
Abstract
Human-induced pluripotent stem cells (hiPSCs) derived from somatic cells of patients have opened possibilities for in vitro modeling of the physiology of neural (and other) cells in psychiatric disease states. Issues in early stages of technology development include (1) establishing a library of cells from adequately phenotyped patients, (2) streamlining laborious, costly hiPSC derivation and characterization, (3) assessing whether mutations or other alterations introduced by reprogramming confound interpretation, (4) developing efficient differentiation strategies to relevant cell types, (5) identifying discernible cellular phenotypes meaningful for cyclic, stress induced or relapsing–remitting diseases, (6) converting phenotypes to screening assays suitable for genome-wide mechanistic studies or large collection compound testing and (7) controlling for variability in relation to disease specificity amidst low sample numbers. Coordination of material for reprogramming from patients well-characterized clinically, genetically and with neuroimaging are beginning, and initial studies have begun to identify cellular phenotypes. Finally, several psychiatric drugs have been found to alter reprogramming efficiency in vitro, suggesting further complexity in applying hiPSCs to psychiatric diseases or that some drugs influence neural differentiation moreso than generally recognized. Despite these challenges, studies utilizing hiPSCs may eventually serve to fill essential niches in the translational pipeline for the discovery of new therapeutics.
Similar content being viewed by others


Introduction: pharmacologic use and discovery in neuropsychiatric disorders
Optimal pharmacologic management of multifactorial disorders depends upon understanding interactions with diverse and complex mechanistic drug targets, subcategorizing patients based on the most informative and drug-responsive phenotypes, and producing the maximal likelihood of therapeutic responses through utilization of complementary classes of pharmacologic agents. However, the treatment of psychiatric disorders has been limited by discovery of very few therapeutic compounds in the past 50 years. For instance, nearly all clinical antipsychotics are thought to work primarily by antagonizing dopamine D2 receptors with variable affinity for serotonin and norepinephrine receptors affording minimal complementarity, and that adding additional antipsychotic medications of the same class are unlikely to produce a significantly improved therapeutic response.1, 2 Essentially all D2 antagonists carry mechanism-related risks ranging from metabolic syndrome, diabetes and extrapyramidal symptoms, and clozapine has been associated with agranulocytosis. In the absence of reliable predictive parameters for assessment of medication responsiveness, patients experience frequent medication changes and polypharmacy including multiple antipsychotics, antidepressants, mood stabilizers, sedatives and off-label medications with uncertain therapeutic benefit.3 Similarly, periods required for stabilizing a psychiatric patient may be excessively or unpredictably long. For instance, the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial found that only about a third of major depression patients remit during their first medication trial, with a similar rate in the second and much lower rates in the third and fourth, typically requiring between 1 and 2 months from the start of each trial. Cumulatively about two-thirds of patients undergo remission during the entirety of multiple trials requiring several months.4 Conversely, those who do not respond may undergo even more lengthy and numerous medication trials. The discrepancies in evolution of psychotropic medications compared with treatments of other multifaceted pathological processes are at least partly attributable to the lack of representative animal models or reliable biomarkers of therapeutic efficacy.5
Another hurdle in psychiatric drug discovery is that despite meticulous characterization of drug interactions with cell surface receptors, the cell-specific physiologically targeted, downstream processes pivotal to efficacy of many psychiatric medicines have remained poorly defined.6 For instance, serotonin reuptake inhibitors such as fluoxetine are well known to block serotonin reuptake by antagonizing presynaptic serotonin receptors; however, how this action fosters slow onset of a clinical benefit has remained vague.7, 8 Indeed, it has also been observed that fluoxetine increases cell proliferation in the rat hippocampus and prefrontal cortex, upregulates brain-derived neurotrophic factor expression and synaptic plasticity in the visual cortex in rats and expands neural progenitors in mouse brains in addition to influencing regulation of ion channels and cell cycle.7, 9, 10, 11, 12 In addition to neuronal effects, fluoxetine has also been observed to influence gene regulation and glucose utilization in astrocytes independent of serotonin effects.11 As another example, the mechanism of action of lithium is perhaps most enigmatic among psychiatric medications. Evidence from murine neuronal culture and brain studies suggests that lithium modulates diverse kinase signaling pathways involving GSK3β, AKT,13 MEK, ERK,14, 15 CaM kinase IV16 and protein kinase C.17 On the other hand, lithium is also known to regulate other processes such as cell cycle progression, calcium homeostasis, apoptosis and oxidative stress.18, 19, 20 With so many putative targets of psychiatric drugs, identification of those most physiologically relevant to the human brain is critical to the informed design of new, more specific and safer drugs.
In addition to the availability of diverse medication choices, another longstanding goal of neuropsychiatry has been to tailor finely pharmacologic treatment to each patient. However, identification of markers predictive of drug responsiveness has been difficult, in part, because multiple genetic loci may be more likely to contribute to responsiveness than any single reliable marker, such as the case of lithium.21 Without reliably predictive genetic markers for drug selection, one alternative may be direct drug testing on patient-derived samples. Initial steps toward utilization of patient material for drug testing have been demonstrated by the field of oncology through culturing human tumor biopsies and testing for sensitivity to various chemotherapeutic agents.22, 23, 24, 25 Indeed, development of many such anticancer agents has resulted from cell culture studies that provide easily interpretable readouts of proliferation, apoptosis and cell cycle arrest.26 Unlike cancer cells, biopsy of many human cell types is potentially harmful to the patient or difficult to culture, particularly those of the nervous system, further impeding studies of the pathophysiology of disease, drug discovery and development of patient-specific treatment.
