
Abstract
Cytochrome P450 3A4 (CYP3A4), is the dominant human liver hemoprotein enzyme localized in the endoplasmic reticulum (ER), and is responsible for the metabolism of more than 50% of clinically relevant drugs. While we were studying CYP3A4 expression and activity in human liver, we found that anti-CYP3A4 antibody cross-reacted with a lower band in liver cytoplasmic fraction. We assessed the activities of CYP3A4 and its truncated form in the microsomal and cytoplasmic fraction, respectively. In the cytoplasmic fraction, truncated CYP3A4 showed catalytic activity when reconstituted with NADPH-cytochrome P-450 reductase and cytochrome b5. In order to determine which site was deleted in the truncated form in vitro, we transfected cells with N-terminal tagged or C-terminal tagged human CYP3A4 cDNA. The truncated CYP3A4 is the N-terminal deleted form and was present in the soluble cytoplasmic fraction. Our result shows, for the first time, that N-terminal truncated, catalytically active CYP3A4 is present principally in the cytoplasm of human liver cells.
Similar content being viewed by others


Introduction
Microsomal cytochrome P450 (CYP) is an integral membrane protein which is localized within the endoplasmic reticulum (ER) (Alston et al., 1991). The various isozymes of CYP operate in conjunction with cytochrome b5 (b5) (Enoch and Strittmatter, 1979) and the FMN/FAD-containing flavoprotein, NADPHcytochrome P-450 reductase (CPR) (Black and Coon, 1982), in the so-called "microsomal electron transport chain" or "P-450 monooxygenase system" (White and Coon, 1980). They are present in liver microsomes and perform a critical function in the metabolism of drugs and other xenobiotics. The hydrophobic amino-terminal region of 20-25 amino acids of these proteins functions as an SRP-dependent insertion signal, and also operates as a stop-transfer signal which anchors the protein to the ER membrane (Sakaguchi et al., 1984). Certain changes in the amino-terminus and/or the adjacent amino acid sequence could be expected to influence the retention of CYP in the microsome, or the correct orientation in the membrane. Furthermore, the membrane-binding domain of the CYP enzymes determines the affinity between CYP enzymes and NADPH-CYP reductase. An excess of NADPH-CYP reductase over CYP in membrane preparations has been shown to enhance the activity of CYP3A4 (Yamazaki et al., 1999). In a similar regard, certain changes in the amino-terminus and/or adjacent amino acid sequence may influence the activities of corresponding CYP enzymes. In fact, the deletion of the proline-proline-glycine-proline sequence, which immediately follows the amino-terminal hydrophobic membrane insertion signal, reduced the stability of the protein, and abolished the enzymatic activity of CYP2C2 (Szczesna-Skorupa et al., 1993). However, no similar studies of CYP3A4 have yet been conducted.
The cytochrome P450 3A4 (CYP3A4) enzyme catalyzes the oxidative metabolism of at least 50% of clinically relevant drugs (Guengerich, 1999; Rendic and Di Carlo, 1997). CYP3A4 is abundant in the liver (the predominant biotransformation site), and exists as an intracellular hemoprotein, similarly to the other CYP enzymes. Western blot analysis, as well as enzymatic activity assays of CYP3A4 has generally been conducted with the microsomal fraction, with a clear band on Western blot analysis (Paine et al., 1999). The different bands were not recognized until Western blot analysis was conducted with the total cell extracts (Harvey et al., 2000). In the total cell proteins, two or more bands were allowed to react with anti-CYP3A or CYP3A4/CYP3A7 antibody, and one of them was identical to the immunoreactive band in the liver microsome. However, other immunoreactive bands detected by the same antibody have yet to be identified (Harvey et al., 2000). In our study of CYP3A4 expression and activity in the human liver, we determined that the anti-CYP3A4 antibody cross-reacted with a lower band than that of intact CYP3A4 in the liver cytoplasmic fraction. Thus, in this study, we compared the activities of CYP3A4 and its truncated form in the microsomal and cytoplasmic fractions, respectively. Moreover, we verified that the N-terminal deleted form of CYP3A4 was present in the soluble cytoplasmic fraction.
