
Abstract
Ataxia is a symptom that is often associated with syndromic inherited diseases. We previously reported the linkage of a novel syndrome, ataxia with blindness and deafness (SCAR3/SCABD, OMIM# 271250), to chromosome 6p21–p23 by linkage mapping of an Arab Israeli consanguineous family. We have now identified by whole-exome sequencing a homozygous missense mutation in the Arab Israeli family in the SLC52A2 gene located in 8qter, therefore excluding linkage of this family to 6p. We confirmed the involvement of SLC52A2 by the identification of a second mutation in an independent family with an identical syndromic presentation, which we suggest to name SCABD2. SCABD2 is therefore allelic to Brown–Vialleto–Van Laere syndrome type 2 defined by prominent motoneuronopathy and deafness, and also caused by SLC52A2 mutations. In the course of this project, we identified a clinically similar family with a homozygous missense mutation in PEX6, which is located in 6p21. Therefore, despite false linkage in the initial family, SCABD1/SCAR3 is located in 6p21 and is caused by PEX6 mutations. Both SLC52A2 and PEX6 should be included in screening panels for the diagnosis of syndromic inherited ataxias, particularly as patients with mutations in SLC52A2 can be ameliorated by riboflavin supplementation.
Similar content being viewed by others


Introduction
Autosomal recessive cerebellar ataxias comprise a clinically and genetically heterogeneous group of neurodegenerative disorders that range from isolated ataxia to syndromic forms associated with neurological and extra-neurological symptoms. Because overall prominent cerebellar and/or spinocerebellar ataxia often occurs when pathways are only ‘mildly’ affected by partial loss of function mutation in genes of recessive diseases,1 ataxia symptoms are frequently found in milder presentation of other disorders and syndromes. For instance, Brown–Vialetto–Van Laere syndrome (BVVLS; MIM 211530), first reported by Brown in 1894,2 is a rare neurological disorder characterized by infancy-onset sensorineural deafness and ponto-bulbar palsy. Two types have been described: BVVLS type 1, caused by mutations in the riboflavin transporter genes SLC52A3 (MIM 613350); and BVVLS type 2 caused by mutations in SLC52A2 (MIM 607882). In the latter, ataxia was presented as one of the potential additional features.3 Peroxisomal biogenesis disorders (PBDs; MIM 601539) are another illustration of autosomal recessive neurological diseases often associated with ataxia. PBDs are indeed characterized by large clinical heterogeneity ranging from severe neurologic dysfunction and death within the first year of life (Zellweger syndrome) to milder disorder, in which patients can survive into young adulthood, but typically exhibit multiple organ dysfunction, sensorineural hearing loss, pigmentary retinal degeneration and psychomotor impairments including ataxia. Very-long-chain fatty acids, phytanic acid, pristanic acid, plasmalogens, pipecolic acid and bile acid levels in blood and urine are good biological markers to show evidence of generalized PBD.4 It has been estimated that over 90% of PBD patients have mutations in the PEX1 (MIM 602136), PEX6 (MIM 601498), PEX10 (MIM 602859), PEX12 (MIM 601758) or PEX26 (MIM 608666) genes.
Nearly hundred genes have been identified causing inherited ataxia. A large part of them has been discovered through the approach of homozygosity mapping. Among these, linkage studies have led to the identification of genes causing many autosomal recessive ataxia such as infantile-onset spinocerebellar ataxia,5 ataxia with isolated vitamin E deficiency,6, 7 ataxia-ocular apraxia 18 and ataxia-ocular apraxia 2,9 for example. However, this approach is not absolutely safe because it is based on statistical methods that identify the chromosomal region with the highest probability to map a disease locus. This is the reason why we reported homozygosity mapping of spinocerebellar ataxia with hearing impairment and optic atrophy, also known as spinocerebellar ataxia autosomal recessive 3 (SCAR3), to chromosome 6p23–p21 (SCABD locus, MIM 271250)10 by linkage studies of a single Arab Israeli family, but we were not able to identify the causal gene in this interval since then. In this study based on whole-exome-sequencing approach, we report that the SCABD gene of the initial family10 is in fact located in 8qter and is allelic to the BVVLS2 gene and we identify PEX6 as the 6p21 SCABD gene in a second independent family. We further report a novel SLC52A2 mutation in a third clinically similar family. Overall, we show here that next-generation sequencing provides valuable differential diagnosis for autosomal recessive syndromic ataxias characterized by marked genetic and phenotypic heterogeneity due to partial loss of function of the mutated genes.
