
Abstract
Background:
Foxp3+ regulatory T (Treg) cells and M2 macrophages are associated with increased tumour progression. However, the interaction between Treg cells and M2 macrophages remains unclear.
Methods:
The expression of FoxP3 and CD163 was detected by immunohistochemistry in 65 cases of laryngeal squamous cell carcinoma (LSCC). In vitro, the generation of activated Treg (aTreg) cells and M2 macrophages by interactions with their precursor cells were analysed by flow cytometry and ELISA. In vivo, the antitumour effects were assessed by combined targeting aTreg cells and M2 macrophages, and intratumoural immunocytes were analysed by flow cytometry.
Results:
In LSCC tissue, accumulation of aTreg cells and M2 macrophages predicted a poor prognosis and were positively associated with each other. In vitro, aTreg cells were induced from CD4+CD25− T cells by cancer cell-activated M2-like macrophages. Consequently, these aTreg cells skewed the differentiation of monocytes towards an M2-like phenotype, thereby forming a positive-feedback loop. Combined targeting aTreg cells and M2 macrophages led to potent antitumour immunity in vivo.
Conclusions:
The positive-feedback loop between aTreg cells and M2 macrophages is essential to maintain or promote immunosuppression in the tumour microenvironment and may be a potential therapeutic target to inhibit tumour progression.
Similar content being viewed by others


Main
Immunotherapy, including anticancer vaccination and adoptive transfer of tumour-specific cytotoxic T lymphocytes, has been considered as a promising treatment for head and neck squamous cell carcinoma (HNSCC), although its effects on tumour regression remain limited (Schuler et al, 2014; Li et al, 2015). The inefficacy of such therapies implies that head and neck carcinoma has developed diverse strategies to evade tumour-specific immunity (Ferris, 2015).
Studies show that regulatory T (Treg) cells and M2 macrophages are crucial players in immune tolerance (Mantovani et al, 2002; Khazaie and von Boehmer, 2006; Zou, 2006; Alhamarneh et al, 2008; Mantovani and Locati, 2013). Recently, to evoke antitumour immunity in vivo, we blocked tumour trafficking of CD45RA−Foxp3high activated Treg (aTreg) cells using a CCR4 antagonist, which led to inhibition of tumour growth and prolonged survival (Sun et al, 2014, 2015, 2016). However, the effect of targeting a single immunosuppressive population is not satisfactory. This prompted us to employ a combined strategy targeting both aTreg cells and M2 macrophages and further investigate their interactions.
In this study, we evaluated the clinical importance of tumour-infiltrating aTreg cells and M2 macrophages in laryngeal squamous cell carcinoma (LSCC) patients and confirmed their positive correlation. We demonstrated that cancer cell-activated M2-like macrophages induced aTreg cells from CD4+CD25− T cells in vitro. Reciprocally, the generated aTreg cells skewed the differentiation of monocytes towards the M2-like phenotype, forming a positive-feedback loop. Blockade of aTreg cell trafficking combined with depletion of tissue macrophages reduced both intratumoural aTreg cells and M2 macrophages to the maximum extent and significantly enhanced antitumour immunity in experimental mice.
Materials and methods
Immunohistochemical analysis
The main clinical and pathological characteristics of LSCC patients are presented in Supplementary Table S1. Clinical staging and the anatomical site of tumours were assessed according to the sixth edition of the Union for International Cancer Control (UICC 2008) tumour-node-metastasis classification of malignant tumours. Paraffin-embedded, formalin-fixed 4 μm-thick tissue sections were processed for immunohistochemical staining with anti-FoxP3 (Ab22510, IgG1, 1 : 300 dilution, Abcam, Cambridge, MA, USA), anti-CD163 (Ab87099, IgG, 1 : 80 dilution, Abcam) and anti-p16/INK4a (Clone EPR1473, 1 : 250 dilution, Epitomics Inc., Burlingame, CA, USA) antibodies. The densities of FoxP3+ or CD163+ cells was evaluated quantitatively by the mean count of four representative fields at 400 × magnification by two independent observers who were blinded to the clinical outcome. Nuclear and cytoplasmic staining of >70% tumour cells was considered p16 positive and believed to correlate with Human papilloma virus (HPV) (Begum and Westra, 2008).
Cell lines and animals
Human LSCC cell lines (SNU46 and SNU899) and a nasopharyngeal squamous cell carcinoma (NPSCC) cell line (HONE1) were kindly provided by Professor Ja-Lok Ku (Seoul National University College of Medicine, South Korea). Tumour culture supernatants (TSNs) were prepared as described previously (Kuang et al, 2007). The mouse squamous cell carcinoma (SCC VII) cell line was kindly provided by Professor Si-Xi Liu (West China Hospital, China). Cells were cultured under standard conditions.