Materials and Methods
Subjects
Liver samples were acquired from four patients who had undergone partial (segmental or lobal) hepatectomy. All subjects provided written informed consent before participating in the present study. Patient 1 (P1) suffered from two intrahepatic stones, the other three (P2-4) from a hepatocellular carcinoma. The P1 intrahepatic stone patient had taken nicardipine, a calcium channel blocker, for the control of hypertension prior to surgery. The other patients had been healthy until the detection of hepatocellular carcinoma. The liver samples from the intrahepatic stone patient evinced a normal appearance, although fatty changes were microscopically detected.
Subcellular fractionation
Liver samples were prepared as described elsewhere (Guengerich, 1994). In brief, the liver specimens were homogenized in ice-cold homogenization buffer containing 100 mM Tris-acetate (pH 7.4), 100 mM KCl, 1 mM EDTA and protease inhibitors (0.1 mM PMSF, 5 µg/ml aprotinin, and 10 µg/ml leupeptin) and centrifuged for 10 min at 7,000 × g. The supernatants were centrifuged for 1 h at 100,000 × g in order to separate the cytoplasmic and microsomal fractions. The protein concentration in the supernatant was assessed using a protein assay kit (BCA kit, Pierce, Rockford, IL) using BSA as a standard.
Western blot analysis
Thirty µg of liver protein from whole lysates, microsomal and cytoplasmic fractions were separated on 10% SDS-polyacrylamide gel and transferred to nitrocellulose membranes. Rabbit anti-human CYP3A4 antibody was used in the immunoblotting experiments. The specificity and properties of similar preparations have been reported elsewhere (Distlerath et al., 1985; Kim et al., 2003). The Western blots were incubated with a 1 : 2,000 dilution of primary antibody, followed by a 1 : 2,500 dilution of alkaline phosphatase-conjugated goat anti-rabbit IgG or a 1 : 5,000 dilution of HRP conjugated goat anti-rabbit IgG. Immunoreactive proteins were visualized with an alkaline phosphatase conjugate substrate kit from Sigma-Aldrich Co. (St. Louis, MO) or an enhanced chemiluminescence solution kit (Pierce, Rockford, IL). In order to monitor the quantity of protein loaded into each lane, the membranes were reprobed with mouse monoclonal anti-actin antibody (Sigma, St. Louis, MO) and processed as described above. The protein bands were analyzed via densitometry.
Plasmid constructs
The cDNA encoding for human CYP3A4 was obtained from Prof. J-G Shin (Inje Univ.) (Lee et al., 2007). Human CYP3A4 cDNA was subcloned into the pECFP-C2 vector (Clontech, CA) for N-terminal tagging or pcDNA 3.1/myc-His (Invitogen, Carlsbad, CA) for C-terminal tagging via polymerase chain reaction. All of the subcloned cDNAs were verified via DNA sequencing.
Cell culture and transfection
HEK293 cells were obtained from the American Type Culture Collection (Rockville, MD) and cultured in DMEM containing 10% FBS at 37℃, in an atmosphere of 5% CO2. The transfection of plasmids into HEK 293 cells was conducted with LipofectAMINE 2000 reagent, (Invitrogen, Carlsbad, CA), in accordance with the manufacturer's instructions. The whole cell lysates were acquired for immunoblot analysis with anti-GFP or c-myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA) after 48 h. The transfected cells were divided into cytoplasmic and microsomal fractions using the fractionation method described for the human liver.
CYP3A4 enzyme activity assay
The enzymatic activity of the same microsome and cytoplasmic fractions prepared for the Western blot analysis were assayed. The P450 contents of the liver microsomes were quantified by Fe2+ - CO versus Fe2+ difference spectroscopy in accordance with the general method described by Omura and Sato (1964). The protein concentrations were determined using a Bradford protein assay kit (Bio-Rad, Richmond, CA) in accordance with the manufacturer's instructions. The CYP3A4 activity assay was conducted in 100 mM potassium phosphate buffer (pH 7.4) including phosphatidylcholine (L-α-dilauroylphosphatidylcholine) (40 µM) as described elsewhere (Kim et al., 2003). The reaction volume was 500 µl. Proteins (50 µg) of the cytoplasmic or microsomal fractions were mixed in the presence of testosterone (100 µM) as a substrate. The reaction was initiated via the addition of an NADPHgenerating system (final conc. 10 mM glucose 6-phosphate, 0.5 mM NADPH, and 1 IU yeast glucose 6-phosphate ml-1). After the incubation of the sample at 37℃ for 30 min, the reaction was halted via the addition of 50 µl of 1.0 N HCl containing 2.0 M NaCl. The resultant product was extracted and analyzed via HPLC with UV detection at 240 nm. Rat NADPH-P450 reductase (CPR) (Hanna et al., 1998) and human cytochrome b5 (b5) (Shimada et al., 1986) were expressed in E. coli and purified as described. To determine the activity of truncated CYP3A4 in the cytoplasmic fraction, CPR (100 pmol) and b5 (50 pmol) were added externally to the cytoplasmic fraction, and the activity assay was conducted as described above.