Subjects and methods
Genetic studies
Blood samples were obtained with informed consent. DNA was extracted by standard procedures.
Whole-exome sequencing was performed for one patient per family, except for family B for which no linkage data were available and for which the two patients were sequenced. Whole-exome sequencing was performed by exon capture with the Agilent (Santa Clara, CA, USA) SureSelect kit and high-throughput sequencing with an Illumina (San Diego, CA, USA) HiSeq2500 sequencer (IGBMC sequencing platform). Reads were mapped to the human reference genome (hg19). Supplementary Table 1 shows the summary of mapping and coverage results for each family exome. dbSNP 137 served as a reference to exclude single-nucleotide polymorphisms (SNPs) with a frequency of 1% or above. Additional search in the 1000 Genome project and the exome variant server (NHLBI GO Exome Sequencing Project) databases was performed to assess the frequency of the selected variants.11 Variants were also analyzed for pathogenicity with in-house pipelines, which combine splice site prediction and protein-coding changes.11 Finally, we used the VaRank program for the automatic ranking of genetic variants. This program relies on the annotation provided by the Alamut Batch software developed by Interactive Biosoftware (Rouen, France) to score each variant.12, 13 VaRank scores lower than 50 correspond to indel in-frame, deep intron–exon boundary mutation away from the consensus splicing sites with a splicing score lower than −10%, and synonymous variant. The score is incremented as following depending on the variant’s type and on a bonus score added when the PhastCons score shows a genomic conservation: intron–exon boundary (50–55); missense (50–65); start or stop loss (80–85); essential splice site (90–95); frameshift (100); and nonsense (100–105). A maximal score of 110 is given to variants reported as probable pathogenic or pathogenic in the dbSNP database (rsClinicalSignificance variant).
Patients from family C were analyzed with Affymetrix 50K Xba SNP arrays according to the protocol provided by the manufacturer (Gene Chip Mapping 100 K Assay manual, Affymetrix Inc., Santa Clara, CA, USA). Homozygous regions shared between the patients were identified with the HomoSNP software14 that was set to identify regions of 25 or more consecutive homozygous SNPs across the 22 autosomes. In order to demonstrate identity by descent, as opposed to identity by state, SNPs of the shared homozygous regions were then individually inspected to verify that the same haplotype was shared between the patients and that, apart from recombination events, genotypes were identical beyond the homozygous regions.
The SLC52A2 and PEX6 variants identified in this study have been submitted to LOVD (patient IDs 00051498, 00051499, 00051501 and 00051502, URL: http://databases.lovd.nl/shared/genes/SLC52A2; patients IDs 00051496 and 00051497, URL: http://databases.lovd.nl/shared/genes/PEX6).
Results
Clinical data
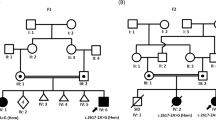
We reported an early-onset recessive ataxia10 in a large Arab Israeli family (family A, Figure 1). Briefly, an uncle and a niece were affected by ataxia starting at 3 years, impaired hearing and visual decline. The uncle became wheelchair bound at about 25 years and was almost deaf at the age of 27 years. The niece had sensory neuropathy on median and tibial nerves. Her muscle biopsy performed at 6 years showed mild fiber disproportion. Blood tests, including free fatty acids, were normal.

