
Abstract
Steroid sulfatase (STS) is responsible for the hydrolysis of aryl and alkyl steroid sulfates and has a pivotal role in regulating the formation of biologically active estrogens. STS may be considered a new promising drug target for treating estrogen-mediated carcinogenesis. However, the molecular mechanism of STS expression is not well-known. To investigate whether tumor necrosis factor (TNF)-α is able to regulate gene transcription of STS, we studied the effect of TNF-α on STS expression in PC-3 human prostate cancer cells. RT-PCR and Western blot analysis showed that TNF-α significantly induced the expression of STS mRNA and protein in a concentration- and time-dependent manner. Treatment with TNF-α resulted in a strong increase in the phosphorylation of Akt on Ser-473 and when cells were treated with phosphatidylinositol (PI) 3-kinase inhibitors such as LY294002 or wortmannin, or Akt inhibitor (Akt inhibitor IV), induction of STS mRNA expression by TNF-α was significantly prevented. Moreover, activation of Akt1 by expressing the constitutively active form of Akt1 increased STS expression whereas dominant-negative Akt suppressed TNF-α-mediated STS induction. We also found that TNF-α is able to increase STS mRNA expression in other human cancer cells such as LNCaP, MDA-MB-231, and MCF-7 as well as PC-3 cells. Taken together, our results strongly suggest that PI 3-kinase/Akt activation mediates induction of human STS gene expression by TNF-α in human cancer cells.
Similar content being viewed by others


Introduction
Steroid sulfatase (STS) is the enzyme responsible for the hydrolysis of steroid sulfates such as estrone sulfate (E1S) and dehydroepiandrosterone sulfate (DHEAS) to their unconjugated, biologically active forms (Reed et al., 2005). Plasma concentrations of E1S or DHEAS are 10-20 times higher than those of the unconjugated forms and the half-life of steroid sulfates (10-12 h) is also much longer than of unconjugated steroids (30-40 min) (Ruder et al., 1972). E1S is considered an important source for the formation of active estrogens which is able to be produced by STS action. The gene for human STS is located on the distal short arm of the X-chromosome and maps to Xp22.3-Xpter. The gene consists of 10 exons and spans 146 kb, with the intron sizes ranging from 102 bp up to 35 kb (Reed et al., 2005). STS is mainly found in steroidogenic and steroid-responsive tissues such as placenta, prostate, testis, ovary, breast, and adrenal gland although tissue distribution of STS is believed to be ubiquitous in small quantities. The functional importance of STS is underlined by X-linked ichthyosis, one of the most prevalent human skin genetic diseases, which is associated with the germ line mutation of STS (Stein et al., 1989; Hernández-Martín et al., 1999).
Metabolic activation of E1S has been suggested to be a major factor in mammary carcinogenesis. STS is responsible for the formation of active estrogens such as estrone (E1) or 17β-estradiol (E2) that can stimulate tumor growth (Reed et al., 2005). Moreover, STS expression is increased in malignant breast tumors and has prognostic importance (Dao et al., 1974; Pasqualini et al., 1986; Naitoh et al., 1989; Utsumi et al., 2000). STS activity is approximately 50 times greater in both premenopausal and postmenopausal breast tumors compared to normal breast tissues (Pasqualini et al., 1996). Because of the postulated significant role of STS on carcinogenicity of estrogens, STS is regarded as a target enzyme for blocking estrogen-mediated carcinogenesis and potent STS inhibitors have been developed and tested in rodents and in postmenopausal women with breast cancer (Purohit et al., 1995; Malini et al., 2000; Stanway et al., 2006; Foster et al., 2008). Although STS expression is mainly considered significant in breast and endometrial cancers, results of another study also suggest the significance of STS expression for estrogen production in human prostate cancer (Selcer et al., 2002).
Although expression and activity of STS is increased in malignant tumors, little is known about the regulation of STS expression. Previous reports indicated that tumor necrosis factor-alpha (TNF-α) increases STS activity in breast cancer cells (Purohit et al., 1996; Newman et al., 2000).