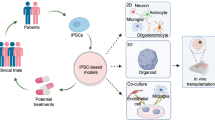
Human-induced pluripotent stem cells as a tool to address some challenges of psychiatric drug discovery
The notion of obtaining neural cells from patients has been entertained for nearly two decades. Neural stem cells (NSCs) were isolated from post-mortem specimens for some diseases. Olfactory epithelium has been proposed as a source of neural cells from living patients. However, none of these sources could optimally address the need for studying specialized neural cells, particularly neurons of the central nervous system, from a broad spectrum of living patients (and their family members) across a range of diseases. A potential tool for addressing not only the obstacles to rational pharmacotherapy discussed in the previous section but also the limitations of obtaining representative neural cells discussed above has been the advent of technology for creating ‘human-induced pluripotent stem cells (hiPSCs)’—the reprogramming of somatic cells (e.g., easily obtainable skin fibroblasts) from a living patient such that they first acquire pluripotence and are then differentiated toward a neural lineage, including patient-specific neurons. ‘Pluripotency’ was initially described in embryonic stem cells (ESCs) derived from mammalian blastocysts which, when cultured, can self-renew and differentiate in vitro into the three primordial germ layers (barring extraembryonic tissues), mesoderm, ectoderm and endoderm.27 However, despite development of differentiation strategies for human ESCs to divergent cell types and the fact that diseased blastocysts could occasionally be obtained from in vitro fertility clinics based on prenatal genetic diagnosis, they were not ideally suited for disease modeling because of the ethical challenges of obtaining them, limited availability and, in normal human ESCs, poor amenability to genetic engineering for simulating disease-related mutations. This obstacle was overcome by development of fate reprogramming through the expression of specific combinations of exogenous factors, c-Myc, Klf4, Oct3/4, Sox2, Nanog, Lin28, in somatic cell types (including fibroblasts, blood cells, umbilical cord vein cells or NSCs to hiPSCs, which show similar propensity for self-renewal and pluripotency as human ESCs.28, 29, 30
The advantage and novelty of hiPSCs is that the cells reflect the genetic signature of the patient from which they are derived, yet can be further differentiated to other physiologically relevant cells types. Such hiPSCs have now been derived from patients with a number of neurological, psychiatric, cardiac and genetic diseases.31 Differentiation of hiPSCs to diverse multipotent precursor cells and eventually to more specialized functional cell types including electrophysiologically active neurons may more closely approximate intracellular signaling, intercellular connectivity and drug sensitivity of the central nervous system than other cells traditionally obtained from patients such as fibroblasts or blood cells. Moreover, the ability to make measurements directly from neural cells with the same genetic background as the patient is likely to allow more precise analysis than the current methods of measuring CSF, imaging and non-invasive electrophysiology. However, as discussed below, a number of obstacles will need to be addressed for realization of practical uses in drug discovery (Tables 1 and 2).
Reprogramming is generally accomplished by introducing exogenous gene products into stable somatic cell lines followed by transition to media preparations conducive to stem cell self-renewal including supplementation with purified fibroblast growth factor 2.32 Although initial studies largely relied on retro- and lenti-virus-mediated transduction of exogenous genetic factors, recent efforts have been made toward non-integrating strategies including episomal-mediated transfection of cells from schizophrenia patients.33 Other strategies applied to disease models have included viral transduction of inducible factors or excisable factors using Cre-lox-mediated recombination.34 Ongoing work has been reported using microRNA, synthetic mRNA and purified protein transduction domain fusion proteins35 (Table 2).
After reprogramming, single colonies exhibiting morphology indicative of stem cells are expanded and stored as frozen stocks. The basic characterization of the cells, although not standardized, typically includes the following confirmations of pluripotency: (1) immunostaining for pluripotency markers Nanog, SSEA3/4, Tra1-60/1-80 and Oct4 among others, (2) spontaneous or directed in vitro differentiation within embryoid bodies (EBs)—cellular aggregates consisting of subsets of cells representing the three fundamental germ layers (affirmed by markers), and (3) in vivo teratoma formation in an immunocompromised. Other informative aspects of characterization have included detection of viral integration sites, expression analysis of exogenous pluripotency factors in hiPSCs and the differentiated derivatives, DNA fingerprinting to confirm origin of the cells, array comparative genomic hybridization, exome sequencing and karyotyping for mutational analysis and mRNA microarray expression analysis for comparison to bioinformatic databanks of pluripotent cellular expression profiles.36, 37 Given the arduous and expensive nature of generating and validating an hiPSC line, studies published to date typically involve a handful of patients with few replicate lines (Tables 1 and 2). In part because of this limitation, there has generally been less than optimal opportunity for replication of results in many independently derived samples from similar patients. Even the reproducibility of experimental results from derivatives of multiple hiPSC lines from the same fibroblast culture or duplicate somatic cell biopsies of the same patient is still uncertain. A recent study reprogramming keratinocytes of schizophrenia patients worked to address the question by repeating the entire hair follicle plucking and reprogramming from a single patient 1 year after initial sample derivation.38 Indeed, any result derived from a given hiPSC line always requires independent confirmation in another hiPSC line.
In studying neuropsychiatric diseases, hiPSCs are generally differentiated to neural cells by one of several strategies. Methods for neural differentiation have traditionally involved coculture with stromal feeder layers coupled with exposure to specific morphogens, media supplements and ECM components that most closely recapitulate the in vivo milieu of NSCs. Alternatively, hiPSCs may be grown in suspension to form embryoid bodies. Within these heterogeneous cell populations, neural rosettes are formed from which neural cells may be separated from other cell types.39, 40 Newer alternative strategies for feeder-free, directed differentiation to NSCs utilizing chemical inhibitors and purified protein activators of specific signaling pathways important in fate determination have also been developed.41, 42, 43 Further differentiation utilizing more specific cocktails of small molecules and purified protein for enrichment of neuronal subtypes such as dopaminergic populations has been applied to hiPSC studies in Parkinson’s disease.44, 45 However, generation of other neuronal subtypes such as serotinergic neurons, and other neural lineages including oligodendrocytes and astrocytes from hiPSCs is still in its earliest stages and therefore has yet to be pursued robustly in hiPSC disease models46 (Table 2).