Identification of SLC52A2 mutation in family A despite initial false linkage. (a) Pedigree showing allele segregation of flanking markers on chromosome 8 in family A. Alleles are indicated by PCR fragment size (in base pair) and correspond to markers that are underlined on the genetic map of chromosome 8 (right). Markers in the pter–qter ordering are shown from top to bottom. Genetic distance (in cM) between markers is indicated. The marker that was not part of the initial study10 is highlighted in yellow. SLC52A2 localization in qter is indicated by a red mark. Note that the affected uncle (V.5) is homozygous neither for the marker closest to SLC52A2 tested in the initial study10 nor with the closest marker tested in this study, indicating the presence of a possibly ancient recombination located very close to SLC52A2. (b) Sanger sequencing of the mutation c.916G>A causing the p.(Gly306Arg) (G306R): the two patients are homozygous for the substitution. The parents of patient VI.1 are heterozygous for the same substitution and their healthy children are either heterozygous (individual VI.2) or wild type (individual VI.4).
In family B (Figure 2), two sisters, now 13 and 9 years old, born for consanguineous Turkish parents were affected with ataxia, impaired hearing and visual decline as they were 10 and 2 years old, respectively. On clinical examination both sisters had ataxic gait, distal weakness and optic atrophy. The younger one had more prominent signs in the form of intrinsic hand and intrinsic foot muscle atrophies with contractures of distal joints. The deep tendon reflexes were absent. She also had intentional tremor and mild dysarthria unlike her sister. Electroneuromyography study showed mild demyelinating motor neuropathy with axonal involvement in both siblings. There was also sensory involvement with no detected sural nerve activity. Audiograms indicated sensorineural deafness. Cerebral MRI revealed mild atrophy of the anterior superior cerebellar vermis in the youngest patient at 9 years (Figure 2). These children are on Riboflavin 400 mg/day for about 4 months now with only subjective benefit so far.

Identification of a novel SLC52A2 mutation in family B. (a) T1-weighted sagittal brain MRI of the youngest patient II.2 at age 9 years, showing mild atrophy of the anterior superior cerebellar vermis. The cerebellum and cerebrum were otherwise normal. (b) Pedigree showing consanguinity and segregation of the disease with mutation c.401C>T causing the p.(Pro134Leu) missense change (P134L). (c) Sanger sequencing of the c.401C>T mutation: the two patients are homozygous for the substitution. The father is heterozygous for the same substitution. Sample of the mother was not available. (d) Amino-acid sequence comparison of orthologous SLC52A2 proteins from different species. Amino acids that are identical to the human SLC52A2 sequence are shown in bold. The position of missense mutation (on the top of the mutated amino acid proline, P, highlighted in yellow) is indicated by an arrow. The mutated amino acid is highly conserved (present in all orthologues of metazoan species).
In family C (Figure 3), two siblings born for consanguineous parents of Turkish origin were affected by early-onset cerebellar ataxia and retinitis pigmentosa. Both had normal birth and normal initial development but experienced delayed walking at 3 years of age with gait ataxia. On clinical examination, the patients presented with cerebellar ataxia, dysmetria, adiadochokinesia, intention tremor and dysarthria. Cerebral MRI showed mild alteration of cerebellar white matter but no cerebellar atrophy (Figure 3). Nerve conduction studies indicated the presence of demyelinating peripheral motor neuropathy with reduced nerve conduction velocity. The older sister had retarded language, brisk reflexes and bilateral extensor plantar response. She was in wheelchair at 16 years and died at the age of 17 years. The brother is 24 years old and has spasticity. He was also followed for left renal stone.

Identification of PEX6 mutation in family C. (a) Whole-genome homozygosity mapping results. SNP genotyping results of the two patients were analyzed with the HomoSNP software in order to map homozygous regions across the 22 autosomes. Results are shown for chromosome 6, which was one of the seven chromosomes that revealed significant regions of homozygosity shared among the two affected children. Each colored line represents one patient, the pedigree is indicated on the left of the chromosome. Homozygous regions are colored according to the number of consecutive homozygous SNP markers: pink, from 20 to 24 markers; purple, from 25 to 29 markers; blue, 30 and more markers. Known loci for genes involved in ataxia are indicated in the line above the mapping results. The localization of PEX6 on the homozygosity map of chromosome 6 is indicated by an arrow. (b) T2-weighted horizontal MRI of patient II.2 at 13 years of age, showing hyper-echogenicity of the cerebellar white matter, suggesting demyelination of the afferent fibers. (c) Pedigree showing consanguinity and segregation of the disease with mutation c.2735C>T causing the p.(Ala912Val) missense change (A912V). Sanger sequencing results are indicated on the right of each corresponding sibling: both patients are homozygous for the mutation c.2735C>T and the parents are heterozygous for the same substitution. (d) Amino-acid sequence comparison of orthologous PEX6 proteins from different species. Amino acids that are identical to the human PEX6 sequence are shown in bold. The position of missense mutation (on the top of the mutated amino acid alanine, A, highlighted in yellow) is indicated by an arrow. The mutated amino acid is conserved in all eukaryotes including plants.