Results
Western blot analysis of microsomal and cytoplasmlic fractions of human liver samples
In the human liver lysates obtained from patient 1 (P1), anti-CYP3A4 antibody cross-reacted with several bands. Among them, the molecular weight of the major band was 50 kDa, which is identical to the size of the standard CYP3A4 protein, and the other was 43 kDa (Figure 1A). In order to assess the subcellular localization of each band, the liver samples were fractionated into cytoplasmic and microsomal fractions by using verified method (Kim et al., 2003). Moreover, in order to prove the purity, another CYP, such as CYP2E1 was detected by specific anti-CYP2E1 antibody. Intact CYP2E1 was only detected in the microsomal fraction not in the cytoplasmic fraction (data not shown). The higher molecular weight bands were detected exclusively in the microsomes, whereas the lower molecular weight bands were detected principally in the cytoplasmic fraction (Figure 1A). In an effort to determine whether there was any differences between the liver disease patients with regard to the quantity of the truncated form, surgically removed human liver samples were obtained from one patient (P1) who suffered from intrahepatic stone, and another three (P2-4) who suffered from hepatocellular carcinoma. As is shown in Figure 1B, although same amount of each fraction was loaded at each well, the intensity of intact CYP3A4 detected in microsomal fraction was different; that of P1 was two-fold higher than that of another three (P2-4). However, there were no differences in the intensity of the truncated CYP3A4 band in the cytoplasmic fraction between each patient and the band size was consistent. These results indicate that the cleavage site of CYP3A4 may be specific in the human liver.

Western blot analysis of CYP3A4 in human liver sample. (A) Human liver tissue from patient 1 (P1) was fractionated into microsome and cytosol. 30 µg of whole lysate (W), cytoplasmic (C) and microsomal (M) proteins were electrophoresed on SDS-PAGE and analyzed via immunoblotting with anti-CYP3A4 antibody. One higher (50 kDa) and one lower (43 kDa) molecular weight band were clearly detected. In the microsomal fraction (M), the higher molecular weight band of the membrane. bound form was exclusively observed, whereas the lower molecular weight band was detected in the cytoplasmic fraction (C). As a loading control, anti-β-actin antibody was used. (B) Liver tissues from P1-4 were fractionated into microsome and cytosol. Each fraction was electrophoresed on SDS-PAGE and analyzed via immunoblotting with anti-CYP3A4 antibody. No differences were detected in the band sizes and intensities in the samples.
Catalytic activity of N-terminal deleted form of CYP3A4 in the human liver cytoplasmic fraction
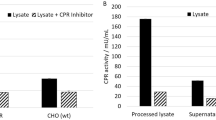
In order to determine whether the truncated form of CYP3A4 in the cytoplasmic fraction has catalytic activity and shows different activity according to liver disease, the liver tissues of patient 3 were separated into two regions; the hepatocellular carcinoma region (L) and the adjacent normal region (N). Each fractionated sample was immunoblotted with anti-CYP3A4 antibody, and the catalytic activities of each fraction were assessed in the presence of testosterone (100 µM), a marker substrate of CYP3A4. We noted no differences in the subcellular localization of the truncated or full-length CYP3A4 between the carcinoma and normal regions (Figure 2A). The average hydroxylation rates of the microsomes from the two liver samples were 1.51 (hepatocellular carcinoma portion) and 1.63 (adjacent tissue) nmol product per min per mg protein (Figure 2B, a and b). As a positive control, the average hydroxylation rate of purified CYP3A4 is 8.1 nmol product/min/ mg protein (Figure 2B, d). In the case of the cytoplasmic fractions, catalytic activity was assessed in the absence or presence of externally added CPR and b5, because there are no redox partners in the cytoplasmic fraction, and b5 has been shown to enhance the rate of CYP3A4 catalysis (Schenkman and Jansson, 1999). In the absence of CPR and b5, we noted no apparent activities in the cytosolic fraction as compared with that of the purified 3A4 (Figure 2B, c). When the redox partners CPR and b5 were added, we were able to detect hydroxylation activity from the HPLC trace; 0.09 (hepatocellular carcinoma portion) and 0.12 (adjacent tissue) nmol product per min per mg protein (Figure 2B, e and f), which suggests that truncated CYP3A4 evidences catalytic activity. Similar to the subcellular localization of the truncated or full-length CYP3A4 between the carcinoma and normal region, the catalytic activity of each liver sample evidenced no differences.