Caveats of interpreting data from hiPSC studies
One current drawback of hiPSC technology is variability imparted at each step of reprogramming and neural differentiation. Despite maintenance of disease-specific polymorphisms, an advantage of hiPSC modeling, numerous studies have reported that the induction of pluripotency also introduces unpredictable new genetic lesions including copy number variants (CNVs), karyotypic abnormalities, deletions and point mutations47, 48, 49 (Table 2). On one hand, earlier passage hiPSCs harbor transient epigenetic and genetic variance alleviated in part by negative growth selection against cells that acquired CNVs.48 Conversely, mutations conferring a selective growth advantage may be preserved, and karyotypic abnormalities, including aneuploidy, may appear in later passages.49, 50 Although early work suggests that at least karyotypic abnormalities may not overtly affect neuralization,50 it is possible that such genetic instability and alteration could obscure patient-specific disease modeling. For instance, CNVs are increasingly recognized to associate with specific diseases such as schizophrenia, although not as much in bipolar disorder. Therefore, reprogramming may diminish the fidelity of some specific models. Although many of the CNVs sustained by hiPSCs are not generally observed in patient populations, the potential for de novo CNVs affecting downstream gene expression and signaling directly or indirectly should be considered when interpreting data from small numbers of iPSc lines.51
There is also evidence that hiPSC lines exhibit variable efficiency of differentiation to neuroectoderm and neural subtypes including motorneurons50, 52, 53 (Table 2). Although it is unclear what factors influence the differentiation of hiPSC lines, neuralization efficiency appears independent of reprogramming strategy, transgene integration and expression, overt karyotypic anomalies or age of the patient at the time of skin biopsy. There is evidence of correlation with donor gender, and importantly, that varying differentiation efficiency is more likely uncovered by less robust differentiation protocols.50, 53 Whether efficiency of differentiation will play out to be a major problem and how to manage or control for these differences has not been established. These considerations are potentially of greatest concern in diseases in which the pivotal cell type is uncertain, preventing informative examination of efficiencies of differentiation to diverse and relevant cellular subtypes. As neuralization and differentiation protocols are better optimized, baseline differences may be standardized sufficiently to make direct comparisons between cell lines more relevant. For example, in one study using hiPSCs from Praeder–Willi syndrome, the authors elected to standardize subsequent assays by choosing to compare only those cell lines that exhibited similar efficiency of differentiation.54
One alternative to reprogramming strategies for differentiation of neurons is ‘transdifferentiation’, the conversion of fibroblasts directly to neurons (the so-called, ‘induced neurons (iN)’) or even dopaminergic neurons (‘iDA’) using exogenous neuronal transgenes.55, 56 Direct conversion may reduce the timeline to experimentation as well as variables of deriving multiple cell-type intermediates. However, it has yet to be addressed how direct neural conversion influences mutational burden and epigenetic signatures, or the extent of variation in differentiation efficiency to neuronal derivatives across many patient lines. One issue with transdifferentiation is that it may require repeated transduction to transdifferentiate enough material for ongoing experiments, in contrast to self-renewing cells like hiPSCs that may be frozen, stored and passaged for later differentiation to neurons without the need for repeated reprogramming. An early attempt for compromise between these two technologies was demonstrated by direct reprogramming of mouse fibroblasts to NSCs, thus bypassing some of the stepwise aspects inherent to use of hiPSC intermediates, but preserving more potential for cellular expansion than direct neuronal conversion.57 Another approach that straddles technologies is called ‘induced conditional self-renewing progenitor (ICSP) cells’, in which multipotent but non-self-renewing progenitor cells are obtained from the organ of therapeutic interest (e.g., the nervous system, without the need for genetic reprogramming) but simply need to have their proliferative potential conditionally augmented in vitro.58
The majority of hiPSC disease studies to date have utilized fibroblasts as the starting material owing to the ease of culture and the ease of reprogramming. However, few studies have been published using fibroblasts from psychiatric patients, and there has been somewhat of a lag in acquiring fibroblasts from informative cases. As a result, the material for many initial studies was obtained from cell banks with only basic diagnostic and demographic information. More recent studies are making efforts to obtain material from patients subtyped genetically and diagnostically, with more comprehensive clinical data as well as ability for future follow-up studies.33
The effects on hiPSC studies of the age of biopsy, passage number of the fibroblasts and the state of the patient at the time of biopsy have yet to be addressed. These questions may be particularly important as the nuances of previous assumptions regarding hiPSCs are beginning to be uncovered. For instance, it has been thought that one advantage of hiPSCs is inherent immunocompatibility with syngeneic donors; however, recent work has shown that this is not necessarily the case, potentially due to the expression of specific immunogenic gene products.59 Efforts are also being made to reprogram nucleated blood cells (e.g., lymphoblasts) more efficiently, given that such material is the most common deposit to most neuropsychiatric patient cell banks.
Finding and modeling neuropsychiatric disease phenotypes with neural cultures from patient-derived hiPSCs
In neuropsychiatric disorders, a key challenge is the lack of translatable animal models, particularly for disorders in which higher function is involved, and the reliance on behavioral models may be limiting.60 Moreover, many of the current animal models are reverse engineered from the consequences of drugs known to be effective such as D2 antagonists, and new medicines have been developed that mimic the behavioral effects of such medicines. As discussed, this limits the range of treatment options. Although behavioral models are appealing in that they may mimic the symptoms or signs of the disease, stem cell-based models could conceivably take us closer to the pathophysiological phenomena at the neural cell level. Uncovering the correlates of disease in cell-based models derived from patients is needed before attempts are made to develop therapeutics based on the underlying pathophysiology, particularly because the current understanding of genetic and epigenetic contributing factors is incomplete. Although the field is still in its infancy, work by Marchetto et al.61 and Brennand et al.34 in psychiatry and a series of papers in genetic neurological disorders illustrate the potential for identifying phenotypes in neurons of patients with neuropsychiatric disease. The potential for screening compounds against such phenotypes in a high-throughput, unbiased manner, and for identifying mechanisms that underlie those phenotypes has yet to be accomplished, however.
One approach toward identifying useful biologic readouts has been broad systematic testing for phenotypes. This was highlighted by observations from basic morphology testing of neurons differentiated from hiPSCs derived from a child with Rett syndrome, a disease cased by a mutation in MecP2. Such neurons showed decreased cell body size and dendritic spine density.61 However, the specificity of such phenotypes for this disease is unclear, as a wide array of psychiatric and neurologic conditions are known to exhibit altered dendritic spine density and morphology including autism, Alzheimer’s, mental retardation and even schizophrenia.62
On the other hand, even nonspecific phenotypes may be relevant for translational research if the readout is amenable to screening for pharmacologically active compounds, which once identified can be secondarily tested for specificity to particular disease-derived cells and processes. This approach has also been useful in the early investigation of schizophrenia, particularly in hiPSCs in which a defined genetic lesion to serve as a starting point is not present. Based on analysis of an array of assays to guide potential experimental directions, hiPSC-derived neurons derived from schizophrenia may bear slightly decreased neurite number and decreased connectivity. As a case-in-point, the antipsychotic drug loxapine may increase connectivity in schizophrenia hiPSC-derived neurons. Whether such assays can be applied to high-content screening approaches for additional therapeutic compounds will likely require further standardization. One practical obstacle is the sheer length of time involved in differentiating the neurons to make them suitable for the assay—approximately 3 months.34 A more recent study attempted to enhance patient selection based on clinical subtyping, a standardized severity rating scale and medication history restricting selection to clozapine-treated patients. Indeed, history of clozapine treatment typically provides an additional specificity to patients largely refractory to most antipsychotics.38 Despite this, without a clear understanding of genetic or biochemical determinants of antipsychotic responsiveness, a step-wise approach to basic characterization is required. Indeed, the authors of this study identified baseline differences in neural differentiation between patients unaffected and with schizophrenia. Although the authors focused on mitochondrial function given its relationship to neuronal function and dopamine signaling, broad baseline characterization and comparison of organellar function in hiPSC-derived models of psychiatric illness including that of the endoplasmic reticulum, lysosomes and golgi apparatus, among others, may provide insights into underlying cell biology or potential for drug target identification and drug discovery.
