
Abstract
The TK-selected chromosome- mediate gene transfer lines were analysed using DNA dot blot method, G—11 banding and in situ hybridization. The results showed that CMGT can provide a wide variety of intermediate size of the transgenome from greater than 80,000kb to less than 2,000 kb. Some of transfectants are intergrated into mouse chromosome which can be detected by G—11 banding and in situ hybridization
Similar content being viewed by others
Introduction
Chromosome-mediated gene transfer (CMGT) can be used to construct hybrid cells that retain fragments of the selected donor chromosome 1. Previously, we reported the generation of transfectants for chromosome 17 by CMGT for linking and ordering genes on chromosome 17 over the whole chromosome 2. This produced the longer distance than the high resolution mapping techniques of DNA-mediated gene transfer or cosmid cloning and at a more sensitive level than can be achieved by somatic cell hybrids or in situ hybridization. This has enabled us to generate a panel of more than 50 transfectants containing different transgenomic fragments of chromosome 17. We have used this panel of transfectants to localise most of the published gene markers and sequences on the chromosome 17. During the course of this study we have obtained some transfectants which contain only the small regions of chromosome 17 and they are particularly relevant to the study of some human genetic disease which are located on chromosome 17. In this paper, we describe some further detailed analysis about transgenome structure and size of these transfectants by using DNA dot blot method, G-11 banding and in situ hybridization. We are able to show that CMGT can provide a wide variety of intermediate size of the transgenome from great than 80,000 kb to loss than 2,000 kb. Some of the transfectants are intergrated into mouse chromosome which can be detected by G-11 banding and in situ hybridization
Material and Methods
Cell lines and culture conditions
Two human/mouse interspecific somatic cell hybrids were used as chromosome donors. PCTBA 1.8 is a mouse 3T3TK-/human somatic cell hybrid3 containing only human chromosome 17. All resultant CMGT transfectants derived from PCTBA 1.8 carry the prefix 'PLT'. GPT 17.3.2 K 41 is a mouse teratocarcinoma/ human somatic cell hybrid4 also containing only human chromosome 17. All resultant CMGT transfectants derived from GPT 17.3.2 K 41 carry the prefix 'KLT'. The mouse cell line LMTK- was used as the thymidine kinase (TK) deficient recipient cell line for all the GMGT experiments. Molt-4 is an Epstein-Barr virus transformed human lymphoblastoid cell line5.
The cells were either cultured in RPMI1640 (Molt-4, PCTBAI.8) at 5% CO2 or in Dulbecco's MEM (GPTK41 and LMTK-) at 10% CO2 and supplemented with 10% fetal calf serum. The PCTBA 1.8 hybrid, and all the CMGT transfectants, PLT and KLT were all routinely maintained in HAT medium (100μM hypoxanthine, 10μM methotrexate and 10μM thymidine) which selects for the presence of cytosal TK (TK1). The hybrids GPT17.3.2K41 were routinely maintained in MX medium (25μg/ml mycophenolic acid, 250μg/ml xanthine) as they contain the selectable marker Ecogpt. Back selection of CMGT transfectants with 5-bromo-2-deoxyuridine (BUdr) was performed by the addition of 50μg/ml BUdr to the culture medium which selects for those CMGT transfectants that have lost their TK1 locus. PLT6. B was one of these cell lines analysized in this study.
Preparation of DNA
Chromosomes hybrids and CMGT transfectants were all prepared by standard laboratory procedures 6. Briefly, two confluent T150 flasks were washed twice with saline and then extracted in 10 mM Tris (pH 8.0), 150mM NaCl, and 1mM EDTA with 0.2% SDS and 300g/ml protease K(type XIV, Sigma) for 4-6h at 37°C with gentle rocking. The DNA was purified by successive organic extractions: twice with buffer-saturated phenol, once with a mixture of buffer-saturated phenol/chloroform/isoamyl alcohol (IAA) at 25:24:1, and lastly with just chloroform/IAA (24; I). Two volumes of cold (−20 °C)ethanol were added to the aqueous phase, and the high-molecular-weight nucleic acids were spooled onto a glass pipette. The DNA was dissolved in 10mM Tris (pH8.0) and 1 mM EDTA at a Concentration of approximately 0.5mg/ml.