Catalytic activity of the N.terminal deleted form of CYP3A4 in the human liver cytoplasmic fraction. (A) Liver tissues of patient 3 were separated into two parts; one is the hepatocellular carcinoma region (L) and another is the adjacent normal region (N). Each tissue was fractionated into microsome (M) and cytosol (C) and those were immunoblotted with anti-CYP3A4 antibody. (B) Catalytic activities of each fraction were assessed in the presence of testosterone. The activity from the microsome fraction of N (a) or L (b) was initiated via the addition of an NADPH-generating system. In the cytoplasmic fractions, the catalytic activity was evaluated in the absence (c) or presence of externally added CPR and b5 in N (e) and L (f). As a positive control, the catalytic activity of purified CYP3A4 was assessed in the presence of CPR and b5 (d). The results shown are the average of two determinations.
Transient expression of CYP3A4
In order to determine which site is deleted in the truncated form in vitro, we constructed human CYP3A4 cDNA into N-terminal CFP or C-terminal myc tagged vectors. Each clone was overexpressed in HEK293 cells and confirmed via immunoblotting with tagging antibodies (anti-GFP and c-myc antibodies) and anti-CYP3A4 antibody in the same blot. Anti-GFP antibody cross-reacted with CFP protein and the size of each protein detected with the tagging antibody were correlated with that of the CYP3A4 antibody (Figure 3A). Then, the N-terminal CFPtagged or C-terminal myc-tagged CYP3A4 overexpressing cells were fractionated into the cytoplasmic and microsomal fractions. As a positive control, human liver lysate showing two bands was used in each blot. In the microsomal fraction of each of the clone-overexpressing cells, the full-length CYP3A4 was detected predominantly. However, even in the cytoplasmic fraction, full-length CYP3A4 was primarily detected (Figure 3B and C), which may have been the result of misfolding and impaired subcellular localization due to the CYP3A4 protein overproduction in the cells. In fact, it has been established that small monomer proteins fold through a succession of a definite number of intermediate steps (Jaenicke, 1991). Moreover, it has been shown that when production of misfolded proteins exceeds the cellular capacity to degrade them, the proteins accumulate in a novel subcellular structure, the aggresome (Johnston et al., 1998).

Transient expression of CYP3A4. (A) Constructs of human CYP3A4 cDNA tagged with N.terminal CFP or C.terminal myc were transfected into HEK293 cells and confirmed using the tag antibodies (anti-GFP and c-Myc antibodies) and anti.CYP3A4 antibody in the same blot. Mock (1), pcDNA 3.1 His-C CYP 3A4 Myc (2), or pECFP-C2 CYP 3A4 (3) DNA was transfected into cells. (B) C.terminal myc or (C) N-terminal CFP CYP3A4 overexpressing cells were fractionated into cytosol and microsome. Thirty µg of whole lysate (W), cytoplasmic (C) and microsomal (M) proteins were electrophoresed on SDS.PAGE and analyzed via immunoblotting with each antibody. Anti-β-actin antibody was used as a loading control. Data were obtained from triplicate experiments.
The truncated form of CYP3A4 was detected only in the cytoplasmic fraction and whole lysates of the C-terminal myc-tagged CYP3A4 overexpressing cells using anti-myc antibody, and was not detected in the N-terminal CFP tagged CYP3A4 overexpressing cells using anti-GFP antibody (Figure 3B and C, arrow indicated). This finding indicates that the truncated form of CYP3A4 is the N-terminal deleted form.