Another potential niche for hiPSC modeling demonstrated byBrennand et al.34 is testing genome-wide association study candidates for altered regulation in schizophrenia-derived cells. Cell sorting or highly directed differentiation strategies may be central to this approach because only a subset of neurons generated may normally express phenotypes of interest. Therefore, expression profiling of heterogeneous cell neural populations may reflect more the subtle differentiation proclivities of particular hiPSC lines than disease-specific gene expression patterns. In addition to attempting to discern disease-specific gene associations, another potential use for hiPSCs would be to monitor disease-specific alterations in function. For instance, it was recently proposed, based on analysis of previously published genome-wide association studies, that patients with schizophrenia show enrichment in regions affecting endosomal trafficking;63 such activity, if true, could be converted into a disease-relevant biological assay performed on patient-derived neurons.
As the relevance of experimentally derived cell populations for functional human neural systems is addressed, a further consideration may be how best to model chronic episodic illnesses such as bipolar disorder or illnesses with defined periods of stability and decompensation common in primary psychotic disorders. Because environmental stimuli may impinge on the severity and timing of these disease states, one approach to modeling them in a dish may be the application of extracellular stress to hiPSC-derived neurons. It has been observed, for instance, that glucocorticoid receptor regulation in response to exogenous hydrocortisone shows greater sensitivity in B-lymphoblastoid cell cultures derived from bipolar disorder patients than unaffected controls.64 As a first test of stress susceptibility of disease-specific neurons, it was shown that Parkinson’s hiPSC dopaminergic-enriched neurons show increased caspase-3 expression during oxidative stress. The authors were able to use this assay to test several compounds, suggesting that such an approach may be eventually extrapolated to drug screening.44 In addition to stress-induced phenotypes, another novel application for hiPSC models may be the study of infectious etiologies of neuropsychiatric diseases. For instance, congenital human cytomegalovirus, a cause of mental retardation, exhibits a significantly higher infectivity rate toward hiPSC neural precursors cells than neurons, but may subsequently inhibit aspects of neuronal differentiation.65
Before the development of hiPSC models, research in understanding psychopharmacologic mechanisms routinely utilized primary neural cell cultures or the culture of neural cell lines. Many of the tools developed using these older strategies can be applied to hiPSC technology. For instance, the effects of such psychotropic drugs as antipsychotics, lithium and valproic acid (VPA) on protein and mRNA regulation, cellular survival, proliferation and differentiation have been examined in cultured murine NSCs and neurons.66, 67, 68, 69 Such metrics should be applicable to hiPSC-derived neurons obtained from actual patients. Another common approach is the use of cells bearing reporter constructs, commonly bioluminescent genes, like luciferase, under the transcriptional control of a functionally relevant gene promoter. Such assays provide a means for tracking gene interactions, pathway activation and drug action. For example, murine glioma cells have been engineered to express a reporter construct in which luciferase was expressed whenever the cyclic AMP response element was activated. Cyclic AMP response element-binding protein activity has been implicated in memory and plasticity. Using such an assay, it was determined that amitryptiline might regulate cyclic AMP response element-binding protein.70 Luciferase constructs have also been utilized to study the effects of genetic polymorphisms on both gene expression and miRNA targeting.71, 72 These and similar reporter constructs could eventually be applied to hiPSC-derived neurons both for pathway analysis and chemical library screening of compounds.
Indeed, utilization of measurable cellular behaviors that may be pertinent to the pathology of the disease-of-interest is another potential advantage of generating relevant cell types from affected patients. For instance, circadian rhythm is a well-known lithium-responsive phenotype and can be measured by gene expression assays in cell culture. Diminished amplitude of expression of circadian rhythm genes has been observed in fibroblasts derived from bipolar patients compared with controls.73 Importantly, such phenotypes can be turned into reporter assays for eventual screening purposes. For instance, BMAL and PER2 promoter activation, regulated cyclically during the circadian rhythm, has been used to study the effects of lithium on circadian cycles in human retinal pigment epithelial cell lines and fibroblasts.74, 75 As proof of principle, it has been shown that circadian rhythm may be similarly studied in neural derivatives of mouse ESCs by mRNA expression and luciferase reporter assays.76 We have also found that expression of circadian genes can be used to trace rhythms in hiPSC-derived neural cells (Tobe BT, Brandel MG, McCarthy M and Snyder EY, unpublished data).
Interestingly, the utility of drug screening in psychiatry has recently been demonstrated serendipitously in cell culture studies. A drug screen designed for modulators of insulin expression in an engineered pancreatic islet cell line, designed to develop therapeutics for diabetes, unexpectedly identified phenothiazine antipsychotics as a major regulator of the insulin promoter. This study may expose a mechanistic explanation of the metabolic side effects observed in neuroleptic-treated patients, and highlight the feasibility of detecting specific activities of psychoactive compounds.77 An early approach toward developing such screening tools for stem cell derivatives was put forth for identifying inhibitors of butulinum toxin. Motor neurons differentiated from mouse ESCs were tested for sensitivity to botulinum toxin and for alleviation of the effect by specific botulinum inhibitors.78
Although iPS-derived neural cells may prove powerful route for revealing cellular phenotypes, animal models will likely still be required to understand the interaction of cellular phenotypes within an intact organism, intereacting with over systems (e.g., vascular, inflammatory, etc.). Interestingly, stem cells can provide an approach to create animal models. The first example of this strategy was the use of globally engrafting NSCs to distribute disease-inducing viruses throughout a normal mouse brain, creating a novel model of acute spongiform encephalopathy;79, 80 it was the first use of stem cell to create a ‘poor man’s transgenic mouse’. A more recent example was the reconstitution of function in Parkinsonian rats with hiPSC-derived neurons from Parkinson’s disease patients.45 One could imagine populating relevant brain regions in rodents with cells from patients with Alzheimer’s disease, mood disorders or schizophrenia in a similar manner and testing relevant circuits for abnormalities in the animals’ behavior. Recreating animal models may be an important step in the validation of a cellular phenotype derived from disease-specific hIPSCs, particularly those modeling psychiatric disorders. Such animal models may then be used for testing novel therapeutics that act on these mechanisms.