In these studies, we explored the effect of TNF-α on STS expression in PC-3 human prostate cancer cells to elucidate whether TNF-α is able to regulate transcription of the STS gene. As TNF-α induces cell survival signals through activation of the PI 3-kinase and its downstream target Akt (Khwaja, 1999; Ozes et al., 1999; Zhou et al., 2000a), evidence for the involvement of PI 3-kinase/Akt signal pathway in STS expression by TNF-α has also been determined.
Results
To determine whether TNF-α regulates STS gene expression in PC3 cells, the mRNA and protein expression of STS were measured by RT-PCR and Western blot, respectively. PC-3 cells were grown for 18 h in the presence of increasing concentrations of TNF-α. TNF-α increased STS mRNA expression in a concentration-dependent manner (Figure 1A). In agreement with increasing mRNA levels, levels of STS protein were also significantly induced by TNF-α (Figure 1A). We compared the time courses for the induction of STS mRNA and protein expression in TNF-α-treated cells (Figure 1B). The induction of STS mRNA was shown after 3 h of treatment with TNF-α (40 ng/ml) and the expression of STS mRNA had increased - 5 fold and - 9 fold by 24 h and 48 h after addition of TNF-α (Figure 1B).

TNF-α induces STS mRNA and protein expression in PC-3 cells. (A) PC-3 cells were treated with various concentrations of TNF-α (0, 10, 20 or 40 ng/ml) for 18 h. Total RNA was isolated and expression of STS mRNA was assessed using RT-PCR. Expression of GAPDH mRNA was determined as RNA control. For Western blot, total cellular lysates were prepared and STS protein expression was assessed by STS antibody. GAPDH was used as a loading control. (B) Cells were incubated at 37℃ with TNF-α (40 ng/ml) for various time intervals. Expression of STS mRNA was assessed using RT-PCR and real-time PCR analysis. The data shown represent the fold increase in the levels of STS mRNA relative to the control after normalization with GAPDH mRNA level. *P < 0.05 compared with the control cells. The protein levels of STS were determined by Western blot analysis.
To explore the molecular basis for TNF-α-induced STS expression, we tested various inhibitors for signal transduction pathways such as LY294002 (PI 3-kinase inhibitor), MG132 (proteasome inhibitor and NF-κB blocker), SB203580 (p38 MAPK inhibitor), or PD168393 (EGFR tyrosine kinase inhibitor). Cells were pretreated with LY294002 (20 µM), MG132 (1 µM), SB203580 (1 µM) or PD168393 (2 µM) or DMSO vehicle for 30 min and then stimulated with 40 ng/ml TNF-α for 18 h. Expression of STS mRNA was assessed using RT-PCR. Figure 2A showed that TNF-α increased STS mRNA and that this induction was almost completely prevented following treatment with LY294002 or MG132. However, neither SB203580 nor PD168393 attenuated TNF-α-induced STS expression. In addition, STS protein expression induced by TNF-α was significantly suppressed by LY294002 pretreatment (Figure 2B). Expression of STS was also investigated by immunofluorescence staining with an antibody against human steroid sulfatase. As shown in Figure 2C, TNF-α strongly stimulated STS expression in PC-3 cells determined by immunofluorescence analysis and pretreatment with LY294002 for 30 min almost completely blocked TNF-α-induced STS expression (Figure 2C). These data indicates that the PI 3-kinase/Akt pathway may be involved in STS gene expression by TNF-α.