Discussion
In the current study, we showed that N-terminal deleted CYP3A4 is present predominantly in human liver cytoplasmic fraction, and has catalytic activity. P450s are integral monotopic ER proteins with their relatively hydrophobic N termini embedded in the ER-membrane bilayer and the bulk of their catalytic domain exposed to the cytosol (Sakaguchi et al., 1984). The deletion of this sequence attenuated the stability of the protein and abolished the enzymatic activity of CYP2C2 (Szczesna-Skorupa et al., 1993). It has been previously reported that N-terminal deleted CYP2E1 localizes within the mitochondria from rat livers in a soluble form, and its catalytic activity remains intact when reconstituted with both adrenodoxin (Adx) and adrenodoxin reductase (AdR) (Neve and Ingelman-Sundberg, 1999). In this study, for the first time, we have described the N-terminal deleted form of CYP3A4 in the human liver. Its molecular weight was similar to that of the N-terminal deleted CYP2E1, but the subcellular localization of N-terminal deleted CYP3A4 was not detected in the mitochondria (data not shown). Because CYP3A4 enzymes bind to the microsomal membrane via the amino-terminus, deletion could result in a loss of the membrane-binding portion of CYP3A4, via an unknown cytoplasmic endoprotease. The similar and lower molecular weight bands detected in the microsomal fraction may be caused by protein instability after fractionation.
The truncated CYP3A4 evidenced detectable enzymatic activity in the presence of the externally-added redox partners, CPR and b5 (Figure 2B). As CYP3A4 functions in conjunction with CPR and b5 in the ER, the truncated form evidenced no apparent activity, due to the inaccessibility of its redox partners (Figure 2B). However, its enzymatic activity was reduced markedly as compared with that of intact CYP3A4 in the microsomal fraction. Thus, our results show that if the levels of N-terminal deleted CYP3A4 are increased in the cytosol, the total catalytic activity of CYP3A4 may be reduced in vivo in the human liver. In fact, the deletion of the proline-prolineglycine-proline sequence, which immediately follows the amino-terminal hydrophobic membrane insertion signal, reduced the stability of the protein and abolished the enzymatic activity of CYP2C2 (Szczesna-Skorupa et al., 1993). The hepatic expression and activity of CYP3A4 varies dramatically from individual to individual and also within the individual subject in a time and tissue-dependent manner (Ozdemir et al., 2000), which could affect the predisposition of an individual to cancers caused by environmental procarcinogens that are bioactivated by CYP3A4 (Kirby et al., 1993). The development of cancer in the liver is a long, chronic process. Many steps in this process are attributable to induce hepatitis and cirrhosis. These steps include the generation of hyperplastic nodules. In the adult liver, cell growth can be induced both in compensatory regeneration and direct hyperplasia (Farber and Sarma, 1987; Columbano and Shinozuka, 1996). In fact, Kondoh et al. (1999) reported that CYP3A4 mRNA is highly induced in nontumorous liver and maintained at variable levels in carcinoma tissues from hepatocarcinoma patients but barely detectable in normal liver from non hepatocarcinoma patients. Moreover, genetic alterations of CYPs have been reported in the development and/or progression of a subset of various cancers (Mochizuki et al., 2005; Shin et al., 2007). However, factors regulating CYP3A activity and expression have been remained limited. In this study, we found a new factor regulating the activity or expression level of CYP3A4. Although, there was no difference in the intensity of the truncated CYP3A4 band in the cytoplasmic fraction between the liver disease patients and hepatocellular carcinoma region and adjacent normal region in the same patient, that is correlated with its catalytic activities. These no difference can be explained that in this study, the catalytic activity and expression level of the truncated CYP3A4 were not compared with normal liver tissues. Thus, these results imply that the protein level of truncated CYP3A4 may be an index to enable comparison of its catalytic activity.
Enhancing effects of cytoplasmic protein on microsomal P450 systems have been reported previously (Mori et al., 1984; Komatsu et al., 2000) and may proceed by a variety of mechanisms, including effects on microsomal enzymes, effects on stability of substrate and/or metabolites, and enhanced affinity of substrate to microsomal P450 enzymes. Thus, it remains to be determined what the functional role of truncated CYP3A4 in the human liver cytosol is.