Using patient-specific hiPSC derivatives for drug discovery
One long-term goal of establishing patient-derived hIPSCs has been to use their cellular derivatives to screen chemical and genetic libraries for agents that might modulate disease-relevant biological readouts;81 the assumption is that aberrantly regulated cellular machinery is present in these cells. Indeed, compounds with potential efficacy may go unrecognized if tested on normal cells or cells derived from a less specific disease condition.
Some early successes with this approach have been in applying drugs to neurons derived from hiPSCs generated from patients with monogenic diseases with the intent of modulating the activity of that single defective gene. It was shown, for instance, that treatment of neurons differentiated from Rett syndrome-derived hiPSCs with gentamicin promoted a read-through of a nonsense premature stop codon mutation in the MECP2 gene leading to increased MECP2 protein levels, and in turn, glutamatergic synapse markers.61 Using hiPSC motor neurons from spinal muscular atrophy patients, it was shown that exposure to tobramycin or VPA increased the expression of disease-ameliorating SMN-2 protein and the presence of intranuclear SMN-containing aggregates known as gems.82
In hiPSC-derived neurons from schizophrenia patients, some preliminary attempts have been made to ‘correct’ cellular abnormalities. Of several antipsychotics tested, loxapine—but not other antipsychotics including olanzapine, clozapine, risperidone or thorazine—appeared to improve neuronal connectivity in vitro based on the above-mentioned rabies virus assay. This result, however, emphasizes the importance of thorough patient histories in hiPSC studies—for instance, the history of responsiveness of the patient under study to different medications. Combining expression analyses with drug testing, it was found that loxapine influenced expression of the previously implicated schizophrenia genome-wide association studies candidate NRG1, but not DISC1. On the other hand, microarray mRNA expression candidates found to be decreased in schizophrenia (compared with control) hiPSC neurons pinpointed other genes as potential direct or indirect targets of loxapine (e.g., GRIK1, ADCY8, WNT7A).34 In this case, drug treatment was applied for 3 weeks, suggesting that expression changes could have resulted from numerous direct and indirect factors. Nevertheless, it is conceivable that genes differentially expressed during disease states or following drug exposure could provide the basis for new reporter-based assays to be used for screening small-molecule compounds.
The approaches to date have involved assessing a few known drugs for their effects on phenotypes of interest. However, the likelihood of finding novel or unexpected drugs or drug targets—or of ‘repurposing’ known drugs—is magnified by the unbiased screening of hundreds of thousands of compounds to find the one or several that exert a specific desired effect upon the phenotype of interest. Such actions may unveil a previously unrecognized pathophysiological mechanism or a lead compound might be suggested, whose structure–activity relationship might then be refined through medicinal chemistry to create an optimal candidate drug. Such an unsupervised high-throughput approach typically involves converting the cell-based phenotype into a scoreable assay that can be accommodated within a microwell, and managed and assessed by robotic systems. To be amenable to such instrumentation, hiPSCs derivatives must meet the following challenges: (1) a need for homogeneity of cell type; heterogeneity reduces the consistency and meaningfulness of a positive hit; (2) a need for the ability to remain healthy and stable in a microwell culture for long periods of time; (3) the need to use high content or complex phenotypic screening methodologies to evaluate meaningful neuronal function (e.g., synaptic spine morphology, neurite configuration, mitochondrial membrane potentials, intracellular aggregates); (4) the lack of known molecular targets that might impede refinement by structure–activity relationship because of the possibility that actions on multiple targets have summed to create the observed cellular effect; (5) the risk that a finding might be particular to the patient from whom the hIPSCs were generated and not universal for the disease-state, making it imperative to re-test any compounds on a panel of patient-derived cells; and (6) the need for animal models that allow validation of phenotypes discovered from the use of hIPSC technology (discussed above).
Psychiatric drugs and their effects on reprogramming: insights and challenges
Surprisingly, the underlying tenets of reprogramming itself may provide insight into the mechanism-of-action of drugs for psychopharmacotherapy. Because of the low efficiency of reprogramming via genetic induction and the potential for mutagenesis through the use of integrating viral and transposon vectors, efforts have been made to identify small molecule compounds that may substitute for these genetic modifications. However, few such molecules have been reported. Strikingly, several of the compounds that have been identified are used routinely for the treatment of psychiatric illness including lithium, VPA and tranylcypromine83, 84, 85 (Figure 1). Although a mechanistic understanding of reprogramming is still developing, chromatin modification may be at least one important aspect.86, 87, 88 How cellular fate may be determined by these alterations has yet to be understood, although it may involve modification and subsequent regulation of discrete sets of genetic loci important for pluripotency and cell cycle regulation versus global epigenetic state changes.89 That certain psychoactive compounds have been found to improve reprogramming coincides with accumulating evidence that chromatin modification may, at least in part, underlie numerous psychiatric conditions and may represent a physiologic target of psychoactive compounds.90, 91, 92

Cellular reprogramming of somatic cells to human-induced pluripotent stem cells (hiPSCs) may be influenced by compounds also used therapeutically in clinical psychiatry. Because introduction of exogenous genetic factors may impart mutagenesis, efforts have been made to identify chemical compounds sufficient to replace genetic factors. Interestingly, to date, few such compounds have been identified, although several are incidentally used clinically in psychiatry. Because the precise mechanism by which such compounds influence either cellular reprogramming or clinical psychiatric symptoms is yet unclear, it is therefore also unknown if there is a common mechanistic component or if dedifferentiation contributes directly to therapeutic action of the drugs. Interestingly, the antidepressant tranylcypromine, and lithium to some degree, is thought to impinge on lysine-specific demethylase 1 in the regulation of histone methylation, which may also influence reprogramming. Lithium, a first-line treatment of bipolar disorder known to exhibit an inhibitory effect on GSK3β, and this in turn has been implicated by numerous studies in reprogramming of mouse cells. Valproic acid, a commonly prescribed mood stabilizer, is a histone deacetylase inhibitor, has also been widely used to increase efficiency of reprogramming (figure illustrations by Rachel Krupa).