Mutation analysis
Because no mutation was found in the 6p21–p23 SCABD locus from the exome-sequencing data of family A, we expanded the analysis to all homozygous variants found on autosomes. After the selection of variants not found in 11 other independent exomes and the exclusion of gene variants found in more than 1% of controls (exome variant server) as well as variants with a VaRank score of 50 or lower, 32 rare variants were retained. Among them, we found the SLC52A2 mutation NM_024531.4: c.[916G>A];[916G>A] causing the p.(Gly306Arg) missense change (G306R, Figure 1), described previously in several reports as the cause of sensory ataxia and sensorineural hearing loss in the context of BVVLS type 2.3, 15, 16 In order to investigate the discrepancy brought by the identification of the causative mutation by whole-exome sequencing in SLC52A2 localized in qter of chromosome 8, we verified the genotype of the four closest 8q markers from the initial ABI PRISM Linkage Mapping Set10 (D8S285, D8S270, D8S284 and D8S272) plus one additional marker closer to SLC52A2 (D8S1836). The results indicate that all markers are non-homozygous in the affected uncle, suggesting the occurrence of a recombination immediately centromeric to SLC52A2 (Figure 1) that precluded the identification of the correct linkage with distantly spaced polymorphic markers.10
In family B, we investigated all homozygous variants shared between the two affected sisters. We identified 9 homozygous variants with an allele frequency lower than 1%, a VaRank score >55 and not found in 11 other independent exomes (Supplementary Table 2). All were missense variants. Three of them were predicted deleterious by the Sift prediction program, including one variant in SLC52A2 NM_024531.4: c.[401C>T];[(401C>T)] (exon 3) causing the p.(Pro134Leu) missense change (P134L, Figure 2; Supplementary Table 2). Proline 134 of SLC52A2 is conserved in all metazoan species (Figure 2), while the other two predicted missense variants of GOLGA8B and SIGLEC6 genes (Supplementary Table 2) are not conserved beyond primate and vertebrate species, respectively (not shown). Sanger sequencing confirmed that the SLC52A2 c.401C>T variant segregates with the disease (homozygous in both patients and heterozygous in the father; Figure 2).
In family C, homozygosity mapping was performed by whole-genome SNP analysis of the patients. SNP analysis revealed nine significant regions of homozygosity on seven chromosomes shared by the two affected siblings: two on chromosome 2, one on 6p21–p23 including the SCABD locus (12.67 Mb; 303 consecutive homozygous SNPs between rs10496793 and rs10497059; Figure 3a), one on chromosome 8, two on chromosome 9, one on chromosome 10, one on chromosome 14 and one on chromosome 15. All regions of shared homozygosity were investigated by whole-exome sequencing. Nine homozygous variations with a frequency of <1% in dbSNP 137 and a VaRank score >50 were found. Among them, we identified a homozygous nucleotide change (hg19 chr6: g.42932599G>A, 59 reads coverage) located in the PEX6 gene (NM_000287.3: c.[2735C>T];[2735C>T], exon 16) and predicted to cause a missense change of alanine 912 into valine (p.(Ala912Val), A912V, Figure 3c). This change was predicted to be very probably pathogenic according to the Sift and Polyphen2 prediction programs (SIFT score: 0.00; Polyphen2 score: 1.0). Alanine 912 is conserved in all eukaryotic orthologues of PEX6 (Figure 3d) and even the semiconservative change to valine is expected to have deleterious consequences. Moreover, the p.(Ala912Val) mutation is reported in the PEX gene database (http://www.dbpex.org/) in a homozygous patient with neonatal adrenoleukodystrophy. Sanger sequencing confirmed that the variant segregates with the disease on chromosome 6p21 (homozygous in both patients, heterozygous in the two parents and heterozygous or wild type in the four healthy siblings; Figure 3c) with a supporting LOD score of 2.3.