Eight-week old male C3H-HeN mice were purchased from Slac Laboratory Animal Co., Ltd. (Shanghai, China). All mice were housed under specific pathogen-free conditions. Experimental procedures were approved by the Institutional Animal Studies Committee and conducted in accordance with Institutional Animal Care and Use Committee guidelines.
Cell preparation
The study protocol was approved by The Ethics Committee of The First Affiliated Hospital of Sun Yat-Sen University and used for research purposes only (Approval No. 2012-349). Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors by Ficoll density gradient centrifugation. CD14+ cells were isolated from PBMCs using CD14 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and cultured with or without 20% TSN for 6 days to obtain macrophages.
To isolate CD4+CD25− T cells and Treg subsets, cells were stained with anti-hCD4-FITC, anti-hCD25-APC, and anti-hCD45RA-eFluor 450 (eBioscience, San Diego, CA, USA) antibodies, and sorted using a BD Influx cell sorter (BD Biosciences). Three Treg subsets were prepared as live cells as described previously (Miyara et al, 2009). Foxp3lowCD45RA+ cells, which were CD25++ (I), Foxp3highCD45RA− cells, which were CD25+++ (II), and Foxp3lowCD45RA− cells, which were CD25++ (III), were prepared by sorting CD25++CD45RA+, CD25+++CD45RA−, and CD25++CD45RA− cells, respectively.
Cell co-culture
Cells were co-cultured in RPMI 1640 medium supplemented with 1% penicillin/streptomycin, and 10% heat-inactivated foetal calf serum. For Treg cell generation, 1 × 106 autologous macrophages were co-cultured with 2 × 106 CD14+-depleted PBMCs in the presence of 0.5 μg ml−l anti-CD3 mAb or with 1 × 106 CD4+CD25− T cells in the presence of 2 μg ml−l anti-CD3 mAb plus 1 μg ml−l anti-CD28 mAb for 4 days. For macrophage differentiation, CD14+ cells were seeded in a round bottom 96-well plate at 2 × 105 cells per well. CD4+CD25− T cells and the three Treg subsets were added to autologous CD14+ cells at a ratio of 1 : 2 and co-cultured for 40 h in the presence of 50 ng ml−l anti-CD3 mAb with or without 20% TSN from SNU899 cells.
For neutralisation experiments, neutralising antibodies against TGF-β1 (5 μg ml−l, mouse IgG1, clone 9016), IL-10 (5 μg ml−l, goat IgG), IL-4 (5 μg ml−l, mouse IgG1, clone 3007), or IL-13 (5 μg ml−l, mouse IgG1, clone 32116) and isotype controls (mouse IgG1 or goat IgG) were added at the start of the co-culture. All from R&D Systems (Minneapolis, MN, USA).
Flow cytometric analysis
To determine the phenotype of macrophages, the following antibodies were used: anti-hCD163-APC, anti-hCD206-Alexa Fluor 488, anti-hHLA-DR-eFluor 450, anti-hCD86-PE, and anti-hCD80-PE-Cy5. To determine the frequency of the three distinct Treg subsets, the following antibodies were used: anti-hCD4-FITC, anti-hCD25-APC, and anti-hCD45RA-eFluor 450 for surface staining; anti-hFoxp3-APC for intracellular staining.
For the intracellular cytokine assay, the sorted CD4+CD25− T cells and each Treg subset were stimulated with a cocktail of phorbol 12-myristate 13-acetate (PMA), ionomycin, and Golgi stop (brefeldin A and monensin; eBioscience) for 5 h. Then, we performed intracellular staining using anti-hIL-4-PE-Cy7, anti-hIL-10-PerCP-eFluor 710, anti-hIL-13-PE (Sony Biotechnology, Inc, San Jose, CA, USA), and anti-hIFN-γ-APC-eFluor 780 antibodies. All antibodies were purchased from eBioscience unless specified otherwise.
Cytokine detection
The concentrations of IL-10, IL-6, IL-12p70, TNF-α, IL-4, IL-5, IL-13, IL-2, IFN-γ, and TGF-β1 in culture supernatants was determined using ELISA kits according to the manufacturer’s instructions (IL-2, IFN-γ, and TNF-α from R&D Systems; others from RayBiotech, Inc, Norcross, GA, USA).
Combined targeting of aTreg cells and M2 macrophages in vivo
Twenty mice were randomly and equally divided into four groups and ear tagged prior to treatment. On day 0, 1 × 105 tumour cells were injected subcutaneously into the right flank. A CCR4 antagonist (87.5 μg; TOCRIS, Minneapolis, MN, USA; Sun et al, 2016), GdCl3 (0.2 mg; Sigma-Aldrich, Saint Louis, MO, USA; Jiang et al, 2005), or CCR4 antagonist+GdCl3 were injected intraperitoneally three times per week for 3 weeks from day 5 after tumour cell injection. A group of five mice for the saline control was set in parallel. Tumour size was measured every 4 days using fine calipers. Tumour volumes were calculated as length × (width)2 × 0.5. All mice were killed at 24 h after the last injection.