Tranylcypromine, an irreversible monoamine oxidase inhibitor, was found sufficient for replacement of KLF4 in reprogramming cocktails applied to murine fibroblasts.84 Tranylcypromine inhibits demethylation of histone H3 at the residue 4 lysine group by covalently binding to two distinct flavin adenine dinucleotide-dependent amine oxidases, lysine-specific demethylase 1 and 2. These enzymes are similar in structure to monoamine oxidases, a main target of the antidepressant effects of tranycypromine.93, 94, 95 Lysine-specific demethylase 1 downregulation may also explain part of the mechanism of the effect of lithium in driving reprogramming.85 Lithium is also thought to increase reprogramming efficiency by modulation of GSK3β, a gene widely implicated in neuropsychiatric illness including bipolar disorder and schizophrenia.85 Indeed, CHIR-99021, a specific inhibitor of Gsk3β, also increases reprogramming efficiency.96
Like tranylcypromine, VPA was initially found to decrease exogenous genetic requirements for reprogramming of fibroblasts. Since this finding, VPA has been used to augment reprogramming protocols. Indeed, work utilizing rat primary astrocytes to assay chromatin modification by psychotherapeutic compounds has shown that VPA increases acetylation of histone H3 lysine 9 and histone H4.97 These data are supported by observations in rat hippocampal neural progenitor cells that VPA increases acetylation of histone H3 and H4, which correlates with altered expression of genetic loci modulated by H4 acetylation, some of which have been implicated in neuronal differentiation.98 Given these observations, it is possible that cellular samples from patients treated with such compounds may show subtle differences in the character of reprogramming fibroblasts to iPSCs or confound interpretation of phenotypes and signatures in relation to underlying mechanisms specific to psychopathology. To date, no studies have examined the effect of systemic drug administration on eventual reprogramming efficiency and differentiation potential of the cells.
Although the majority of reprogramming experiments have utilized fibroblasts as the starting material, several studies have reported the dedifferentiation of neural cells to hiPSCs. Moreover, human NSCs have been reprogrammed via introduction of OCT4 alone.30, 99 This may be partially or completely explained by the observation that human NSCs, unlike most cell types, show endogenous production of Sox2, c-Myc and Klf4, which may confer enhanced susceptibility to dedifferentiation. Akin to stochastic theories of in vitro dedifferentiation and resulting low efficiency of such processes, how molecular therapeutics might alter neural differentiation in vivo is difficult to delineate, particularly given the subtle subtype distinctions within cellular lineages and varying influences of the microenvironmental intercellular milieu. However, if neuronal differentiation is particularly susceptible to exogenous factors for differentiation, this may have profound implications for psychotherapeutic drug development. That psychoactive compounds are now being observed to alter broad cellular fates suggests that this may be an aspect of the effects of psychotropic medication. Indeed, recent attention to the potential of psychiatric medication for neuroprotection and the emerging importance of cell generation and neuronal differentiation in adult brains may complement these observations. Future studies examining the effects of long-term administration of drugs on chromatin modification in animal models and in human iPSC-derived models or accessible human tissue samples may provide insight into the potential epigenetic effects of medication administration. Examination of correlations between epigenetic and clinical efficacy data may also aid in determining if epigenetic mechanisms contribute directly to therapeutic effects. Indeed, understanding how medications alter cellular fate via chromatin modification may lend insight into determinants of latency in early stages of psychopharmacologic treatment, or how second or third trials of a medication may be efficacious even in patients after a failed therapy using alternate therapy of the same drug class.
Conclusion
The ability to generate neurons from the reprogramming of non-neural cells that are readily obtainable from living patients and their relatives provides new tools for drug testing and screening in psychiatric disorders. However, major hurdles have yet to be overcome. These issues include reproducibility of results; fidelity of reprogrammed genomes to the source cells; specificity of findings for a particular disease state; methods of validation; development of physiologically relevant assays and feasibility of large-scale studies given the high financial costs. Utilization of hiPSCs, like any biological model, depends heavily on being combined with other research tools; however, some methods most pivotal to realizing the potential of hiPSCs are also in their infancy. In particular, neural differentiation protocols for generating even early stage neurons, not to mention the most clinically relevant specialized cell types, are generally cumbersome, lengthy, expensive and usually incapable of yielding large populations of homogeneous cell types in a state amenable to high-throughput screening or large-scale comprehensive profiling. Routine generation of oligodendrocytes, astrocytes and neuronal subtypes such as serotinergic neurons are particularly difficult. However, cellular reprogramming is a rapidly developing field as numerous methodologies have been developed over a short period of time; concurrently, better differentiation methods—including direct transdifferentiation of non-neural cells to neural lineages—are being rapidly developed. Once these and the other limitations discussed in the previous sections are addressed, the opportunity for modeling psychiatric disease ‘in-a-dish’ sufficient for the first steps in unveiling novel pathophysiological mechanisms, prognostics, diagnostics, drug targets and the drugs suited to those targets should herald a new era in psychopharmacological research.
References
Carpenter WT, Koenig JI . The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology 2008; 33: 2061–2079.
Barnes TR, Paton C . Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs 2011; 25: 383–399.
Zink M, Englisch S, Meyer-Lindenberg A . Polypharmacy in schizophrenia. Curr Opin Psychiatry 2010; 23: 103–111.
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163: 1905–1917.
Becker RE, Greig NH . Lost in translation: neuropsychiatric drug development. Sci Transl Med 2010; 2: 61rv6.
Lacasse JR, Leo J . Serotonin and depression: a disconnect between the advertisements and the scientific literature. PLoS Med 2005; 2: e392.
Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O'Leary OF et al. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 2008; 320: 385–388.
Cowen PJ . Serotonin and depression: pathophysiological mechanism or marketing myth? Trends Pharmacol Sci 2008; 29: 433–436.
Castren E, Voikar V, Rantamaki T . Role of neurotrophic factors in depression. Curr Opin Pharmacol 2007; 7: 18–21.
Encinas JM, Vaahtokari A, Enikolopov G . Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci USA 2006; 103: 8233–8238.
Mostert JP, Koch MW, Heerings M, Heersema DJ, De Keyser J . Therapeutic potential of fluoxetine in neurological disorders. CNS Neurosci Ther 2008; 14: 153–164.
Krishnan A, Hariharan R, Nair SA, Pillai MR . Fluoxetine mediates G0/G1 arrest by inducing functional inhibition of cyclin dependent kinase subunit (CKS)1. Biochem Pharmacol 2008; 75: 1924–1934.
Beaulieu JM, Caron MG . Looking at lithium: molecular moods and complex behaviour. Mol Interv 2008; 8: 230–241.
Pardo R, Andreolotti AG, Ramos B, Picatoste F, Claro E . Opposed effects of lithium on the MEK-ERK pathway in neural cells: inhibition in astrocytes and stimulation in neurons by GSK3 independent mechanisms. J Neurochem 2003; 87: 417–426.
Catapano LA, Manji HK . Kinases as drug targets in the treatment of bipolar disorder. Drug Discov Today 2008; 13: 295–302.
Tardito D, Tiraboschi E, Kasahara J, Racagni G, Popoli M . Reduced CREB phosphorylation after chronic lithium treatment is associated with down-regulation of CaM kinase IV in rat hippocampus. Int J Neuropsychopharmacol 2007; 10: 491–496.