Of the eight remaining variants that did not appear to be disease causing, one was a frameshift (c.876delC, exon 1, p.(Gly293Alafs*151)) located in the FOXD4L1 gene (chromosome 2), which is a member of a presumably redundant family of transcription factors (seven FOXD4-like genes)17 and four were missense changes predicted neutral by Polyphen2 (Supplementary Table 3). The three remaining variants were missense changes predicted to be deleterious by Sift and Polyphen2: one was in SEPT10 (p.(Leu186Val), exon 5), which encodes for 1 of the 13 septins,18 the second was located in the major histocompatibility complex class II DN alpha gene HLA-DOA (rs141887419, p.(Gly231Val), exon 4) and the last one was located in HSD17B8 (c.523G>A, p.(Gly175Arg), exon 5), a gene implicated in mitochondrial fatty-acid synthesis19 making it a good candidate for being defective in family C. Defect of the encoded protein is expected to cause a reduction of lipoic acid levels.19 Whereas Sanger sequencing confirmed that the c.523G>A variant segregates with the disease on chromosome 6p21, no decrease in lipoic acid level was found in the fibroblasts of the affected boy (not shown).
Biochemical analyses of patients from family C
The fact that the c.2735C>T; p.(Ala912Val) missense change in PEX6 was the only mutation of family C located in a gene involved in syndromic ataxia prompted us to (re-)evaluate peroxisomal function in the patients. A slightly increased serum phytanic acid level (5.7 μg/ml for a normal upper limit of 5.0 μg/ml for the reference laboratory) found in the hospital records of the deceased affected girl was compatible with a very mild partial loss of function of a peroxisomal protein. The fibroblasts of the affected boy were therefore sent for extensive peroxisomal enzyme testing, which revealed mild but clear alterations: the C26:0 level was high but just within the normal range in two independent measurements, however the C26/C22 ratio was increased (Supplementary Table 4), indicating mildly impaired peroxisomal oxidation. Dihydroxyacetonephosphate-acyltransferase (the first enzyme in the plasmalogen biosynthesis pathway and located in the peroxisome) activity was normal. Immunofluorescence microscopy revealed the absence of peroxisomal staining with antibodies against catalase (a peroxisomal matrix protein), indicating the lack of import-competent peroxisomes, while staining with antibodies against peroxisomal membrane protein 70 revealed the presence of peroxisomal membrane structures (ghosts) with abnormal morphology. Immunoblot analysis of peroxisomal proteins revealed normal intra-peroxisomal processing of acyl-CoA oxidase 1 (ACOX1; 20- and 50-kDa bands) but abnormal processing of 3-oxoacyl-CoA thiolase (reduced 41-kDa band of the mature thiolase and presence of the unprocessed 44-kDa band). These results support the view that the p.(Ala912Val) missense change is pathogenic but causes only a very partial loss of function (hypomorphic mutation) in cultured fibroblasts.