After killing the mice, tumours and spleens were isolated and prepared as single cell suspensions. Mononuclear cells were isolated using a Ficoll density gradient and analysed for aTreg cells, M2 macrophages, CD4+ and CD8+ T cells, and Th1 cytokines by flow cytometry using anti-mouse CD4-eFluor 450, anti-mouse CD8-Alexa Fluor 700, anti-mouse CD25-APC, anti-mouse CD45RA-FITC (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-mouse CD206-PE-Cy7 (Sony Biotechnology, Inc), anti-mouse F4/80-PE-Cy5.5, anti-mouse FoxP3-PE, anti-mouse IL-2-PE, anti-mouse IFN-γ-PE-Cy5.5, and anti-mouse TNF-α-PE-Cy7 antibodies, and a fixation/permeabilisation kit. Antibodies were purchased from eBioscience unless specified otherwise. Immunohistochemical staining of CD3 (Ab16669, IgG, 1 : 100 dilution, Abcam) was also performed to assess intratumoural CD3+ T cells. Another randomly divided four groups of mice were used for survival analysis. Observation was aborted on day 50.
Statistical analysis
All statistical analysws were performed with SPSS for Windows version 13.0 (IBM, Chicago, IL, USA). For Foxp3 and CD163, the cutoff to define the subgroups was the median value. Analysis of the correlation between variables was conducted using Spearman’s rank coefficient tests. Survival variables were estimated using the Kaplan–Meier method and compared using log-rank tests. Differences between groups were assessed using the Mann–Whitney U-test, Student’s t-test, or Kruskal–Wallis test. Multivariate analysis using the Cox proportional hazard model was used to determine the influence of each variable, when adjusted to the others, on overall survival. P-values of <0.05 were considered as statistically significant.
Results
Accumulation of aTreg cells and M2 macrophages is positively associated with each other in LSCC tissue and predicts poor survival
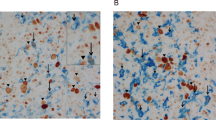
We first examined the prevalence of tumour-infiltrating aTreg cells and M2 macrophages in 65 patients with LSCC, the common type of HNSCC (Supplementary Table S1). Immunohistochemical staining of Foxp3 and CD163 was performed in paraffin-embedded tissues. Both Foxp3+ aTreg cells and CD163+ macrophages had substantially infiltrated into the stroma, but scarcely into tumour sites (Figure 1A). To determine the clinical importance of Foxp3+ aTreg cells and CD163+ macrophages in stromal of tumours, we analysed their associations with clinicopathological characteristics and overall survival. The levels of Foxp3+ aTreg cell and CD163+ macrophage infiltration in patients at late clinical stages (III and IV) were significantly higher than those at early clinical stages (I and II; P<0.01 and P<0.05, respectively; Figure 1B and C) (Supplementary Table S2). Expression of Foxp3 and CD163 increased with the pathological stage, although no significant difference was found in Foxp3 expression (Figure 1D and E; Supplementary Table S2). Patients with high numbers of intratumoural Foxp3+ aTreg cells/CD163+ macrophages had shorter overall survival (medians: 74/78 months) than those with low numbers of Foxp3+ aTreg cells/CD163+ macrophages (medians: 94/90 months; Figure 1F and G). Furthermore, there was a significant difference between the group with high expression of both Foxp3 and CD163 and the group with high expression of either Foxp3 or CD163 and the group with low expression of both Foxp3 and CD163 (medians: 71, 87, and 94 months, respectively; Figure 1H). These data supported important roles of both aTreg cells and M2 macrophages in accelerating tumour progression and predicting poor survival. In a Cox Multivariate analysis (Supplementary Table S3), clinical stage, nodal status, tobacco and alcohol consumption were associated with overall survival. The presence of high numbers of intratumoural Foxp3+ aTreg cells in combination with high numbers of intratumoural CD163+ macrophages was an independent prognostic marker for overall survival, while the HPV status had no independent prognostic value (Supplementary Figure S1).

Clinical implications and correlation of tumour-infiltrating aTreg cells and M2 macrophages. (A) Representative images of immunohistochemical staining of Foxp3+ and CD163+ cells: Foxp3 expression in the tumour (a), low and high Foxp3 expression in the stroma (b and c), CD163 expression in the tumour (d), low and high CD163 expression in the stroma (e and f). Positive cells are stained brown (× 400 magnification). Lower panels are magnified images of the boxed areas in the corresponding upper panels. (B and C) Levels of Foxp3+ and CD163+ cell infiltration in patients with early stage tumours were lower than those in patients with late stage tumours. Statistical differences between the two groups were analysed using the Mann-Whitney U-test. (D and E) Comparison of the levels of Foxp3+ and CD163+ cell infiltration between three groups with different degrees of differentiation. Statistical differences were analysed using the Kruskal–Wallis test. (F–H) Kaplan–Meier analysis of overall survival in terms of intratumoural Foxp3+ aTreg cells (F), CD163+ macrophages (G), and their combination (H) in LSCC samples (n=65). High numbers of Foxp3+ aTreg cells with concomitant high numbers of CD163+ macrophages were associated with poor survival. (I) Spearman’s rank correlation coefficient was calculated to assess the relationship between Foxp3+ and CD163+ cells. The density of CD163+ macrophages was positively correlated with that of Foxp3+ aTreg cells in tumour tissue.