Hahn CG, Friedman E . Abnormalities in protein kinase C signaling and the pathophysiology of bipolar disorder. Bipolar Disord 1999; 1: 81–86.
Quiroz JA, Machado-Vieira R, Zarate CA Jr, Manji HK . Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology 2010; 62: 50–60.
McCarthy MJ, Leckband SG, Kelsoe JR . Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics 2010; 11: 1439–1465.
Camins A, Crespo-Biel N, Junyent F, Verdaguer E, Canudas AM, Pallas M . Calpains as a target for therapy of neurodegenerative diseases: putative role of lithium. Curr Drug Metab 2009; 10: 433–447.
Perlis RH, Smoller JW, Ferreira MA, McQuillin A, Bass N, Lawrence J et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am J Psychiatry 2009; 166: 718–725.
Antonelli A, Ferrari SM, Fallahi P, Berti P, Materazzi G, Barani L et al. Primary cell cultures from anaplastic thyroid cancer obtained by fine-needle aspiration used for chemosensitivity tests. Clin Endocrinol (Oxf) 2008; 69: 148–152.
Bagriacik EU, Baykaner MK, Yaman M, Sivrikaya G, Durdag E, Emmez H et al. Establishment of a primary pleomorphic xanthoastrocytoma cell line: in vitro responsiveness to some chemotherapeutics. Neurosurgery 2011; 70: 188–197.
Basu D, Montone KT, Wang LP, Gimotty PA, Hammond R, Diehl JA et al. Detecting and targeting mesenchymal-like subpopulations within squamous cell carcinomas. Cell Cycle 2011; 10: 2008–2016.
Suzuki M, Ishikawa H, Tanaka A, Mataga I . Heterogeneity of anticancer drug sensitivity in squamous cell carcinoma of the tongue. Hum Cell 2011; 24: 21–29.
Liu R, Hsieh CY, Lam KS . New approaches in identifying drugs to inactivate oncogene products. Semin Cancer Biol 2004; 14: 13–21.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282: 1145–1147.
Cai J, Li W, Su H, Qin D, Yang J, Zhu F et al. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J Biol Chem 2010; 285: 11227–11234.
Kaneko S, Otsu M, Nakauchi H . Reprogramming adult hematopoietic cells. Curr Opin Hematol 2010; 17: 271–275.
Kim JB, Greber B, Arauzo-Bravo MJ, Meyer J, Park KI, Zaehres H et al. Direct reprogramming of human neural stem cells by OCT4. Nature 2009; 461: 649–3.
Unternaehrer JJ, Daley GQ . Induced pluripotent stem cells for modelling human diseases. Philos Trans R Soc Lond Ser B 2011; 366: 2274–2285.
Takahashi K, Okita K, Nakagawa M, Yamanaka S . Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2007; 2: 3081–3089.
Chiang CH, Su Y, Wen Z, Yoritomo N, Ross CA, Margolis RL et al. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol Psychiatry 2011; 16: 358–360.
Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011; 473: 221–225.
Sidhu KS . New approaches for the generation of induced pluripotent stem cells. Expert Opin Biol Ther 2011; 11: 569–579.
Muller FJ, Schuldt BM, Williams R, Mason D, Altun G, Papapetrou EP et al. A bioinformatic assay for pluripotency in human cells. Nat Methods 2011; 8: 315–317.
Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc Natl Acad Sci USA 2011; 108: 6537–6542.
Robicsek O, Karry R, Petit I, Salman-Kesner N, Muller FJ, Klein E et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol Psychiatry 2013; 18: 1067–1076.
Yeo GW, Coufal N, Aigner S, Winner B, Scolnick JA, Marchetto MC et al. Multiple layers of molecular controls modulate self-renewal and neuronal lineage specification of embryonic stem cells. Hum Mol Genet 2008; 17: R67–R75.
Dhara SK, Stice SL . Neural differentiation of human embryonic stem cells. J Cell Biochem 2008; 105: 633–640.
Li W, Sun W, Zhang Y, Wei W, Ambasudhan R, Xia P et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci USA 2011; 108: 8299–8304.
Lee G, Chambers SM, Tomishima MJ, Studer L . Derivation of neural crest cells from human pluripotent stem cells. Nat Protoc 2010; 5: 688–701.
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L . Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009; 27: 275–280.
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011; 8: 267–280.
Hargus G, Cooper O, Deleidi M, Levy A, Lee K, Marlow E et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc Natl Acad Sci USA 2010; 107: 15921–15926.
Emdad L, D'Souza SL, Kothari HP, Qadeer ZA, Germano IM . Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev 2012; 21: 404–410.
Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011; 471: 63–67.
Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Narva E et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011; 471: 58–62.
Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010; 7: 521–531.
Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ et al. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol 2011; 29: 279–286.
Stankiewicz P, Lupski JR . Structural variation in the human genome and its role in disease. Annu Rev Med 2010; 61: 437–455.
Hu BY, Weick JP, Yu J, Ma LX, Zhang XQ, Thomson JA et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci USA 2010; 107: 4335–4340.
Kim JE, O’Sullivan ML, Sanchez CA, Hwang M, Israel MA, Brennand K et al. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc Natl Acad Sci USA 2011; 108: 3005–3010.
Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc Natl Acad Sci USA 2010; 107: 17668–17673.
Caiazzo M, Dell'anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011; 476: 224–227.
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M . Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010; 463: 1035–1041.
Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S et al. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci USA 2011; 108: 7838–7843.
Kim KS, Lee HJ, Jeong HS, Li J, Teng YD, Sidman RL et al. Self-renewal induced efficiently, safely, and effective therapeutically with one regulatable gene in a human somatic progenitor cell. Proc Natl Acad Sci USA 2011; 108: 4876–4881.
Zhao T, Zhang ZN, Rong Z, Xu Y . Immunogenicity of induced pluripotent stem cells. Nature 2011; 474: 212–215.
Nestler EJ, Hyman SE . Animal models of neuropsychiatric disorders. Nat Neurosci 2010; 13: 1161–1169.
Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010; 143: 527–539.
Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM . Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 2011; 14: 285–293.
Ryder PV, Faundez V . Schizophrenia: the ‘BLOC’ may be in the endosomes. Sci Signal 2009; 2: pe66.
Henning U, Krieger K, Loeffler S, Rivas F, Orozco G, de Castro MG et al. Increased levels of glucocorticoid receptors and enhanced glucocorticoid receptor auto-regulation after hydrocortisone challenge in B-lymphoblastoids from patients with affective disorders. Psychoneuroendocrinology 2005; 30: 325–332.