DNA Dot blot analysis
The Schleicher and Schuell sample filtration unit was used with the protocol recommended by the manufacturer 7. Purified cellular DNA was diluted in 100mM Tris pH7.4, to a volume of 180μl. Then the solution were added to a Falcon Microtest plate. To denture DNA, 20μl of 3M NaOH and 100μl of 20×SSC was added. The plates were heated to 80 °C for 10 minutes, cooled and then neutralised with 40μl of 2M Tris pH7.4. The DNA was applied to the nitrocellulose filters as recommended by Schleicher and Schuell. The filters were baked for 2 hours at 80 °C and hybridized as described below.
Radioactive labelling of DNA
DNA probe was boilling for 7 min. and radiolabelled with [32P]dCTP (Amersham, southern blot) or 3HdNTP (in situ hybridization)by the random priming method 8. Reactions was stopped by adding of TES (TE pH8, 0.1%SDS) to a volume of 150μl. Labelled DNA was then seperated from unincorporated nucleotides by a spin-celumn:a 1 ml plastic syringe was plugged with silicon glass wool and filled with Sephadex G50. The column was equilibrated with TES before application of the labelling reaction. Radiolabelled DNA was collected in an eppendorf tube by centrifugatioa of the column at 1000 rpm for 3 mins. Incorporation was routinely at least 2×108cpm/μg DNA, as measured by scintillation counting. The radioactive probe was boiled for 10 mins followed by rapid cooling on ice before hybridization.
Filter hybridization
The baked filters were prehybridised in 50% formamide, 5×SSC, 5×Denhardts solution, 0.1% SDS and 100μg/ml of sonicated salmon sperm DNA for 2 hours at 42 °C. Hybridization was carried out in fresh buffer with the addition of 0.25-1.00×106 cpm/ml [32P]dCTP labelled probe for 16 hours at 42 °C. The filters were washed to a stringency of 0.1 × SSC; 0.1% SDS at 65 °C. She filters were then exposed to Kodak XAR-5 film at −70 °C with intensifying screen for 1—5 days.
The G-11 differential staining
The G-11 differential staining technique was used to distinguish human chromosome and mouse chromosomes 9. Firstly, treated the slides aged chromosomes 7 days in 2×SSC for 5 min at 56 °C. Then, a stock solution of 0.7M NaOH was prepared. After diluting the NaOH stock 1:50 in the distilled water, make up Giemsa solution using 1:20 dilution. Stained the slides for 20 min in the diluted Giemsa and then rinse briefly in PBSA (pH6.8) and dry.
In situ hybridization
Cell lines are used to grow until a healthy dividing population is obtained, with around 5×105 cells per 10ml of culture medium. After carried out chromosome isolation according to the routine procedure, in situ hybridizations were performed essentially as described by Harper and Saunders 10.
Results
A series of 9 transfectants generated by chromosome-mediated gene transfer 2 were surveyed. The more detailed analysis of these transfectants and other transfectants can be found in reference 2. Table 1 has shown the co–transfer of syntenic gene markers in TK-1 selected CMGT.
Quantitative studies were designed to determine the amount of human genomic material contained in these CMGT lines. Kafatos demonstrated that spoting of known quantities of DNA onto nitrocellulose and probing for specific sequences could be used to quantitate the latter11. Duplicated “dot” blots of serially diluted(1:10) DNA samples (10μg to 10−6μg) from each CMGT lines were prepared using a Schleicher and Schuell filtration unit. These were probed with 32P-labled human Molt-4 cell line DNA in order to detect the human repetitive component in the mouse LMtk background. We used the condition that minimize hybridization to mismatching repetitive elements: (1) stringent hybridization: 50% formamide at 42 °C; (2) stringent washing: low salt (0.1 × SSC) and high temperatures (65 °C). (3) short hybridization time: 16 hours. (4) highly radioactive probe: >6×l08 cpm/μg. These conditions have been estimated to permit less than a 10% mismatch 12.