Moreover, Spearman’s rank correlation coefficient was calculated to assess the relationship between Foxp3+ aTreg cells and CD163+ macrophages. The densities of the two markers were positively correlated with each other in LSCC specimens (Figure 1I). This quantitative relationship encouraged us to further investigate their biological interactions.
Macrophages exposed to tumour culture supernatant from LSCC-derived cell lines exhibit an M2-like phenotype and augment the Foxp3+ Treg population in peripheral blood mononuclear cells
To investigate whether cancer cells with a distinct epithelial phenotype have a different ability to activate macrophages, we cultured freshly isolated human monocytes in medium with 25% tumour culture supernatant (TSN) from a nasopharyngeal carcinoma cell line (HONE1) or laryngeal carcinoma cell lines (SNU46 or SNU899) for 6 days to obtain macrophages. Figure 2A shows that, relative to untreated macrophages, TSN-treated macrophages displayed strong up-regulation of typical M2 macrophage markers CD163 and CD206 (data not shown) and reduced expression of HLA-DR, CD86, and CD80, which are associated with immunosuppression. They also produced higher amounts of an M2 macrophage cytokine (interleukin (IL)-10) and lower levels of M1 macrophage cytokines (tumour necrosis factor-α (TNF-α), IL-6, and IL-12p70) (Figure 2B), indicating that macrophages were activated to an ‘M2-like state’ by TSN. Moreover, the level of activation by TSN from SNU899 cells, as measured by cytokine production and surface marker expression of M2 macrophages, was the highest among all groups, suggesting that SNU899-derived TSN was a strong macrophage activator.

Macrophages exposed to TSNs from LSCC-derived cell lines exhibit an M2-like phenotype and augment the Foxp3+ Treg population in PBMCs. Human monocytes were cultured for 6 days with medium alone (Mϕ) or with TSNs from a nasopharyngeal carcinoma cell line (MϕHONE1) or laryngeal carcinoma cell lines (MϕSNU46 and MϕSNU899). Thereafter, Mϕ or TSN-exposed Mϕ were co-cultured with autologous responder cells and stimulated with anti-CD3/CD28 mAbs for 4 days. (A) Expression of CD163/HLA-DR/CD86/CD80 in macrophages. The histograms are representative of five independent experiments of macrophages from five different donors. Numerical values denote the mean fluorescence intensity (MFI). (B) On day7, Mϕ and TSN-exposed Mϕ were washed and then cultured for 24 h with fresh medium alone, then the cytokine levels in the media of macrophages were analysed (mean±s.e.m., n=5 independent experiments; *P<0.05, **P<0.01 compared with Mϕ). (C) Representative data showed that MϕSNU899 increased the Foxp3+ Treg frequency among CD4+ T cells more than either MϕSNU46 or MϕHONE1 when co-cultured with autologous CD14+-depleted PBMCs. Data are mean values±s.d. of triplicate determinations. (D) Statistical analysis revealed that the levels of Th2 cytokines (IL-5 and IL-13) were most increased in the co-culture media of MϕSNU899 and PBMCs (mean±s.e.m., n=3; P<0.05; Δ, compared with PBMCs; Θ, compared with PBMCs+Mϕ). TSNs, tumour culture supernatants.
To determine whether these activated macrophages induced Foxp3+ Treg cells, they were co-cultured with autologous peripheral blood mononuclear cells (PBMCs). Consistent with the changes in surface markers and cytokines, more Foxp3+ Treg cells were derived from macrophages treated with TSN from SNU899 (MϕSNU899: 82.0±4.7%) than untreated macrophages (Mϕ: 36.5±2.9%) or those treated with TSNs from other HNSCC cell lines (MϕSNU46: 68.2±3.2%; MϕHNOE1: 64.0±3.4%; Figure 2C). In addition, to investigate the overall effect of macrophages on the immune response, we examined cytokines in the co-cultures by immunosorbent assays (ELISAs). The levels of Th2 cytokines (IL-5 and IL-13) were correspondingly increased in the culture supernatants (Figure 2D), while the production of Th1 cytokines (interferon-γ (IFN-γ), IL-2, and TNF-α) was suppressed to undetectable levels (data not shown), implying that the TSN-exposed macrophages mediated a tumour-promoting effect.
