) values for β-actin. The gene-specific primers were as follows: for β-actin, 5′-GCCGACAGGATGCAGAAGGAGATCA-3′ and 5′-AAGCATTTGCGGTGGACGATGGA-3′; for CTGF, 5′-CCAATGACAACGCCTCCTG-3′ and 5′-TGGTGCAGCCAGAAAGCTC-3′; for Cyr61, 5′-AGCCTCGCATCCTATACAACC-3′ and 5′-TTCTTTCACAAGGCGGCACTC-3′; for ANKRD1, 5′-CACTTCTAGCCCACCCTGTGA-3′ and 5′-CCACAGGTTCCGTAATGATTT-3′; for TAGLN, 5′-CCCGAGAACCCACCCTCCA-3′ and 5′-AAAGCCATCAGGGTCCTCTGC-3′; and for EDN1, 5′-TGTGTCTACTTCTGCCACCT-3′ and 5′-CCCTGAGTTCTTTTCCTGCTT-3′
small interfering RNA treatment
Cells were transfected with small interfering RNA (siRNA) against YAP or TAZ (Santa Cruz Biotechnology) using Lipofectamine 2000 reagent (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions.
Statistical analysis
The band intensities were measured with ImageJ software (Bethesda, MD, USA), and statistical analysis was performed using Excel 2003. All data are expressed as the means±s.e.m. Statistical significance was evaluated using Student’s unpaired t-test. A probability below 0.05 (P<0.05) was considered significant.
Results
Functional expression of LPA receptors in HSG cells
We first identified the types of LPA receptors that are expressed in HSG cells using reverse transcriptase–PCR. The mRNA transcripts for all LPA receptor subtypes, from 1 to 6, were detected, and the expected size of the PCR products was 362 bp, 342 bp, 426 bp, 437 bp, 402 bp, 409 bp and 458 bp, respectively (Figure 1a). We then tested whether LPA could increase the intracellular free Ca2+ ([Ca2+]i) level, which has a vital role in salivary secretion. Application of 1 μM LPA consistently induced an increase in the [Ca2+]i (Figure 1b), and this increase was also observed in [Ca2+]i-free medium (gray line in Figure 1b). However, preincubation of the cells with 10 μM U73211 or 50 μM 2-APB for 10 min significantly inhibited the LPA-induced [Ca2+]i increase (gray lines in Figure 1c and d). After LPA stimulation, subsequent stimulation of cells with 100 μM CCh, a typical muscarinic agonist, or with LPA after CCh stimulation did not increase the [Ca2+]i (Figures 1e and f).

Functional expression of lysophosphatidic acid (LPA) receptors in HSG cells. (a) Expression of mRNA transcripts for all LPA receptor subtypes, from 1 to 6, using reverse transcriptase–PCR. Lanes ‘M and N’ indicate the kb marker and negative control, respectively. β-actin was used as the positive control. (b) LPA-induced a transient [Ca2+]i increase in the presence or absence (gray line) of [Ca2+]i in the medium. (c and d) LPA-induced the [Ca2+]i response before and after (gray line) pretreatment of cells with 10 μM U-73211 and 10 μM 2-APB. (e) Subsequent stimulation of the cells with 100 μM carbachol (CCh) after LPA stimulation. (f) Subsequent stimulation of the cells with 1 μM LPA after CCh stimulation.