D'Aiuto L, Di Maio R, Heath B, Raimondi G, Milosevic J, Watson AM et al. Human induced pluripotent stem cell-derived models to investigate human cytomegalovirus infection in neural cells. PLoS One 2012; 7: e49700.
Basta-Kaim A, Budziszewska B, Jaworska-Feil L, Tetich M, Kubera M, Leskiewicz M et al. Antipsychotic drugs inhibit the human corticotropin-releasing-hormone gene promoter activity in neuro-2A cells-an involvement of protein kinases. Neuropsychopharmacology 2006; 31: 853–865.
Kurosawa S, Hashimoto E, Ukai W, Toki S, Saito S, Saito T . Olanzapine potentiates neuronal survival and neural stem cell differentiation: regulation of endoplasmic reticulum stress response proteins. J Neural Transm 2007; 114: 1121–1128.
Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang DM . Lithium stimulates progenitor proliferation in cultured brain neurons. Neuroscience 2003; 117: 55–61.
Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H et al. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J Neurochem 2004; 91: 238–251.
Hisaoka K, Maeda N, Tsuchioka M, Takebayashi M . Antidepressants induce acute CREB phosphorylation and CRE-mediated gene expression in glial cells: a possible contribution to GDNF production. Brain Res 2008; 1196: 53–58.
Zhou X, Barrett TB, Kelsoe JR . Promoter variant in the GRK3 gene associated with bipolar disorder alters gene expression. Biol Psychiatry 2008; 64: 104–110.
Huang W, Li MD . Differential allelic expression of dopamine D1 receptor gene (DRD1) is modulated by microRNA miR-504. Biol Psychiatry 2009; 65: 702–705.
Yang S, Van Dongen HP, Wang K, Berrettini W, Bucan M . Assessment of circadian function in fibroblasts of patients with bipolar disorder. Mol Psychiatry 2009; 14: 143–155.
McCarthy MJ, Wei H, Marnoy Z, Darvish R, McPhie D, Cohen B et al. Genetic and clinical factors predict lithium's effects on PER2 gene expression rhythms in cells from bipolar disorder patients. Translational Psychiatry 2013 (in press).
Yoshikawa A, Shimada H, Numazawa K, Sasaki T, Ikeda M, Kawashima M et al. Establishment of human cell lines showing circadian rhythms of bioluminescence. Neurosci Lett 2008; 446: 40–44.
Kowalska E, Moriggi E, Bauer C, Dibner C, Brown SA . The circadian clock starts ticking at a developmentally early stage. J Biol Rhythms 2010; 25: 442–449.
Kiselyuk A, Farber-Katz S, Cohen T, Lee SH, Geron I, Azimi B et al. Phenothiazine neuroleptics signal to the human insulin promoter as revealed by a novel high-throughput screen. J Biomol Screen 2010; 15: 663–670.
Kiris E, Nuss JE, Burnett JC, Kota KP, Koh DC, Wanner LM et al. Embryonic stem cell-derived motoneurons provide a highly sensitive cell culture model for botulinum neurotoxin studies, with implications for high-throughput drug discovery. Stem Cell Res 2011; 6: 195–205.
Lynch WP, Sharpe AH, Snyder EY . Neural stem cells as engraftable packaging lines can mediate gene delivery to microglia: evidence from studying retroviral env-related neurodegeneration. J Virol 1999; 73: 6841–6851.
Lynch WP, Snyder EY, Qualtiere L, Portis JL, Sharpe AH . Late virus replication events in microglia are required for neurovirulent retrovirus-induced spongiform neurodegeneration: evidence from neural progenitor-derived chimeric mouse brains. J Virol 1996; 70: 8896–8907.
Tobe BT, Snyder EY, Nye JS . Modeling complex neuropsychiatric disorders with human induced pluripotent stem cells. Curr Opin Pharmacol 2011; 11: 521–527.
Ebert AD, Yu J, Rose FF Jr., Mattis VB, Lorson CL, Thomson JA et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009; 457: 277–280.
Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 2008; 26: 1269–1275.
Lyssiotis CA, Foreman RK, Staerk J, Garcia M, Mathur D, Markoulaki S et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA 2009; 106: 8912–8917.
Wang Q, Xu X, Li J, Liu J, Gu H, Zhang R et al. Lithium, an anti-psychotic drug, greatly enhances the generation of induced pluripotent stem cells. Cell Res 2011; 21: 1424–1435.
Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol 2009; 27: 353–360.
Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 2007; 1: 55–70.
Yamanaka S . Elite and stochastic models for induced pluripotent stem cell generation. Nature 2009; 460: 49–52.
Plath K, Lowry WE . Progress in understanding reprogramming to the induced pluripotent state. Nat Rev Genet 2011; 12: 253–265.
Renthal W, Nestler EJ . Histone acetylation in drug addiction. Semin Cell Dev Biol 2009; 20: 387–394.
Covington HE III, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci 2009; 29: 11451–11460.
Pidsley R, Mill J . Epigenetic studies of psychosis: current findings, methodological approaches, and implications for postmortem research. Biol Psychiatry 2011; 69: 146–156.
Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R . Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol 2006; 13: 563–567.
Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M et al. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 2007; 46: 8058–8065.
Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem 2009; 284: 17775–17782.
Li W, Zhou H, Abujarour R, Zhu S, Young Joo J, Lin T et al. Generation of human-induced pluripotent stem cells in the absence of exogenous Sox2. Stem Cells 2009; 27: 2992–3000.
Perisic T, Zimmermann N, Kirmeier T, Asmus M, Tuorto F, Uhr M et al. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology 2010; 35: 792–805.
Yu IT, Park JY, Kim SH, Lee JS, Kim YS, Son H . Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 2009; 56: 473–480.
Hester ME, Song S, Miranda CJ, Eagle A, Schwartz PH, Kaspar BK . Two factor reprogramming of human neural stem cells into pluripotency. PLoS One 2009; 4: e7044.
Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 2011; 8: 409–412.
Acknowledgements
BT funded by CIRM fellowship grant. Other work was funded by UCSD psychiatry department T32, RC2 MH090011. Special thanks to Jeff Sanders for thoughtful discussions and review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tobe, B., Brandel, M., Nye, J. et al. Implications and limitations of cellular reprogramming for psychiatric drug development. Exp Mol Med 45, e59 (2013). https://doi.org/10.1038/emm.2013.124
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.124
Keywords
This article is cited by
-
The genetics of bipolar disorder
Molecular Psychiatry (2020)