Taken together, these results indicate that macrophages activated by TSN from LSCC cell lines have an M2-like phenotype and may serve as an important initiator of Treg cell induction.
TSN-exposed M2-like macrophages induce an aTreg cell subset from CD4+CD25− T cells
To eliminate the possibility that TSN-exposed macrophages were merely expanding the pre-existing naturally occurring Treg cells present at the initiation of culture, we sorted autologous CD4+CD25− T cells as responder cells before adding macrophages (n=3). A clear conversion from CD4+CD25− Foxp3− T cells to CD4+CD25+ Foxp3+ Treg cells was observed by flow cytometric analysis (Figure 3Aa and Ab). We further identified the functionally distinct subsets of these newly generated Treg cells (Figure 3Ac), because Treg cells with a phenotype of CD45RA+Foxp3low or CD45RA−Foxp3high can exhibit immunosuppressive functions (Miyara et al, 2009). We found a dominant increase in CD45RA−Foxp3high aTreg cells of the suppressive Treg subsets (Figure 3B and C), indicating that the Treg cells induced by M2-like macrophages may directly mediate an inhibitory effect on tumour immunity.

TSN-exposed M2-like macrophages significantly induce aTreg cells from CD4+CD25− T cells. CD14+ monocytes were cultured with TSNs from a laryngeal carcinoma cell line (MϕSNU899), followed by coculture with autologous CD4+CD25− T cells for 4 days with or without neutralising mAbs against TGFβ-1, IL-10, or TGFβ-1/IL-10 in the presence of 2 μg ml−l anti-CD3 mAb plus 1 μg ml−1 anti-CD28 mAb. (A) Foxp3+CD25+ Treg cells were actively induced by MϕSNU899 from CD25-depleted cells (a and b). Flow cytometry of each subset of induced Treg cells (c). (B) The frequency of the induced Treg cells from CD4+CD25− T cells was compared with that of the pre-existing naturally occurring Treg cells in autologous CD4+ T cells. Representative plots of the frequency of Fr. I and Fr. II Treg cells among CD4+ T cells and in the co-culture of MϕSNU899 with CD4+CD25− T cells. (C) Statistical analysis showed that the percentage of Fr. II aTreg cells was markedly increased in the co-culture of MϕSNU899 with CD4+CD25− T cells (mean±s.d., n=3; **P<0.01 compared with CD4+ T cells). (D) Representative plots of the frequency of Fr. II aTreg cells assessed after 4 days of co-culture with or without the indicated neutralising mAbs. (E) A histogram revealed that the increase of Fr. II aTreg cells was partially blocked by an anti-IL-10 antibody (mean±s.d., n=3; *P<0.05). (A–E) I, CD45RA+Foxp3low resting Treg (rTreg) cells; II, CD45RA−Foxp3high activated Treg (aTreg) cells; III, CD45RA−Foxp3lowCD4+ T cells. Both of Fr. I and Fr. II were immunosuppressive, and Fr. III was non-suppressive. TSNs, tumour culture supernatants.
To determine the nature of the soluble factors involved in the induction of aTreg cells, neutralising antibodies against transforming growth factor (TGF)-β1, IL-10, or TGF-β1/ IL-10 were added to the cocultures. Our results showed that neutralising IL-10 partially attenuated the typical increase in aTreg cell frequency resulting from co-culture of MϕSNU899 with CD4+CD25− T cells, whereas blocking TGF-β1 had no significant effect (Figure 3D and E). Therefore, IL-10 attributed to MϕSNU899 was involved in the induction of aTreg cells from CD4+CD25− T cells.
aTreg cells skew the differentiation of monocytes toward M2 macrophages in the TSN
To investigate whether the MϕSNU899-induced Treg subsets skew the differentiation of monocytes to M2-like macrophages, CD14+ monocytes were cultured alone, with CD4+CD25− T cells, or with each sorted Treg subset (n=5). An anti-CD3 monoclonal antibody (mAb) was present in all conditions to stimulate the T cells. After 40 h, cells were collected for flow cytometry. Interestingly, compared with control monocytes, monocytes co-cultured with aTreg cells, but not CD45RA+Foxp3lowCD4+ resting Treg (rTreg) cells or CD45RA−Foxp3lowCD4+ T cells, displayed strong up-regulation of the typical M2 macrophage marker CD163. In addition, reduced expression of HLA-DR, CD86, and CD80 was observed (Figure 4A). The differences in the modulation of monocyte/macrophage functions may lie in the distinct cytokine patterns of these Treg subsets. Of note is that the aTreg subset was the highest producer of IL-10, CD45RA−Foxp3lowCD4+ T cells produced high amounts of IL-4, IL-13, and IFN-γ, while the rTreg subset was a poor producer of these cytokines (Figure 4B and C). To validate the cytokine effect on the aTreg cell-M2 macrophage induction, neutralising antibodies against IL-10, IL-4/IL-13, or IL-10/IL-4/IL-13 were added to the co-cultures. Unsurprisingly, neutralising IL-10 was sufficient to reverse the upregulation of CD163 and the down-regulation of HLA-DR (data not shown), whereas neutralising IL-4/IL-13 had no effect (Figure 4D).