Serum and LPA-induced Hippo pathway activation
We next determined whether serum and LPA could activate the Hippo pathway in HSG cells. In serum-free conditions, most YAP proteins were phosphorylated in these cells (second lane in the upper panel in Figure 2a). However, addition of 2 or 5% FBS into the extracellular medium gradually but significantly dephosphorylated YAP, and 10% FBS completely dephosphorylated YAP (first lane in the upper panel in Figure 2a). The lower panel shows a summary of the results. The amount of phosphorylated YAP significantly decreased when 2, 5 and 10% serum were added. Figure 2b shows the translocation of YAP from the cytoplasm to the nucleus. At the resting state, in serum-free media, YAP (red color) did not localize within the nucleus (blue color); however, addition of 10% serum markedly caused the translocation of YAP from the cytosol to the nucleus (noted by the merged, purple color). After confirming the activation of the Hippo pathway by serum, we then examined whether LPA could activate the Hippo pathway. We determined that 0.1 μM of LPA was associated with minimal levels of dephosphorylated YAP, but 1 μM of LPA significantly dephosphorylated YAP (the top row in Figure 2c). Although the dephosphorylation effect was not prominent compared with that of YAP, 1 μM LPA also dephosphorylated TAZ proteins (the third row in Figure 2c). The lower histograms summarize the results and show the effects of various LPA concentrations on YAP and TAZ dephosphorylation. We next examined whether this LPA-induced dephosphorylation of YAP/TAZ was dependent on Ca2+. Preincubation of cells with 10 μM BAPTA-AM for 30 min did not inhibit the LPA-induced YAP/TAZ dephosphorylation (the upper panel in Figure 2d). The lower histograms summarize the effects of various LPA concentrations on YAP and TAZ dephosphorylation after preincubation with BAPTA-AM. The LPA-induced translocation of YAP/TAZ from the cytosol to the nucleus is shown in Figure 2e. At the resting state, TAZ (green) and YAP (red) did not localize within the nucleus; however, 1 μM LPA markedly translocated YAP and TAZ from the cytosol to the nucleus (note the merged, purple and cobalt colors, respectively). Preincubation of cells with BABTA-AM had little effect on the LPA-induced YAP/TAZ translocation into the nucleus (the lower panels in Figure 2e), which confirms our western blot data in Figure 2d.

Serum and lysophosphatidic acid (LPA)-induced dephosphorylation of the YAP/TAZ proteins. (a) Dephosphorylation of YAP/TAZ by the addition of serum. Normal culture media (with 10%) was replenished with serum-free or media containing 2, 5 or 10% fetal bovine serum (FBS) for 2 h before western blot analysis. The p-YAP intensities in the upper rows were quantified and normalized to that of Yes-associated protein (YAP). The data are represented as the means±s.e.m. (n=3). *P<0.05. (b) Representative images for immunocytochemistry showing the translocation of YAP from the cytosol to the nucleus (merged purple color) after the addition of 10% serum. Bar plots show the distribution of YAP (Nuc: predominantly in the nucleus; Cyto: predominantly in the cytoplasm) from over 100 cells. (c) LPA-induced YAP/TAZ dephosphorylation. LPA (1 μM) dephosphorylated the YAP and transcriptional co-activator with a PDZ-binding motif (TAZ) proteins. (d) The effect of 10 μM BAPTA-AM on LPA-induced YAP/TAZ dephosphorylation. The densities of p-YAP and p-TAZ from the upper rows were quantified and normalized to that of YAP and TAZ, respectively. The data are represented as the means±s.e.m. (n=3). *P<0.05. (e) Representative images for the immunocytochemistry analysis showing the translocation of YAP/TAZ from the cytosol to the nucleus after LPA treatment (merged purple and cobalt color). Cells were fixed and immunostained for YAP (red), TAZ (green), and DAPI (blue). Representative data from three independent experiments are shown. ‘F’, ‘L’ and ‘B’ on the vertical axis indicate FBS, LPA and BAPTA, respectively. Bar plots represent the distribution of YAP/TAZ (Nuc: predominantly in the nucleus; Cyto: predominantly in the cytoplasm) from over 100 cells. The underlined scale bar is 30 μm.
Hippo signaling pathway mediated by Lats and RhoA/ROCK
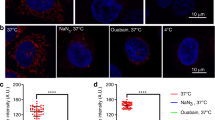
We next examined whether LPA could activate Lats, a signaling molecule in the pathway upstream of YAP/TAZ. LPA dephosphorylated Lats and Mob kinase activator 1 (Mob1), which interacts with Lats, in a concentration-dependent manner (first to third rows in Figure 3a). We finally tested the involvement of RhoA/ROCK in the LPA-induced Hippo pathway. Cells were preincubated with 10 μM Y27632, a Rho-kinase (ROCK) inhibitor, which inhibited both the LPA-induced YAP/TAZ dephosphorylation (Figure 3b) and the translocation of YAP/TAZ into the nucleus based on the western blot experiment (Figure 3b) and immunocytochemistry (Figure 3c). Our data demonstrate that LPA induces dephosphorylation of YAP/TAZ mediated by both the Lats and RhoA/ROCK pathways.