Abbreviations
- b 5 :
-
cytochrome b5
- CYP3A4:
-
cytochrome P450 3A4
- CPR:
-
NADPH-cytochrome P-450 reductase
- ER:
-
endoplasmic reticulum
References
Alston K, Robinson RC, Park SS, Gelboin HV, Friedman FK . Interactions among cytochromes P-450 in the endoplasmic reticulum. Detection of chemically cross-linked complexes with monoclonal antibodies . J Biol Chem 1991 ; 266 : 735 - 739
Black SD, Coon MJ . Structural features of liver microsomal NADPH-cytochrome P-450 reductase. Hydrophobic domain, hydrophilic domain, and connecting region . J Biol Chem 1982 ; 257 : 5929 - 5938
Columbano A, Shinozuka H . Liver regeneration versus direct hyperplasia . FASEB J 1996 ; 10 : 1118 - 1128
Distlerath LM, Reilly PE, Martin MV, Davis GG, Wilkinson GR, Guengerich FP . Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism . J Biol Chem 1985 ; 260 : 9057 - 9067
Enoch HG, Strittmatter P . Cytochrome b5 reduction by NADPH-cytochrome P-450 reductase . J Biol Chem 1979 ; 254 : 8976 - 8981
Farber E, Sarma DSR . Hepatocarcinogenesis: a dynamic cellular perspective . Lab Investig 1987 ; 56 : 4 - 22
Guengerich FP, Hayes AW . Analysis and characterization of enzymes . Principles and Methods of Toxicology 1994 ; : 1259 - 1313
Guengerich FP . Cytochrome P-450 3A4: regulation and role in drug metabolism . Annu Rev Pharmacol Toxicol 1999 ; 39 : 1 - 17
Hanna M, De Biasi V, Bond B, Salter C, Hutt AJ, Camilleri P . Estimation of the partitioning characteristics of drugs: a comparison of a large and diverse drug series utilizing chromatographic and electrophoretic methodology . Anal Chem 1998 ; 70 : 2092 - 2099
Harvey JL, Paine AJ, Maurel P, Wright MC . Effect of the adrenal 11-beta-hydroxylase inhibitor metyrapone on human hepatic cytochrome P-450 expression: induction of cytochrome P-450 3A4 . Drug Metab Dispos 2000 ; 28 : 96 - 101
Jaenicke R . Protein folding: local structures, domains, subunits, and assemblies . Biochemistry 1991 ; 30 : 3147 - 3161
Johnston JA, Ward CL, Kopito RR . Aggresomes: a cellular response to misfolded proteins . J Cell Biol 1998 ; 143 : 1883 - 1898
Kim KH, Ahn T, Yun CH . Membrane properties induced by anionic phospholipids and phosphatidylethanolamine are critical for the membrane binding and catalytic activity of human cytochrome P450 3A4 . Biochemistry 2003 ; 42 : 15377 - 15387
Kirby GM, Wolf CR, Neal GE, Judah DJ, Henderson CJ, Srivatanakul P, Wild CP . In vitro metabolism of aflatoxin B1 by normal and tumorous liver tissue from Thailand . Carcinogenesis 1993 ; 14 : 2613 - 2620
Komatsu T, Yamazaki H, Asahi S, Gillam EM, Guengerich FP, Nakajima M, Yokoi T . Formation of a dihydroxy metabolite of phenytoin in human liver microsomes/cytosol: roles of cytochromes P450 2C9, 2C19, and 3A4 . Drug Metab Dispos 2000 ; 28 : 1361 - 1368
Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, Goseki N, Matsubara O, Takenaka K, Shichita M, Tanaka K, Shuda M, Yamamoto M . Identification and characterization of genes associated with human hepatocellular carcinogenesis . Cancer Res 1999 ; 59 : 4990 - 4996
Lee SJ, Lee SS, Jeong HE, Shon JH, Ryu JY, Sunwoo YE, Liu KH, Kang W, Park YJ, Shin CM, Shin JG . The CYP3A4*18 allele, the most frequent coding variant in asian populations, does not significantly affect the midazolam disposition in heterozygous individuals . Drug Metab Dispos 2007 ; 35 : 2095 - 2101