aTreg cells skew the differentiation of monocytes partly mediated via IL-10 towards M2 macrophages in the TSN. (A) Monocytes were cultured without T cells, with CD4+CD25− T cells, or with each sorted Treg subset in the presence of an anti-CD3 mAb (50 ng ml−l). Monocyte phenotypes were assessed after 40 h of culture by flow cytometry (average mean fluorescence intensity, MFI±s.e.m., n=5; P<0.05; Δ, no T vs CD25−; Θ, no T vs I/II/III; #, CD25− vs I/II/III). (B) Production of IL-4, IL-10, IL-13, and IFN-γ by CD4+CD25− T cells and each Treg subset after stimulation with PMA+ionomycin. The percentage of cytokine-secreting cells in each cell population is shown. Data are representative of three separate experiments. (C) The histogram represents the cytokine expression profiles of CD25− T cells and each Treg subset (means±s.d., n=3). (D) Monocytes and Fr. II aTreg cells were co-cultured with or without neutralising mAbs against IL-10, IL-4/IL-13, or IL-10/IL-4/IL-13. CD163 expression was assessed by flow cytometry at 40 h after the start of the co-culture (MFI±s.e.m., n=3; **P<0.01). (E) Monocytes were cultured for 40 h without or with Fr. II aTreg cells in the absence or presence of TSN from the SNU899 laryngeal carcinoma cell line. Monocytes cultured with medium alone were set as the control. CD163 and HLA-DR expression is shown. (MFI±s.e.m., n=3; *P<0.05, **P<0.01). (A–E) I, CD45RA+Foxp3low resting Treg (rTreg) cells; II, CD45RA−Foxp3high activated Treg (aTreg) cells; III, CD45RA−Foxp3lowCD4+ T cells. TSNs, tumour culture supernatants.
Next, to examine this aTreg-mediated effect on monocyte/macrophage functions in a tumour context, we added SNU899 cell-derived TSNs to the co-cultures. Interestingly, CD163 expression was increased further and accompanied by a further decline in the HLA-DR level. As a control, TSN alone could only induce a very small increase in the expression of CD163 and a marginal decrease in the HLA-DR level (Figure 4E). These data imply that the tumour cell induced soluble microenvironment may enhance the ability of aTreg cells to convert monocytes into M2 macrophages.
The positive-feedback loop between tumour-infiltrating aTreg cells and M2 macrophages contributes to immunosuppression
Because TSN-exposed M2-like macrophages induced aTreg cells that in turn promoted induction of M2 macrophages in vitro, we investigated the feedback relationship between aTreg cells and M2 macrophages in vivo. After administration of a C–C chemokine receptor type 4 (CCR4) antagonist to specifically block tumour recruitment of aTreg cells, we observed that the percentage of tumour-infiltrating M2 macrophages was decreased by 27.17% (Figures 5Ab and Bb; 61.1±4.10% in control vs 44.50±3.54% in CCR4 antagonist, n=5, paired t-test, P<0.05). In turn, treatment with gadolinium chloride (GdCl3) to deplete tissue macrophages attenuated the proportion of aTreg cells by 28.20% (Figures 5Aa and Ba; 30.85±1.91% in control vs 22.15±2.05% in GdCl3, n=5, paired t-test, P<0.05), suggesting mutually dependent decreases or increases of aTreg cells and M2 macrophages in tumours.

Combined targeting of aTreg cells and M2 macrophages has a superior effect on deregulating the two intratumoural populations and significantly increasing antitumour immunity in mice. (A) Representative plots of the frequency of aTreg cells (a), M2 macrophages (b), and CD4+ and CD8+ T cells (c) in the tumour masses of the control, CCR4 antagonist-, GdCl3-, and CCR4 antagonist+GdCl3-treated groups. (B) Comparison of the percentage of aTreg cells (a), M2 macrophages (b), and CD4+ and CD8+ T cells (c and d) in the tumour tissues of the various groups. (C) Production of IFN-γ, IL-2, and TNF-α by intratumoural CD4+ T cells was detected by intracellular staining. Representative data of each cytokine are shown (a–c). (D) The frequency of Th1 cytokine (IFN-γ, IL-2, and TNF-α)-positive cells was calculated as the percentage of CD4+ T cells. Histograms show the frequencies of each Th1 cell subset in the four groups (a–c). Data in B and D represent the mean±s.d. (n=5). *P<0.05, **P<0.01 compared with the control. Statistical comparisons were performed using the Student’s t-test. M2, type 2 macrophages.