Effects of specific inhibitors on TNF-α induced STS mRNA expression in PC-3 cells. (A) Cells were pretreated with 20 µM LY294002 (LY), 1 µM MG132 (MG), 1 µM SB203580 (SB), or 2 µM PD168393 (PD) for 30 min before incubation with 40 ng/ml of TNF-α for 18 h. Total RNA was isolated and expression of STS mRNA was assessed using RT-PCR. Expression of GAPDH mRNA was determined as RNA control. The intensities of the PCR products were quantified using a densitometer. The bar graph was obtained by calculating the percentages of the intensity of each PCR products of STS gene relative to GAPDH controls, defined as 100%. The experiments were carried out in triplicates. *P < 0.05 compared with the TNF-α only-treated cells. (B) Cells were pretreated with 20 µM LY294002 for 30 min and then incubated with 40 ng/ml of TNF-α for 18 h. Total cellular lysates were prepared and STS protein expression was determined by Western blot analysis. GAPDH was used as a loading control. (C) Cells were preincubated with 20 µM LY294002 for 30 min before incubation with TNF-α (40 ng/ml) for 18 h. Cells were fixed and incubated with STS antibody. Cells were then stained with Alexa Fluor 594-labeled secondary antibody and fluorescence was determined by fluorescence microscopy.
To study whether TNF-α induces STS expression through a pathway that involves the PI 3-kinase/Akt pathway, the pharmacological inhibitors of PI 3-kinase/Akt were utilized (Figure 3A). All of the PI 3-kinase/Akt inhibitors such as LY294002 (PI 3-kinase inhibitor, 20 µM), wortmannin (PI 3-kinase inhibitor, 10 µM), or Akt inhibitor IV (10 µM) blocked STS mRNA expression by TNF-α. Since TNF-α has been found to activate PI 3-kinase/Akt pathway in various cells (Ozes et al., 1999; Madge and Pober, 2000), we determined the effect of TNF-α on phosphorylation of Akt on Ser-473. As shown in Figure 3B, an increase in Akt phosphorylation was observed at 18 h following TNF-α treatment (40 ng/ml) in association with increased STS level. To determine if activation of the PI 3-kinase/Akt pathway by TNF-α is required for STS induction, we carried out experiments with a dominant negative plasmid against Akt. When cells were transfected with the plasmid for dominant negative Akt (DN-Akt), phosphorylation of Akt on Ser-473 was almost completely blocked and TNF-α-induced STS expression also decreased to the control level (Figure 3B). To confirm that Akt phosphorylation is required for STS gene expression, the plasmid for a constitutively active form of Akt1 (Myr-Akt1) was transiently transfected into PC-3 cells and the status of Akt phosphorylation (on Ser-473) and STS expression were examined after 48 h (Figure 3C). We found that enhanced Akt phosphorylation on Ser-473 by myristoylated Akt1 transfection increased STS expression.

Activation of Akt causes TNF-α-induced STS gene expression in PC-3 cells. (A) Effects of PI 3-kinase/Akt inhibitors. PC-3 cells were pretreated for 30 min with 20 µM LY294002 (LY), 10 µM Akt inhibitor IV (AktIV), or 10 µM wortmannin (Wort) and subsequently incubated with 40 ng/ml of TNF-α for 18 h. Total RNA was isolated and expression of STS mRNA was assessed using RT-PCR. Expression of GAPDH mRNA was determined as RNA control. (B) Effect of dominant-negative Akt expression. Cells were transfected with DN-Akt plasmid and incubated with 40 ng/ml of TNF-α for 18 h. The expression of phospho-Akt, total Akt, and STS were determined by Western blot analysis. Expression of GAPDH was determined as a loading control. (C) Effect of constitutively active Akt1 expression. Cells were transfected with Myr-Akt1 plasmid and total cellular lysates were prepared and STS protein expression was determined by Western blot analysis.
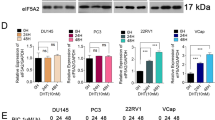
To determine whether induction of STS mRNA expression by TNF-α is limited only to PC-3 prostate cancer cells, we used three additional cancer cell lines. In addition to PC-3 (androgen-independent prostate cancer cell), LNCaP (androgen-dependent prostate cancer cell), MDA-MB-231 (estrogen-independent breast cancer cell), or MCF-7 cells (estrogen-dependent breast cancer cell) were incubated with TNF-α (40 ng/ml) for 18 h. In agreement with results for the PC-3 cells, the three different cancer cell lines also showed induction of STS mRNA by TNF-α (Figure 4).