We have used dot blot analysis to determine the relative amount of human DNA in each of the transfectants. The co-transfer of markers known to be syntenic with thymidine kinase on chromosome 17 in these transfectants listed in Table 1.
Controls for these experiments were the human/mouse parent cell line, PCTBA 1.8, which only has one human chromsome 17 in the mouse background. Mouse cell line 1R was used as negative control.
The resulting autoradiographs from these experiments are presented in fig. 1. Dilution blots of the appropriate control (Molt-4, 1R 14, and PCTBA 1.8) were hybridized in the same solution. Quantitation of the result was done by densitometric scans of 1-day autoradiographs.

Dot blot analysis of nine CMGT transfectants containing micro-transgenomes. The relative amount of DNA from each cell lines were given by scanning the autoradio-graph shown in the Figure.
Using the hybridization signal obtained from 1μg of DNA from PCTBA 1.8 as an R=1.00, the CMGT lines can be ranked according to their average R value. From these results, one can seen that there are no disconcordancies with the DNA markers and isozyme data (Figure 1). The two cell lines, PLT-13 and KLT-6, were known to contain large human fragments as they contained all of the markers so far detected on chromosome 17. In addition, KLT-6 showed excessive DNA content even higher than single human chromsome 17; later we are able to show by in situ hybridization that it has two copies of human chromosome fragments attached to the mouse chromosome.
Using the donor chromosome 17 only hybrid PCTBA1.8 as a standard, the amount of chromosome 17 in each transfectant has been calculated as a proportion of the whole chromosome. The results shown in Table 2 indicate a good correlation of the intensity of the signal obtained in the dot blot with the number of chromosome 17 markers present (as shown in Table 1). The KLT6 transfectant probably contains more than one copy of chromosome 17 in some cells as it has a greater human DNA content than the PCTBA1.8 hybrid which has been shown to only have one chromosome 17 per cell.
G-11 differential staining
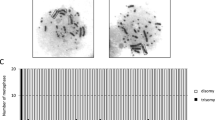
G-11 differential staining was used to detect the human fragment attached to a meuse chromosome in two of the very large transfectants containing all of the genes analysed (PLTI3 and KLT6). We also detected a human fragment using G11 staining with KLTI2. From this transfectant, one can clearly see that the human fragment is attached in a mouse chromosome (Fig. 2). However most of our transfectants do not show any cytologically deteebable human material using G11.

G-11 banding of transfectant KLT-12. Arrow indicates human fragment.
In situ hybridisation
Human fragments can easily be detected, using total human genomic DNA ss a probe for in situ hybridisation, in the relatively large transfectants such as PLT6 and PLT6.B. Figure 3 shows PLT6.B where the human fragment was integrated into one of the mouse chromosomes. Such a fragment can be seen in most of the nuclei of this transfectant. Interestingly, the human centromeric probe for the D17Z1 locus hybridises to the same region as the total human DNA. A similar phenomenon also appears to have occurred in KLT6 and KLT12. But in the former transfectants in which have shown to have whole human chromsome 17 markers, there are two duplicated human chromosomes which are attached to mouse's chromsomes (Figure 4). No obvious human fragment can be seen in tranfectants such as PLT8, which only contain GAA, UMPH, GHC, TK1 and GALK using in situ hybridisations with total human genomic DNA as a probe. This is probably due to insufficient human repeat sequences in what may be quite a small region of the chromsome.

In situ hybridization of transfectant PLT-6B. Arrow indicates human fragment.

In situ hybridization of transfectant KLT-6. Arrow indicates human fragment.
Discussion
Technically, chromosome mediated gene transfer (CMGT) is quite similiar to the more familiar technique of DNA mediated gene transfer. The real difference between these two methods is that in CMGT the donor genetic material is in the form of isolated chromosomes rather than naked DNA, so it is postulated that CMGT can create far more large fragment than that of DNA transfection. Since normal DNA extraction procedure is quite harsh to keep the DNA molecular longer than 200kb, so the sequences longer than that are rarely transferred intact in DNA transfection. Instead, in this study, we demonstrated that CMGT can create a wide variety of intermediate size of the transgenome from greater than 80,000kb to less than 2,000 kb. So CMGT has the potential for filling an important gap in the resolving power of somatic cell and molecular genetie techniques.