Both aTreg cells and M2 macrophages have a key role in subversion of anti-tumour immunity. We found that targeting aTreg cells or M2 macrophages resulted in expansion of CD4+ and CD8+ T cells in tumours (Figures 5Ac, Bc and Bd). Moreover, T cells in CCR4 antagonist- and GdCl3-treated groups secreted higher amounts of IFN-γ, IL-2, and TNF-α than T cells in the control group (Figure 5C and D), which is consistent with previous reports (Noy and Pollard, 2014; Sun et al, 2016). Therefore, the positive-feedback loop between aTreg cells and M2 macrophages by promoting induction of each cell type may significantly contribute to immunosuppression in the TME.
Combined targeting aTreg cells and M2 macrophages hampers their positive-feedback loop and evokes a potent antitumour immune response in vivo
This aTreg cell-M2 macrophage loop by enhancing tumour-induced immunosuppression represents a barrier to successful immunotherapy. Therefore, combined targeting of the generation of the two suppressive cell populations is a desirable goal in chemo- and immuno-therapeutic approaches. We first observed that tumour growth was significantly inhibited and the survival of mice was significantly prolonged in the CCR4 antagonist+GdCl3-treated group compared with the PBS control group and single-treated groups (Figure 6A–C; Supplementary Table S4). The weight of tumours, which were excised at 28 days after tumour cell injection, was significantly lower in the combined treatment group than in other groups (Figure 6D and E; Supplementary Table S4), whereas spleen weights were higher in this group (Figure 6F; Supplementary Table S4). These results suggest the effectiveness and practicability of this combined strategy targeting both aTreg cells and M2 macrophages.

Blockade of aTreg cell trafficking combined with depletion of M2 macrophages exerts a marked inhibitive effect on tumour growth. (A) Representative photographs of tumour-bearing mice in control, CCR4 antagonist-, GdCl3- and CCR4 antagonist+GdCl3-treated groups at 4 weeks after tumour cell injection. (B) Tumour sizes were measured every 4 days after tumour cell injection. The tumour volume was calculated using the formula: width2 × length × 0.5 (length>width). *P<0.05, **P<0.01. (C) Survival is depicted as a percentage of surviving mice on the indicated day after tumour cell injection. Statistical comparisons were performed using the Kaplan–Meier method. n=6; *P<0.05, **P<0.01. (D and E) Four weeks after tumour cell injection, the tumour mass was excised and its weight was measured. *P<0.05, **P<0.01. (F) Comparison of the mean spleen weight among groups assessed at 4 weeks after tumour cell injection. *P<0.05. (G and H) Immunohistochemical staining (original magnification: × 200) of the tumour mass in the four groups (n=5 for each group) using an anti-CD3 mAb. The number of T cells was manually counted and is presented as the ratio of T cells to non-T cells including tumour cells. Data are the mean±s.d. of CD3 ratios. *P<0.05, **P<0.01. (I) The frequency of CD4+ or CD8+ T cells was determined by flow cytometry and calculated as the percentage of CD4+ or CD8+ cells in splenocytes from mice with no statistical significance among the four groups (P>0.05).
To investigate whether the inhibition of tumour growth in the combined treatment group was associated with T-cell-mediated antitumour immunity, we analysed the composition of T cells that had infiltrated into the tumour tissues of mice. Immunohistochemical staining and flow cytometric analysis showed that the frequencies of CD3+, CD4+, and CD8+ T cells and Th1 cells were significantly higher in tumour tissues of the combined treatment group than in those of the other groups (Figure 6G and H and Figure 5Ac, Bc–d, C and D). These results indicated that the CCR4 antagonist+GdCl3 treatment evoked the most potent T-cell responses in the TME. Notably, this treatment had no obvious effect on T cells in the spleen (Figure 6I), which supports its potential therapeutic implications.
We hypothesised that the combined treatment targeting aTreg cells and M2 macrophages hampers their positive-feedback loop inducing each other, leading to enhanced T-cell populations (Supplementary Figure S2). Our results showed that, after the combined treatment, the frequency of tumour-infiltrating aTreg cells was lower than that induced by blockage of aTreg cells alone (Figures 5Aa and Ba; 9.85±2.05% vs 17.30±3.54%), while the M2 macrophage content was also decreased to 21.5±2.12% from 37.80±2.55% of single M2 macrophage depletion (Figures 5Ab and Bb), indicating an effect on the feedback loop. Therefore, this combined strategy reduces suppressor cells to the least extent by targeting them directly and interfering with their interactions in the TME.
Discussion
In this study, we show that M2-polarised macrophages in the tumour environment promote the differentiation of CD4+CD25−T cells into aTreg cells. In turn, these generated aTreg cells skew the differentiation of monocytes toward M2 macrophages, forming a positive-feedback loop. This M2 macrophage-aTreg cell loop contributes to immunosuppression and is associated to advanced clinical stage and poor survival in LSCC patients.