Discussion and Conclusion
In the study of Bomont et al.,10 homozygosity mapping conducted in family A, affected by an early-onset recessive ataxia with hearing impairment and optic atrophy, suggested linkage over a 17-cM region at 6p21–p23 with a LOD score of 3.25.10 In present study, exome sequencing of the same family revealed the homozygous mutation p.(Gly306Arg) in SLC52A2, localized in chromosome 8qter, as the cause of the family disease. We also identified a second missense mutation p.(Pro134Leu) in SLC52A2 of an independent family with exactly the same clinical presentation (progressive ataxia, sensorimotor neuropathy, bilateral optic atrophy and sensorineuronal deafness) therefore confirming the syndromic association. SLC52A2 and SLC52A3 belong to a family of genes encoding for mitochondrial riboflavin transporters that, when mutated, cause BVVLS, a severe subgroup of autosomal recessive childhood-onset motor neuron diseases associated with sensorineural deafness. Recently, two studies have reported patients with p.(Gly306Arg) homozygous mutation and having not only sensorineural deafness and muscle weakness and wasting with respiratory insufficiency but also sensory ataxia and optic atrophy suggesting that mutations in SLC52A2 may be quite specific for the SCABD phenotype.3, 16
The published segregation of the disease with the 6p21–p23 chromosome interval in family A appears to be due to chance and not to linkage, as a LOD score of 3.25 corresponds to a probability of observing a false linkage of 0.05 after Bonferoni correction for multiple testing (whole-genome analysis). Correct linkage of the disease to 8qter was missed in this early study due to the high recombination rate of telomeric regions and to the absence of 8qter polymorphic markers in the initial Linkage Mapping Set used for whole-genome analysis.
We also identified in the present study an independent consanguineous family with ataxia and retinitis pigmentosa and linkage to the 6p21–p23 chromosome interval with a LOD score of 2.3 and confirmed the linkage by identification of the causal mutation p.(Ala912Val) in PEX6, located in 6p21. PEX6 encodes for peroxin 6, a component of the peroxisomal protein import machinery. PEX6 belongs to the AAA ATPase family and contains two AAA cassettes and a AAA protein family signature.20 We could demonstrate involvement of the p.(Ala912Val) mutation in family C thanks to abnormal biochemical and morphological studies of peroxisomes in the fibroblasts. Although mutations in PEX6 are know to cause Zellweger syndrome, ‘neonatal adrenoleukodystrophy’ or ‘infantile Refsum disease’ identified by serum biochemical evidence of peroxisomal disorder, two other reports identified mutations in PEX6 in patients with a less severe phenotype consistent with SCABD diagnosis. Najmabadi et al. found a homozygous missense mutation (p.(Leu534Pro)) in the PEX6 gene in five siblings from a large consanguineous family with moderate intellectual disability, retinitis pigmentosa, hearing loss and ataxia.21 In this family, no peroxisomal serum parameters were reported. In the second report, two missense mutations (c.1802G>A, p.(Arg601Gln) and c.2356C>T, p.(Arg786Trp)) were identified in compound heterozygous in the PEX6 gene in a patient with ataxic gait, bilateral sensorineural hearing loss, diplopia and cognitive dysfunction. Additional studies in cultured skin fibroblasts were consistent with late-onset Zellweger spectrum disorder.22
We therefore postulate that PEX6 is the gene defective at the SCABD1/SCAR3 locus, defined by chromosomal localization at 6p21 (OMIM# 271250), and we propose that patients with ataxia, deafness, optic atrophy and mutations in SLC52A2 present with the SCABD type 2 entity (SCABD2 locus at 8q24.3). On the basis of Anheim et al. classification,1 SCABD1 and SCABD2 belong to the group of cerebellar ataxia with sensorimotor neuropathy. Distinguishing the two entities at the molecular level is of high importance as patients with mutations in SLC52A2 can be ameliorated by riboflavin supplementation.3 Furthermore, these two families illustrate the concept of spinocerebellar ataxia due to distinct mildly affected metabolic pathways,1 as we demonstrate that partial loss of function in peroxisomes or mitochondria may lead to strikingly overlapping syndromes.
References
Anheim M, Tranchant C, Koenig M : The autosomal recessive cerebellar ataxias. N Engl J Med 2012; 366: 636–646.
Brown CH : Infantile amyotrophic lateral sclerosis of the family type. J Nerv Ment Dis 1894; 19: 707–716.
Foley AR, Menezes MP, Pandraud A et al: Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 2014; 137: 44–56.
Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW : Peroxisome biogenesis disorders. Biochim Biophys Acta 2006; 1763: 1733–1748.
Nikali K, Suomalainen A, Saharinen J et al: Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet 2005; 14: 2981–2990.