Generally, there are good correlation of DNA contents of transfectants with the number of chromosome 17 markers present (as shown in Table 1). However, there are some exceptions. Far example, KLT-6 and PLT-13 have been shown to have all the markers detected on chromosome 17, but there is a big difference with regard to their DNA contents. The KLT-6 DNA content was as high as 2.5 times than that of PLT-13. This difference could be due to that: in in situ hybridization, there are two copies of chromosome 17 fragments in KLT-6, while in PLT-13 there is only one (data not shown).
Possibly the most useful aspect of GMGT is that it generates hybrid cell lines which are enriched for certain small regions of the human chromosome. This is of particular value when the genes of interest are located close to the selectable marker. For example, in the study of eystic fibrosis, the selectable transforming oncogene Met was used to generate a series of transfectants containing small regions of chromosome 7 which were used as a source of probes forthe study of this disease 16. Since the gene for von Recklinghausen neurofibromatosis (NF-1) has been shown to be close to the centromere of chromosome 17 17, we should be able to use the chromosome 17 transfectants that have retained the centromere to obtain probes which may prove useful in the study of NFI. We are currently making cosmid libraries from some of these transfectants and beginning to obtain the cosmids to study these regions 18.
Such enrichment procedure also will provide rich resources for other genetic disease involved with structural abnormalibilities of human chromosome 17, such as acute promyelocytic leukaemia (APL, FAB claasification M3) having reciprocal translocation involving the long arm of chromosomes 15 and 17 t(15:17) (q22; q11.2—12) 19, and loss of heterozygosity of chromosome 17p markers reported in the progression of colorectal carcinomas 20. Deletions of chromosome 17 also have been associated with mental retardation syndromes such as Miller-Dicker syndrome (del 17p13) 21 and Smith-Magenis syndrome (del 17p11) 22 et al. Transfectants of our CMGT will be useful in those genetic disease studies as well.
Until recent1y, the physical nature of 4he transgenomes created by GMGT has been a major area of uncertainiy. We are trying to use cytogenetic technique to detect or resolve transgenome structure. But, more often, the histochemical techniques used were too insensitive to detect transgenome fragment. We first demonstrated that G11 staining method can be used to detect relatively large transfeetants. The more sensitive method is to use in situ hybridization with total human DNA as a probe. Usually discrete fragments of human DNA are inserted into mouse chromosome, even in some very largo ones which have all the markers doteeted on chromosome 17. In a transfectants, KLT-6, we observed that the transgenomes can undergo duplication forming two identical chromosomes.
Interesting enough, when we use chromosome 17 centromeric specific probes, D17Z1 23, to hybridize the trasnfectants which have human centromere alpha sattelites (data not shown), we observed that a number of transfectants have both human and mouse centromeres, but human centromere apparent1y lost its phenotypo of constriction. This phenomenon is quite similar to the so called bicentromerics chromosomes in which one centromere has undergone centromere suppression. It is possibly that in the mouse cytoplasm background, the human centromere is not as fit as mouse centromere and this could be an explanation of frequently lost human chromosomes in human/mouse hybrids. The detailed analysis of the structure and behaviour of transgenomes in CMGTs suggests an alternative and complementary approach to the molecular understanding of those basic questions of chromosome form and function.
References
Goodfellow PN, Pritchard CA . Chromsome fragmentation by chromosome mediated gene transfer. Cancer Survey 1988; 17(2): 265.
Xu W, Gorman PA, Rider SH, Hedge P J, Moore G, Pritchard C, Sheer D, Solomon E . Construction cf a genetic map of human chromosome 17 by use of chromosome-me diated gene transfer. Proc Natl Acad Sci USA 1988; 85: 8563–8567.
Bai Y, Sheer D, Hiorns L, Knowles RW, Tunnacliffe A . A monoclonal antibody recognizing a cell snrface antigen coded for by a gene on human chromosome 17. Ann Hum Genet Lond 1982; 46: 337–347.