Induction of STS mRNA expression by TNF-α in various cancer cell lines. Cells were incubated at 37℃ with TNF-α (40 ng/ml) for 18 h. Total RNA was isolated and expression of STS mRNA was assessed using RT-PCR. Expression of GAPDH mRNA was determined as RNA control.
Discussion
Because steroids such as estrogens play a key role in the development of hormone-dependent cancers, understanding biosynthetic mechanisms and related enzymes is important. Because E1S is considered a reserved form of estrogens in cells, the interest in the STS that initiates the generation of E1 from E1S rose to identify a target enzyme for blocking hormone-related tumor formation.
Although the roles of STS on E2-related carcinogenesis have been studied mainly in breast cancer cells, STS may also control the formation of estrogens from E1S in prostate cancer cells. STS has been found in 85% of malignant prostate cancer tissue but absent in the normal tissues (Nakamura et al., 2006).
Previous studies have reported that inflammatory cytokines such as TNF-α, interleukin-6 (IL-6), and interleukin-1β (IL-1β) exist at high concentrations in mammary tumor cells and stimulate STS activity (Purohit et al., 1996; Newman et al., 2000). In MCF-7 cells, TNF-α (7.5 ng/ml) increased the activity of steroid sulfatase by 116% compared with untreated cells (Purohit et al., 1996). Moreover, STS mRNA expression in breast tumor-derived fibroblasts was approximately 2.4-fold higher than in fibroblasts from proximal tissues (Newman et al., 2000). Although TNF-α increases the activity of STS in MCF-7 cells, molecular mechanisms of STS expression by TNF-α are not completely understood yet.
In this study, we demonstrated whether human STS gene expression might be controlled by TNF-α. Our data clearly show that TNF-α is able to induce STS mRNA expression in PC-3 cells in a concentration and time-dependent manner. A previous report indicated that the ability of TNF-α to increase STS activity in MCF-7 cells might result from a post-transcriptional effect (Newman et al., 2000). Thus, we suggest that transcriptional control is also important for STS expression as well as post-transcriptional regulation.
We observed that the PI 3-kinase/Akt pathway positively regulated TNF-α-induced STS gene expression because PI 3-kinase inhibitors (LY294002 and wortmannin), or Akt inhibitor (Akt inhibitor IV) suppressed STS mRNA expression by TNF-α. The importance of Akt in TNF-α-induced STS expression is confirmed by the observation that downregulation of Akt by dominant-negative Akt effectively suppressed STS induction by TNF-α and that overexpression of myristoylated Akt1 increased STS gene expression. These results suggest that Akt activation is essential for TNF-α-mediated STS expression.
Akt has been identified as a key regulator of the survival signaling pathways mediated by growth factors and cytokines, which exerts antiapoptotic effects (Kennedy et al., 1997; Zhou et al., 2000b; Lawlor and Alessi, 2001). Several studies have implicated that Akt is overexpressed in various tumor types including prostate and breast cancers (Nakatani et al., 1999). Furthermore, TNF-α has been found to stimulate the phosphorylation of Akt and this phosphorylation is dependent on PI 3-kinase activation (Madge and Pober, 2000; Radeff-Huang et al., 2007). Therefore, it is likely that the ability of TNF-α to induce STS expression is mediated by PI 3-kinase/Akt activation.
Akt inhibitor IV is a cell-permeable benzimidazole compound that inhibits Akt phosphorylation by targeting the ATP binding site of a kinase upstream of Akt but it does not affect PI 3-kinase activity. Akt inhibitor IV is known to strongly inhibit phosphorylation of Ser-473 for Akt1. Data from our study with Akt inhibitor IV indicate that the phosphorylation of Akt on Ser-473 may be necessary for the TNF-α effect. Moreover, decreased phosphorylation of Akt on Ser-473 by dominant-negative Akt prevents TNF-α to induce STS expression and enhanced phosphorylation of Ser-473 by the activated form of Akt stimulates STS expression.