lysophosphatidic acid (LPA)-induced YAP/TAZ dephosphorylation and translocation via the RhoA/ROCK pathway. (a) LPA-induced dephosphorylation of Lats and Mob activator kinase 1 (Mob1) in a concentration-dependent manner. (b) The effect of Y-27632 for 1 h prior to stimulation with LPA (1 μM, 2 h) after fetal bovine serum (FBS) starvation. The intensities of p-YAP and p-TAZ from the upper rows were quantified and normalized to that of Yes-associated protein (YAP) and transcriptional co-activator with a PDZ-binding motif (TAZ), respectively. The data are represented as the means±s.e.m. (n=3). *P<0.05. (c) The effect of Y-27632 on the LPA-induced translocation of YAP/TAZ to the nucleus. Cells were fixed and stained for YAP (red), TAZ (green) and nucleus (blue). ‘F’, ‘L’ and ‘Y’ on the vertical axis indicate FBS, LPA and Y-27632, respectively. Representative data from three independent experiments are shown. The underlined scale bar is 30 μm.
Localization of YAP/TAZ in the subcellular fractionation
We next determined the subcellular localization of YAP/TAZ using cell fractionation and western blotting. The cytoplasmic localization of YAP/TAZ in LPA-treated cells was decreased compared with that of untreated cells, whereas the nuclear localization of YAP/TAZ in LPA-treated cells was increased compared with that of nontreated cells (Figure 4). These results are consistent with Figure 2, showing that LPA induces the dephosphorylation of YAP/TAZ and induces YAP/TAZ to move into the nucleus.

The accumulation of YAP/TAZ in the subcellular fractionation to generate cytoplasmic and nuclear fractions. The sample was fractionated to generate cytoplasmic and nuclear fractions in control conditions or after treatment for 2 h with 1 μM LPA. The presence of the cytoplasmic marker β-tubulin and the nuclear marker lamin A/C in the appropriate fraction was confirmed by western blotting with the corresponding antibodies. The results are representative of three experiments. LPA, lysophosphatidic acid; TAZ, transcriptional co-activator with a PDZ-binding motif; YAP, Yes-associated protein.
Relative expression levels of genes by LPA stimulation
YAP/TAZ is a cofactor for TEA domain family member transcription factors, and the activation/inhibition of YAP/TAZ may have an important role in the regulation of various genes. To identify whether LPA treatment can induce gene expression, we quantified the mRNA levels of connective tissue growth factor (CTGF), ankyrin repeat domain-containing protein 1 (ANKRD1), cysteine-rich angiogenic inducer 61 (CYR61), transgelin (TAGLN) and endothelin 1 (EDN1). As shown in Figure 5, the mRNA levels of CTGF, ANKRD1, and CYR61, but not of TAGLN or EDN1, increased in response to LPA treatment.

The lysophosphatidic acid (LPA)-stimulated mRNA levels of CTGF, ANKRD1, CYR61, TAGLN and EDN1. HSG cells were treated with 1 μM LPA for 2 h, followed by quantification of the mRNA levels of CTGF, ANKRD1, CYR61, TAGLN and EDN1 by real-time PCR and normalization to the level of β-actin. *P<0.05 and **P<0.01 when compared with the control. The bar represents the means±s.e.m. (n=3).
Involvement of YAP and TAZ in TNF-α and LPA-induced apoptosis
We finally investigated the roles of YAP and TAZ in cell apoptosis using siRNA. Figure 6a shows the decreased expression levels of YAP and TAZ after siRNA treatment. Treatment of the cells with 20 nM TNF-α increased the expression of caspase-3 and its substrate, cleaved PARP (the upper panels in Figures 6b and c). We then investigated the effect of LPA on TNF-α-induced caspase-3 activation. Simultaneous application of 1 μM LPA and TNF-α further increased the expression level of caspase-3 and cleaved PARP. Pretreatment of the cells with YAP or TAZ siRNA significantly decreased expression levels of these two proteins (the third columns in Figures 6b and c).