Ben Hamida C, Doerflinger N, Belal S et al: Localization of Friedreich ataxia phenotype with selective vitamin E deficiency to chromosome 8q by homozygosity mapping. Nat Genet 1993; 5: 195–200.
Ouahchi K, Arita M, Kayden H et al: Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet 1995; 9: 141–145.
Moreira MC, Barbot C, Tachi N et al: The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 2001; 29: 189–193.
Moreira M-C, Klur S, Watanabe M et al: Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 2004; 36: 225–227.
Bomont P, Watanabe M, Gershoni-Barush R et al: Homozygosity mapping of spinocerebellar ataxia with cerebellar atrophy and peripheral neuropathy to 9q33-34, and with hearing impairment and optic atrophy to 6p21-23. Eur J Hum Genet 2000; 8: 986–990.
DePristo MA, Banks E, Poplin R et al: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Geoffroy V, Pizot C, Redin C et al: VaRank: a simple and powerful tool for ranking genetic variants. PeerJ 2015; 3: e796.
Redin C, Le Gras S, Mhamdi O et al: Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alström syndromes. J Med Genet 2012; 49: 502–512.
Lagier-Tourenne C, Tazir M, López LC et al: ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet 2008; 82: 661–672.
Haack TB, Makowski C, Yao Y et al: Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J Inherit Metab Dis 2012; 35: 943–948.
Johnson JO, Gibbs JR, Megarbane A et al: Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012; 135: 2875–2882.
Klein SL, Neilson KM, Orban J et al: Conserved structural domains in FoxD4L1, a neural forkhead box transcription factor, are required to repress or activate target genes. PLoS One 2013; 8: e61845.
Hall PA, Jung K, Hillan KJ, Russell SEH : Expression profiling the human septin gene family. J Pathol 2005; 206: 269–278.
Chen Z, Kastaniotis AJ, Miinalainen IJ, Rajaram V, Wierenga RK, Hiltunen JK : 17beta-hydroxysteroid dehydrogenase type 8 and carbonyl reductase type 4 assemble as a ketoacyl reductase of human mitochondrial FAS. FASEB J 2009; 23: 3682–3691.
Collins CS, Kalish JE, Morrell JC, McCaffery JM, Gould SJ : The peroxisome biogenesis factors pex4p, pex22p, pex1p, and pex6p act in the terminal steps of peroxisomal matrix protein import. Mol Cell Biol 2000; 20: 7516–7526.
Najmabadi H, Hu H, Garshasbi M et al: Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478: 57–63.
Tran C, Hewson S, Steinberg SJ, Mercimek-Mahmutoglu S : Late-onset Zellweger spectrum disorder caused by PEX6 mutations mimicking X-Linked adrenoleukodystrophy. Pediatr Neurol 2014; 51: 262–265.
Acknowledgements
We acknowledge Bernard Jost, Serge Vicaire, Stéphanie Legras, Michael Dumas, Véronique Geoffroy and Jean-Philippe Villemin for next-generation sequencing and analysis. Funding: This study was supported by funds from the Institut National de la Santé et de la Recherche Médicale (INSERM), the Agence Nationale pour la Recherche—Maladies Neurologiques et Psychiatriques (ANR-09-MNPS-001-01 to M.K.), the ANR/E-rare JTC 2011 ‘Euro-SCAR’ (2011-RARE-004-01 to M.K.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Guissart, C., Drouot, N., Oncel, I. et al. Genes for spinocerebellar ataxia with blindness and deafness (SCABD/SCAR3, MIM# 271250 and SCABD2). Eur J Hum Genet 24, 1154–1159 (2016). https://doi.org/10.1038/ejhg.2015.259
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.259
This article is cited by
-
ATP8A2-related disorders as recessive cerebellar ataxia
Journal of Neurology (2020)
-
The Classification of Autosomal Recessive Cerebellar Ataxias: a Consensus Statement from the Society for Research on the Cerebellum and Ataxias Task Force
The Cerebellum (2019)
-
Successful treatment of a genetic childhood ataxia due to riboflavin transporter deficiency
Cerebellum & Ataxias (2018)