The mammalian target of rapamycin (mTOR) is a member of the phosphatidylinositol kinase-related kinase family that assembles into two functionally distinct complexes such as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Feldman et al., 2005). Akt stimulates mTORC1 to directly phosphorylate the ribosomal S6 kinases and the eIF4E-binding proteins. mTORC2 is comprised of mTOR, Rictor, mSIN1 and mLST8 and the best-characterized function of mTORC2 is in the phosphorylation of Akt on Ser-473 (Sarbassov et al., 2005; Wullschleger et al., 2006). Because our data strongly suggest that Akt signaling may actively participate in STS expression, the possibility that the involvement of mTOR as a downstream effector of Akt in the mechanism of TNF-α-induced STS expression needs to be elucidated.
In this article, we showed that the proteasome inhibitor MG-132 (1 µM) significantly suppressed TNF-α-induced STS mRNA (Figure 2A). Because MG-132 is able to inhibit NF-κB activity by preventing the proteasomal degradation of IκBα, NF-κB signaling pathway may also play an important role in STS gene expression by TNF-α. Previous report indicated that 1α,25-dihydrovitamin D3 stimulates steroid sulfatase activity through NF-κB pathway in myeloid leukemic cells such as HL-60 and THP-1 (Hughes et al., 2008). Because NF-κB signaling pathway may control STS gene expression by TNF-α as well as 1α,25-dihydrovitamin D3, it will be necessary to determine what mechanism is involved in this event. Recently, we found that insulin-like growth factor II (IGF-II), which is known to activate NF-κB pathway, is able to induce STS gene expression in PC-3 cells and the proteasome inhibitors such as MG-132 and bortezomib completely prevented IGF-II-mediated STS mRNA expression (data not shown).
The ability of TNF-α to induce STS expression is not limited to PC-3 cells, because we found that TNF-α also increases STS mRNA expression in LNCaP, MDA-MB-231, and MCF-7 cells. There are three mammalian isoforms of Akt, which are Akt1, Akt2, and Akt3. Nakatani et al. (1999) demonstrate that Akt1 and Akt3 were present in PC-3 cells, whereas little Akt3 was found in LNCaP cells. Similarly, Akt1 and Akt3 were present in MDA-MB-231 cells, while only Akt1 was detected in MCF-7 cells by Western blotting. Our results that TNF-α is able to induce STS expression in all these cancer cell lines suggest that Akt1 is likely to mediate TNF-α-induced STS expression.
In conclusion, the present findings indicate that TNF-α-mediated induction of human STS occurs by transcriptional activation via a mechanism involving PI 3-kinase/Akt pathway in human prostate cancer cells as well as in human mammary cancer cells. Induction of STS gene expression by inflammatory cytokines such as TNF-α has profound implications on estrogen-mediated carcinogenesis.
Methods
Reagents
Human recombinant TNF-α was purchased from PeproTech (Rocky Hill, NJ). RPMI 1640 medium, penicillin or streptomycin were obtained from Welgene (Daegu, Korea). Fetal bovine serum (FBS) or enhanced chemiluminescence detecting reagent was purchased from Thermo Fisher Scientific (Rockford, IL). LY294002, MG132, PD168393, and Akt inhibitor-IV were obtained from Calbiochem (San Diego, CA). SB203580 was obtained from Tocris (Bristol, UK). Wortmannin was obtained from Enzo Life Science (Plymouth Meeting, PA). Maloney murine leukemia virus (M-MLV) reverse transcriptase or RNase inhibitor (RNasin) was purchased from Promega (Madison, WI). Taq DNA polymerase was obtained from TaKaRa Bio (Shiga, Japan). Goat polyclonal antibodies for STS or HRP-conjugated mouse anti-goat IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa Fluor® 594-conjugated donkey anti-goat IgG was from Invitrogen (Eugene, OR). Other chemicals and reagents were of the highest grade commercially available.
Cell culture
Cell lines were obtained from American Type Culture Collection (ATCC, Manassa, VA) and cultured in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS, 100 unit/ml penicillin, and 100 µg/ml streptomycin. Cells were maintained at 37℃ in a humidified atmosphere of 5% CO2.