Involvement of YAP/ TAZ in TNF-α and LPA-induced apoptosis. (a) Reduced levels of YAP and TAZ proteins after YAP and TAZ siRNA treatment. (b and c) Cells were transfected with YAP and TAZ siRNA. After 24 h, the cells were treated with 20 nM TNF-α in the presence or absence of 1 μM LPA for 48 h. LPA, lysophosphatidic acid; TAZ, transcriptional co-activator with a PDZ-binding motif; TNF-α, tumor necrosis factor-α; YAP, Yes-associated protein.
Discussion
LPA receptors, which are G protein-coupled receptors, affect diverse physiological and pathophysiological functions, including cancer, fibrosis, bone metabolism and autoimmune disease.1, 16 To date, six distinct G protein-coupled receptors (LPA1–6) have been identified and have been shown to be differentially expressed at varying levels in epithelial cells.16 In HSG cells, LPA, epidermal growth factor and basic fibroblast growth factor stimulate Rho proteins, which are essential for acinar formation.2 Recent reports have indicated that the Hippo signaling pathway affects salivary gland development and is associated with Sjogren’s syndrome.12 However, the functional expression of LPA receptors and the associated signaling pathway have not been rigorously studied in secretory epithelia, including salivary glands.
In our study, mRNA transcripts for all subtypes of the LPA receptors were detected in HSG cells. LPA receptors in HSG cells appeared to function because LPA treatment consistently increased the [Ca2+]i. Such an LPA-induced [Ca2+]i increase was also shown in Ca2+-free medium, although it had a fast [Ca2+]i decay without the [Ca2+]i plateau that occurred in Ca2+ containing medium (Figure 1b). The LPA-induced [Ca2+]i increase was inhibited by U73211, a PLC inhibitor, and 2-APB, an IP3-sensitive Ca2+ store blocker. Thus, our results suggest that LPA-induced [Ca2+]i increases are due to both [Ca2+]i influx and [Ca2+]i release from the intracellular Ca2+ stores, particularly from IP3-sensitive Ca2+ stores. Because no [Ca2+]i response to the subsequent stimulation of cells with CCh after LPA stimulation was detected, this further result confirms that LPA uses the same intracellular Ca2+ stores as CCh, a typical muscarinic agonist.
Next, we examined whether the Hippo pathway exists in HSG cells. Serum is a complex component that consists of growth factors, nutrients, lipids and other factors which can regulate YAP/TAZ activation. Among these factors, serum lipid-containing S1P and LPA potently decreases YAP/TAZ phosphorylation.8 Therefore, we first examined whether serum could activate the Hippo pathway before LPA application. Incubation of cells with FBS dephosphorylated the YAP protein in a concentration-dependent manner, which resulted in the translocation of YAP from the cytoplasm to the nucleus (Figure 2b). After confirming the dephosphorylation effect of serum on YAP/TAZ, we tested the effect of LPA. As expected, LPA also activated the dephosphorylation of YAP/TAZ in a concentration-dependent manner. Preincubation of the cells with BAPTA-AM, a strong Ca2+ chelator, had little effect on LPA-induced YAP/TAZ activation (Figure 2d); however, extracellular Ca2+ depletion impaired cell tight junctions and triggered the shift of YAP/TAZ from the cytoplasm to the nucleus in Eph4 mouse mammary epithelial cells.17
Reports have indicated that the main Hippo pathway is a serine/threonine kinase cascade that involves MST1/2 protein kinase and large tumor suppressor homologs (Lats1/2).6 MST1/2 phosphorylates Lats1/2 and scaffolding proteins such as Mob.18 After Mob1 is phosphorylated by MST1/2, Mob1 interacts with Lats1/2, which in turn leads to an increase in their kinase activity.19 We examined whether two signaling molecules, the Lats and Mob1 were involved in the upstream pathway involved in dephosphorylating YAP/TAZ and found that LPA dephosphorylated the Lats and Mob1 (Figure 3a). In our experiments, inhibition of ROCK decreased LPA-induced YAP/TAZ dephosphorylation. To date, many regulators that regulate the core of the Hippo pathway (YAP and TAZ) at different levels have been identified.6 In particular, LPA and S1P have been reported as the main G protein-coupled receptors that regulate the Hippo pathway.10, 20 Generally, LPA and S1P regulate YAP and TAZ via the RhoA/ROCK pathway and/or the MST/Lats pathway, causing the dephosphorylation and nuclear accumulation of YAP and TAZ.6, 8 However, the upstream core Hippo signaling pathway can differ according to the LPA receptor subtypes that are expressed in each cell. For example, LPA acts through LPA2 and LPA3 to induce YAP activity mediated by RhoA/ROCK/PP1A, but not by Lats kinase, in OVCA433 EOC cells, whereas LPA acts through LPA1 and an LPA3 receptor to stimulate YAP activity mediated by Lats pathways in HEK293 cells.8, 11 In our experiments, we found that mRNA transcripts for all LPA subtype receptors were expressed in HSG cells (Figure 1a) and that LPA dephosphorylated YAP/TAZ through both the Lats and RhoA/ROCK pathways (Figures 2 and 3), suggesting that more than two types of LPA receptors are functionally expressed in HSG cells.