Mochizuki J, Murakami S, Sanjo A, Takagi I, Akizuki S, Ohnishi A . Genetic polymorphisms of cytochrome P450 in patients with hepatitis C virus-associated hepatocellular carcinoma . J Gastroenterol Hepatol 2005 ; 20 : 1191 - 1197
Mori Y, Niwa T, Yamazaki H, Nii H, Toyoshi K, Hirano K, Sugiura M . Influence of microsomal and cytosolic fractions from the liver of 4 animal species and man on the mutagenicity of carcinogenic aminoazo dyes and nature of the mutagenicity-enhancing factor in the cytosol from rat liver . Chem Pharm Bull 1984 ; 32 : 3641 - 3650
Neve EP, Ingelman Sundberg M . A soluble NH(2)-terminally truncated catalytically active form of rat cytochrome P450 2E1 targeted to liver mitochondria(1) . FEBS Lett 1999 ; 460 : 309 - 314
Omura T, Sato R . The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature . J Biol Chem 1964 ; 239 : 2370 - 2378
Ozdemir V, Kalow W, Tang BK, Paterson AD, Walker SE, Endrenyi L, Kashuba AD . Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method . Pharmacogenetics 2000 ; 10 : 373 - 388
Paine MF, Schmiedlin Ren P, Watkins PB . Cytochrome P-450 1A1 expression in human small bowel: interindividual variation and inhibition by ketoconazole . Drug Metab Dispos 1999 ; 27 : 360 - 364
Rendic S, Di Carlo FJ . Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors . Drug Metab Rev 1997 ; 29 : 413 - 580
Sakaguchi M, Mihara K, Sato R . Signal recognition particle is required for co-translational insertion of cytochrome P-450 into microsomal membranes . Proc Natl Acad Sci USA 1984 ; 81 : 3361 - 3364
Schenkman JB, Jansson I . Interactions between cytochrome P450 and cytochrome b5 . Drug Metab Rev 1999 ; 31 : 351 - 364
Shimada T, Misono KS, Guengerich FP . Human liver microsomal cytochrome P-450 mephenytoin 4-hydroxylase, a prototype of genetic polymorphism in oxidative drug metabolism. Purification and characterization of two similar forms involved in the reaction . J Biol Chem 1986 ; 261 : 909 - 921
Shin A, Kang D, Choi JY, Lee KM, Park SK, Noh DY, Ahn SH, Yoo KY . Cytochrome P450 1A1 (CYP1A1) polymorphisms and breast cancer risk in Korean women . Exp Mol Med 2007 ; 39 : 361 - 366
Szczesna-Skorupa E, Straub P, Kemper B . Deletion of a conserved tetrapeptide, PPGP, in P450 2C2 results in loss of enzymatic activity without a change in its cellular location . Arch Biochem Biophys 1993 ; 304 : 170 - 175
White RE, Coon MJ . Oxygen activation by cytochrome P-450 . Annu Rev Biochem 1980 ; 49 : 315 - 356
Yamazaki H, Nakajima M, Nakamura M, Asahi S, Shimada N, Gillam EM, Guengerich FP, Shimada T, Yokoi T . Enhancement of cytochrome P-450 3A4 catalytic activities by cytochrome b (5) in bacterial membranes . Drug Metab Dispos 1999 ; 27 : 999 - 1004
Acknowledgements
We are highly thankful to Professor Shin J-G (Inje Univ), who kindly gave the human CYP3A4 cDNA construct. This study was supported by a grant from the Korea Health 21 R&D Project. Ministry of Health and Welfare, R. O. K (03-PJ10-PG13-GD01-0002).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jeon, S., Kim, KH., Yun, CH. et al. An NH2-terminal truncated cytochrome P450 CYP3A4 showing catalytic activity is present in the cytoplasm of human liver cells. Exp Mol Med 40, 254–260 (2008). https://doi.org/10.3858/emm.2008.40.2.254
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2008.40.2.254
Keywords
This article is cited by
-
What is the Functional Role of N-terminal Transmembrane Helices in the Metabolism Mediated by Liver Microsomal Cytochrome P450 and its Reductase?
Cell Biochemistry and Biophysics (2012)