RT-PCR and quantitative real-time PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA). Total RNA (500 ng) was transcribed at 37℃ for 1 h in a volume of 20 µl containing 5 × RT buffer, 10 mM dNTPs, 40 units of RNase inhibitor, 200 units of M-MLV reverse transcriptase, and 100 pmole of oligo-dT primer. Subsequently, 0.8 µl of the reaction mixture from each samples was amplified with 10 pmole of each oligonucleotide primers, 0.2 mM dNTPs, 1.5 mM MgCl2 and 1.25 units of Taq DNA polymerase in a final volume of 25 µl. PCR was performed as follows: One cycle of 95℃ for 2 min, followed by 35 cycles of denaturation at 95℃ for 10 s, annealing at 61℃ for 15 s, and extension at 72℃ for 15 s. Human STS cDNA was amplified using a sense primer (5'-CCTCCTACTGTTCTTT CTGTGGG-3') and an antisense primer (5'-GGTCGATATT GGGAGTCCTGATA-3'). Human GAPDH was amplified using a sense primer (5'-TGAACGGGAAGCTCACTGG-3') and an antisense primer (5'-TCCACCACCCTGTTGCTGT A-3'). The number of amplification cycles was optimized in preliminary experiments to ensure that the PCR did not reach a plateau. PCR products were subjected to a 2% (w/v) agarose gel electrophoresis, and analyzed by ChemiDoc XRS (Bio-Rad, Hercules, CA). Real-time PCR was performed using SYBR® Premix Ex Taq (Takara) and the MyiQ2 Two-Color Real-Time PCR detection system (Bio-Rad). The PCR analyses for human STS was done as follows: after an initial denaturation at 95℃ for 10 min, the amplification was done by denaturation at 95℃ for 10 s, annealing at 58℃ for 15 s, and extension at 72℃ for 15 s for 40 cycles.
Western blot
Cells were solubilized with ice-cold lysis buffer (pH 7.4) containing 25 mM HEPES, 1% Triton X-100, 50 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, and 1 µg/ml leupeptin. Extracted proteins (30 µg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% polyacrylamide gels, and were electrophoretically transferred onto polyvinylidene difluoride (PVDF) membrane. Membranes were blocked in 5% (w/v) nonfat dried milk in Tris-buffered saline for 2 h at 4℃. Membranes were then incubated overnight with anti-STS polyclonal antibodies as a 1:500 dilution in 5% (w/v) non-fat dried milk in Tris-buffered saline containing 0.1% Tween-20. The membrane was incubated with a horseradish peroxidase (HRP)-conjugated goat anti-goat IgG antibody. Proteins were visualized by an enhanced chemiluminescence method and the band intensity was analyzed by ChemiDoc XRS densitometer and quantified by Quantity One software (Bio-Rad). Protein concentrations were estimated using the bichinchoninic acid method according to the supplier's recommendation (Thermo) using bovine serum albumin as a standard.
Transient transfection
For transient transfection, cells were transfected with WelFect-EX transfection reagent (Welgene) using pECE myr-Akt-HA or kinase-deficient dominant-negative pCMV6-Akt-K179M mutant plasmids as recommended by the manufacturer. Briefly, one day prior to transfection, approximately 5 × 105 cells per 60 mm-plate were seeded in RPMI 1640 medium containing 10% FBS. Cells were washed in PBS and then resuspended in Opti-MEM serum free medium. Transfection was carried out with 3 µg of plasmid DNA and 12.5 µl of transfection reagent for 5 h at 37℃. After transfection, cells were maintained in RPMI medium containing 10% FBS for 36 h.