To the best of our knowledge, this is the first study reporting the expression of LPA receptors and Hippo signaling pathway activation by LPA in salivary gland epithelial cells. LPA also enhanced TNF-α-induced caspase-3 and cleaved PARP, which was prevented by YAP and TAZ siRNA. This result demonstrates that the Hippo signaling pathway affects the apoptotic pathway, TNF-induced caspase-3 and cleaved PARP, which significantly decreased the downregulation of YAP and TAZ (Figures 6b and c). It has been reported that the Hippo pathway promotes cell survival and differentiation.21 However, our data show that stimulation of YAP/TAZ by LPA is required for TNF-α-induced LPA synergistic apoptosis (Figure 6) and that the mRNA of YAP/TAZ-dependent target genes, including CTGF, ANKRD1 and CYR61, was upregulated in LPA-treated HSG cells (Figure 5). CTGF is an inflammatory mediator that increases interleukin-6 production.22, 23 CYR61 promotes the apoptotic function of inflammation cytokines such as TNF-α,24, 25 whereas ANKRD1 is induced during wound repair to enhance healing.26 Therefore, it is possible that YAP/TAZ activated by LPA enhances the production of more CTGF and CYR61 mRNA than of survival factors such as ANKRD1. This result implies that upregulation of genes markedly increases the susceptibility of HSG cells to apoptosis. In conclusion, our results strongly suggest that LPA has an important role in apoptotic cell death through YAP and TAZ in the Hippo pathway.
References
Mutoh T, Rivera R, Chun J . Insights into the pharmacological relevance of lysophospholipid receptors. Br J Pharmacol 2012; 165: 829–844.
Crema VO, Hamassaki DE, Santos MF . Small Rho GTPases are important for acinus formation in a human salivary gland cell line. Cell Tissue Res 2006; 325: 493–500.
Noguchi Y, Okamoto A, Kasama T, Imajoh-Ohmi S, Karatsu T, Nogawa H . Lysophosphatidic acid cooperates with EGF in inducing branching morphogenesis of embryonic mouse salivary epithelium. Dev Dyn 2006; 235: 403–410.
Sekiguchi M, Iwasaki T, Kitano M, Kuno H, Hashimoto N, Kawahito Y et al. Role of sphingosine 1-phosphate in the pathogenesis of Sjogren's syndrome. J Immunol 2008; 180: 1921–1928.
Seo J, Koo NY, Choi WY, Kang JA, Min JH, Jo SH et al. Sphingosine-1-phosphate signaling in human submandibular cells. J Dent Res 2010; 89: 1148–1153.
Johnson R, Halder G . The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 2014; 13: 63–79.
Codelia VA, Irvine KD . Hippo signaling goes long range. Cell 2012; 150: 669–670.
Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012; 150: 780–791.
Oka T, Sudol M . Nuclear localization and pro-apoptotic signaling of YAP2 require intact PDZ-binding motif. Genes Cells 2009; 14: 607–615.
Jeong GO, Shin SH, Seo EJ, Kwon YW, Heo SC, Kim KH et al. TAZ mediates lysophosphatidic acid-induced migration and proliferation of epithelial ovarian cancer cells. Cell Physiol Biochem 2013; 32: 253–263.