Tunnacliffe A . Parkar M, Povey S, Bengtsson Be, Stanley K, Solomon E, Goodfellow PINT . Integration of Ecogpt and SV40 early region sequences into human chromosome 17: a dominant selection system in whole cell and micro-cell human-mouse hybrids. EMBO J 1983; 2: 1577–1584.
Minowada MD, Ohnuma T, Moore GE . Rosette-forming human lymphoid cell lines. 1. Establishment and evidence for origin of thymus-derived lymphocytes. J Natl Cancer Inst 1972; 49: 891–895.
Gross-Bellard M, Oudet P, Chambon P . Isolation of high molecular weight DNA from mammalian cells. Eur J Bioehem 1973; 36: 32–38.
Murphy PD ., Miller CL . and Ruddle FH . Quantitation of the transgenome size in chromosome-mediated gene transfer lines. Cytogenet. Cell Genet 1985; 39: 125–133.
Feinberg AP . and Vogelstein B . A technique for radiolabelling DNA restriction endonucease fragments to high specific activity. Anal .biochem 1984; 137: 266–267.
Bobrow M . and Cross J . 1974; Differential staining of human and mouse chromosomes in interspecific cell hybrids. Nature (London) 251: 77–79.
Harper ME .,and Saunders GF . Localization of single copy DNA sequences on G-banded human chromosomes by in situ hybridization. Chromosma 1981; 83: 431–439.
HMG9. Cytogenet. Cell Genet. HMG 1987; 9: 46.
Kafatos FC, Jones WC ., Efstratiadis A . Determination of nuclei acid sequence homologies and relative concentrations by a dot hybridization procedure. Nucl Acids Res 1979; 7: 1541–1542.
Jabs EW, Wolf SF, Migeon BR . Gharacterization of reiterated human DNA with respect to mammalian X chromosome homology. Somat Cell Molecu Genet 1984; 10: 93–103.
Nabholz M . Miggiano V, Bodmer W . Genetic analysis with human-mouse somatic cell hybrids. Nature (London) 1969; 223: 358–363.
Mendelsohn ML . Mayall BH, Bogart E, Moore II, Dan H & Perry BH . DNA content and DNA-based centromeric index of the 24 human chromosomes. Science 1973; 179: 1126–1129.
Eetivill X, et el. A canaictate for the cystic fibrosis locus isolated by selection for methylation-free islands. Nature 1987; 326: 840–845.
Barker D, et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 1987; 236: 1100–1102.
Xu W, Liu L, Bao Y, Yagle M, Solomon E . Enrichment of regional cosmid clones from a human chromosome 17 transfectant PLT6B constructed by chromosome mediated gene transfer. Abstract accepted in HGM10 Yale USA 1989.
Rowley JD, Golomb HM, Vardiman J, Futuhara S, Dougherty C, Potter D . Further evidence for a non-random chromosomal abnormality in acute promyelocytic leukaemia. Int J Cancer 1977; 20: 869–872.
Fearon ER, Hamilton SR, Vogelstein B . Clonal analysis of human colorectal tumors. Science 1987; 218: 193–196.
Stratton RF, Dobyns WB, Airhart SD, Ledbetter DH . New chromosomal syndrome: Moller-Dieker syndrome ann monosomy 17p13. Hum Genet 1984; 67: 193–200.
Smith ACM et al. Interstitial deletion of 17 (p11.2p11.2)in nine patients. Amer J Med Genet 1986; 24: 421–32.
Waye JS . and Willard HF . Structure, organization and sequence of alpha satellite DNA from human chromosome 17: Evidence for evolution by unequal crossing-over and an ancesteral pentamer repeat shared with the human X chromosome. Mol Cell Biol 1986; 6: 3156–3165.
Acknowledgements
I would like to thak Dr. Ellen Solomen for her guidance and support.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Xu, W. Genetic analysis of transgenome structure and size of chromosome-mediated gene transfer lines. Cell Res 2, 15–23 (1992). https://doi.org/10.1038/cr.1992.2
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/cr.1992.2