Immunofluorescence staining
Cells grown on coverslips were treated for 18 h with TNF-α in RPMI 1640 medium with 10% FBS, rapidly washed with PBS, then fixed with 3.7% (w/v) paraformaldehyde in phosphate buffer containing 0.1 M sodium phosphate, pH 7.4, for 30 min at room temperature. After washing with PBS, the cells were blocked for 15 min in PBS containing 5% donkey serum and 0.2% Triton X-100, then incubated with anti-STS antibody (1:500) for 1 h, washed extensively, and stained for 1 h with Alexa Fluor 594-labeled donkey anti-goat IgG antibody (1:1000). After further washes, the coverslips were mounted on glass slides using Ultra Cruz Mounting Medium (Santa Cruz). Fluorescence signals were analyzed on a DM 2500 fluorescence microscope (Leica, Wetzlar, Germany).
Data analysis
Statistical analysis was performed by using one-way analysis of variance, followed by Dunnett's pairwise multiple comparison t-test with GraphPad Prism software (GraphPad Software Inc., San Diego, CA) when appropriate. The difference was considered statistically significant at P < 0.05.
Abbreviations
- PI:
-
phosphatidylinositol
- STS:
-
steroid sulfatase
- TNF:
-
tumor necrosis factor
References
Dao TL, Hayes C, Libby PR . Steroid sulfatase activities in human breast tumors . Proc Soc Exp Biol Med 1974 ; 146 : 381 - 384
Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D, Biroc SL, Alicke B, Bryant J, Yuan S, Buckman BO, Lentz D, Ferrer M, Whitlow M, Adler M, Finster S, Chang Z, Arnaiz DO . Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1 . J Biol Chem 2005 ; 280 : 19867 - 19874
Foster PA, Woo LW, Potter BV, Reed MJ, Purohit A . The use of steroid sulfatase inhibitors as a novel therapeutic strategy against hormone-dependent endometrial cancer . Endocrinology 2008 ; 149 : 4035 - 4042
Hernández-Martín A, Gonzalez-Sarmiento R, De Unamuno P . X-linked ichthyosis: an update . Br J Dermatol 1999 ; 141 : 617 - 627
Hughes PJ, Lee JS, Reiner NE, Brown G . The vitamin D receptor-mediated activation of phosphatidylinositol 3-kinase (PI3Kα) plays a role in the 1α,25-dihydroxyvitamin D3-stimulated increase in steroid sulphatase activity in myeloid leukaemic cell lines . J Cell Biochem 2008 ; 103 : 1551 - 1572
Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N . The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal . Genes Dev 1997 ; 11 : 701 - 713
Khwaja A . Akt is more than just a Bad kinase . Nature 1999 ; 401 : 33 - 34
Lawlor MA, Alessi DR . PKB/Akt: a key mediator of cell proliferation, survival and insulin responses ? J Cell Sci 2001 ; 114 : 2903 - 2910
Madge LA, Pober JS . A phosphatidylinositol 3-kinase/Akt pathway, activated by tumor necrosis factor or interleukin-1, inhibits apoptosis but does not activate NFkappaB in human endothelial cells . J Biol Chem 2000 ; 275 : 15458 - 15465
Malini B, Purohit A, Ganeshapillai D, Woo LW, Potter BV, Reed MJ . Inhibition of steroid sulphatase activity by tricyclic coumarin sulphamates . J Steroid Biochem Mol Biol 2000 ; 75 : 253 - 258
Naitoh K, Honjo H, Yamamoto T, Urabe M, Ogino Y, Yasumura T, Nambara T . Estrone sulfate and sulfatase activity in human breast cancer and endometrial cancer . J Steroid Biochem 1989 ; 33 : 1049 - 1054
Nakamura Y, Suzuki T, Fukuda T, Ito A, Endo M, Moriya T, Arai Y, Sasano H . Steroid sulfatase and estrogen sulfotransferase in human prostate cancer . Prostate 2006 ; 66 : 1005 - 1012
Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W, Weigel RJ, Roth RA . Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines . J Biol Chem 1999 ; 274 : 21528 - 21532