Cai H, Xu Y . The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal 2013; 11: 31.
Enger TB, Samad-Zadeh A, Bouchie MP, Skarstein K, Galtung HK, Mera T et al. The Hippo signaling pathway is required for salivary gland development and its dysregulation is associated with Sjogren's syndrome. Lab Invest 2013; 93: 1203–1218.
Barrera MJ, Bahamondes V, Sepulveda D, Quest AF, Castro I, Cortes J et al. Sjogren's syndrome and the epithelial target: a comprehensive review. J Autoimmun 2013; 42: 7–18.
Kramer JM . Early events in Sjogren's syndrome pathogenesis: the importance of innate immunity in disease initiation. Cytokine 2014; 67: 92–101.
Parameswaran N, Patial S . Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr 2010; 20: 87–103.
Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T et al. LPA receptors: subtypes and biological actions. Ann Rev Pharmacol Toxicol 2010; 50: 157–186.
Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell 2010; 19: 831–844.
Li C, Srivastava RK, Elmets CA, Afaq F, Athar M . Arsenic-induced cutaneous hyperplastic lesions are associated with the dysregulation of Yap, a Hippo signaling-related protein. Biochem Biophys Res Commun 2013; 438: 607–612.
Yu FX, Guan KL . The Hippo pathway: regulators and regulations. Genes Dev 2013; 27: 355–371.
Jans R, Mottram L, Johnson DL, Brown AM, Sikkink S, Ross K et al. Lysophosphatidic acid promotes cell migration through STIM1- and Orai1-mediated Ca2+(i) mobilization and NFAT2 activation. J Investig Dermatol 2013; 133: 793–802.
Menzel M, Meckbach D, Weide B, Toussaint NC, Schilbach K, Noor S et al. In melanoma, Hippo signaling is affected by copy number alterations and YAP1 overexpression impairs patient survival. Pigment Cell Melanoma Res 2014; 27: 671–673.
Liu SC, Hsu CJ, Chen HT, Tsou HK, Chuang SM, Tang CH . CTGF increases IL-6 expression in human synovial fibroblasts through integrin-dependent signaling pathway. PLoS ONE 2012; 7: e51097.
Booth AJ, Bishop DK . TGF-beta, IL-6, IL-17 and CTGF direct multiple pathologies of chronic cardiac allograft rejection. Immunotherapy 2010; 2: 511–520.
Juric V, Chen CC, Lau LF . Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol Cell Biol 2009; 29: 3266–3279.
Juric V, Chen CC, Lau LF . TNFalpha-induced apoptosis enabled by CCN1/CYR61: pathways of reactive oxygen species generation and cytochrome c release. PLoS ONE 2012; 7: e31303.
Almodovar-Garcia K, Kwon M, Samaras SE, Davidson JM . ANKRD1 acts as a transcriptional repressor of MMP13 via the AP-1 site. Mol Cell Biol 2014; 34: 1500–1511.
Acknowledgements
This work was supported by a National Research Foundation of Korea Grant through the Oral-Maxillofacial Dysfunction Research Center for the Elderly (No. 2013-070465) at Seoul National University in Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Hwang, SM., Jin, M., Shin, Y. et al. Role of LPA and the Hippo pathway on apoptosis in salivary gland epithelial cells. Exp Mol Med 46, e125 (2014). https://doi.org/10.1038/emm.2014.77
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.77
This article is cited by
-
Verteporfin reverses progestin resistance through YAP/TAZ-PI3K-Akt pathway in endometrial carcinoma
Cell Death Discovery (2023)
-
YAP and TAZ play a crucial role in human erythrocyte maturation and enucleation
Stem Cell Research & Therapy (2022)
-
Hippo-YAP/MCP-1 mediated tubular maladaptive repair promote inflammation in renal failed recovery after ischemic AKI
Cell Death & Disease (2021)
-
BMP7 plays a critical role in TMEM100-inhibited cell proliferation and apoptosis in mouse metanephric mesenchymal cells in vitro
In Vitro Cellular & Developmental Biology - Animal (2018)
-
Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells
Scientific Reports (2016)