Newman SP, Purohit A, Ghilchik MW, Potter BV, Reed MJ . Regulation of steroid sulphatase expression and activity in breast cancer . J Steroid Biochem Mol Biol 2000 ; 75 : 259 - 264
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB . NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase . Nature 1999 ; 401 : 82 - 85
Pasqualini JR, Gelly C, Lecerf F . Estrogen sulfates: biological and ultrastructural responses and metabolism in MCF-7 human breast cancer cells . Breast Cancer Res Treat 1986 ; 8 : 233 - 240
Pasqualini JR, Chetrite G, Blacker C, Feinstein MC, Delalonde L, Talbi M, Maloche C . Concentrations of estrone, estradiol, and estrone sulfate and evaluation of sulfatase and aromatase activities in pre- and postmenopausal breast cancer patients . J Clin Endocrinol Metab 1996 ; 81 : 1460 - 1464
Purohit A, Williams GJ, Roberts CJ, Potter BV, Reed MJ . In vivo inhibition of oestrone sulphatase and dehydroepiandrosterone sulphatase by oestrone-3-O-sulphamate . Int J Cancer 1995 ; 63 : 106 - 111
Purohit A, Reed MJ, Morris NC, Williams GJ, Potter BV . Regulation and inhibition of steroid sulfatase activity in breast cancer . Ann NY Acad Sci 1996 ; 784 : 40 - 49
Radeff-Huang J, Seasholtz TM, Chang JW, Smith JM, Walsh CT, Brown JH . Tumor necrosis factor-alpha-stimulated cell proliferation is mediated through sphingosine kinase-dependent Akt activation and cyclin D expression . J Biol Chem 2007 ; 282 : 863 - 870
Reed MJ, Purohit A, Woo LW, Newman SP, Potter BV . Steroid sulfatase: molecular biology, regulation, and inhibition . Endocr Rev 2005 ; 26 : 171 - 202
Ruder HJ, Loriaux L, Lipsett MB . Estrone sulfate: Production rate and metabolism in man . J Clin Invest 1972 ; 51 : 1020 - 1033
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM . Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex . Science 2005 ; 307 : 1098 - 1101
Selcer KW, Kabler H, Sarap J, Xiao Z, Li PK . Inhibition of steryl sulfatase activity in LNCaP human prostate cancer cells . Steroids 2002 ; 67 : 821 - 826
Stanway SJ, Purohit A, Woo LW, Sufi S, Vigushin D, Ward R, Wilson RH, Stanczyk FZ, Dobbs N, Kulinskaya E, Elliot M, Potter BV, Reed MJ, Coombes RC . Phase I study of STX 64 (667 Coumate) in breast cancer patients: the first study of a steroid sulfatase inhibitor . Clin Cancer Res 2006 ; 12 : 1585 - 1592
Stein C, Hille A, Seidel J, Rijnbout S, Waheed A, Schmidt B, Geuze H, von Figura K . Cloning and expression of human steroid-sulfatase. Membrane topology, glycosylation, and subcellular distribution in BHK-21 cells . J Biol Chem 1989 ; 264 : 13865 - 13872
Utsumi T, Yoshimura N, Takeuchi S, Maruta M, Maeda K, Harada N . Elevated steroid sulfatase expression in breast cancers . J Steroid Biochem Mol Biol 2000 ; 73 : 141 - 145
Wullschleger S, Loewith R, Hall MN . TOR signaling in growth and metabolism . Cell 2006 ; 124 : 471 - 484
Zhou BP, Hu MC, Miller SA, Yu Z, Xia W, Lin SY, Hung MC . HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-κB pathway . J Biol Chem 2000a ; 275 : 8027 - 8031
Zhou H, Li XM, Meinkoth J, Pittman RN . Akt regulates cell survival and apoptosis at a postmitochondrial level . J Cell Biol 2000b ; 151 : 483 - 494
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology of Republic of Korea (No. 2010-0008189).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Suh, BY., Jung, JJ., Park, N. et al. Induction of steroid sulfatase expression by tumor necrosis factor-α through phosphatidylinositol 3-kinase/Akt signaling pathway in PC-3 human prostate cancer cells. Exp Mol Med 43, 646–652 (2011). https://doi.org/10.3858/emm.2011.43.11.073
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2011.43.11.073