Tumour-infiltrating Treg cells promote local tumour growth by exerting immunosuppressive effects against tumour-associated antigen (TAA) T-cell responses (Curiel et al, 2004; Sato et al, 2005; Khazaie and von Boehmer, 2006; Zou, 2006; Liu et al, 2009). Yet Treg cells have been reported to be both negative and positive prognostic factors for cancers (Curiel et al, 2004; Wolf et al, 2005; Badoual et al, 2006; Hiraoka et al, 2006; Fu et al, 2007; Frey et al, 2010; Bron et al, 2013; Jie et al, 2015; Kindt et al, 2017). This contradiction regarding the role of Treg cells in cancer prognosis might because Treg cells in humans are functionally heterogeneous, and include suppressive and non-suppressive cell subsets (Miyara et al, 2009; DeLeeuw et al, 2012). Tumour-infiltrating suppressive aTreg cells were proved to be the real Treg subset controlling the poor prognosis of cancers (Sun et al, 2014, 2015, 2016; Saito et al, 2016). Recently, to evoke antitumour immunity in vivo, we blocked tumour trafficking of aTreg cells using a CCR4 antagonist, which led to inhibition of tumour growth and prolonged survival (Sun et al, 2016). However, the effect was not satisfactory, indicating that a single target in the treatment of HNSCC has a limitation.
Macrophages are a major component of the leukocyte infiltrate in tumours and considered to be another crucial mediator of tumour immunity (Balkwill and Mantovani, 2001; Qian and Pollard, 2010; Noy and Pollard, 2014). Different types of macrophages, including M1 and M2, are of high scientific importance for subtyping. M1 macrophages are immunostimulatory and anti-tumoural, while M2 macrophages are immunosuppressive and pro-tumoural. Thus far, studies have been mainly based on whole intratumoural macrophages and have not reached a consensus on their effects on tumours. In the thyroid, lungs, oral cavity, and hepatocellular cancers, an increased macrophage density is associated with poor survival (Marcus et al, 2004; Chen et al, 2005; Ryder et al, 2008; Zhu et al, 2008). However, in pancreatic and gastric cancers, high intratumoural macrophages correlate with increased survival (Kim et al, 2008; Wang et al, 2011). The causes of such conflicting results may lie in the fact that whole macrophages were taken into account and not the subsets of macrophages, especially M2, because the phenotype of macrophages can vary in different tumour types. In our study, the tumour-infiltrating macrophages in LSCC tissues were a distinct M2-polarised population and negatively associated with overall survival.
Different types of malignant tumours, including HNSCC, have various abilities to polarise macrophages, which is characterised by the expression of M2 macrophage surface markers and the ability to secrete M2 cytokines. Our study showed that, compared with HNSCC cell lines SNU46 and HONE1, laryngeal carcinoma cell line SNU899-derived TSNs had the most significant influence on the differentiation of monocytes toward M2 macrophages, resulting in the highest expression of M2-specific marker and cytokine levels. The varied degree of polarisation may lead to a difference in the function of macrophages, such as their interaction with lymphocytes (Biswas and Mantovani, 2010).
Our study demonstrated that the high intratumoural M2 macrophage density was positively correlated with the elevated intratumoural Foxp3+ Treg population. Accumulation of M2 macrophages and/or Foxp3+ Treg cells in LSCC tissue is associated to advanced T stage and poor survival. We hypothesised that these cells cooperate with each other to enhance immunosuppression in tumours. Our in vitro results revealed that TSN-exposed macrophages with the most typical M2 features had the strongest ability to induce Foxp3+ Treg cells by acting on responder PBMCs. To identify whether the induced Tregs were converted from CD4+CD25−T cells, rather than expansion of previous Tregs in the culture system, we replaced the responder cells with CD4+CD25−T cells and observed clear upregulation of Treg cell surface antigens by flow cytometry, thereby confirming their interactions. Previous study has demonstrated that Foxp3+ Treg cells are composed of three phenotypically and functionally distinct subpopulations: CD45RA+Foxp3low rTreg cells and CD45RA−Foxp3high aTreg cells, both of which are suppressive in vitro, and cytokine-secreting CD45RA−Foxp3 low nonsuppressive T cells (Miyara et al, 2009). Based on this classification of human Tregs, our study provided evidence to support the notion of heterogeneous Treg subsets in HNSCC patients (Sun et al, 2014, 2015, 2016, ). We showed that aTreg cells, as the predominant cell population among tumour-infiltrating Foxp3+ Treg cells, accelerate disease progression by suppressing TAA immunity in patients with HNSCC (Sun et al, 2016; Wei et al, 2016). To determine whether TSN-exposed M2-like macrophages contribute to an increase in aTreg cells, we analysed the subsets of induced Treg cells. Interestingly, the aTreg cell frequency was dominantly increased in the suppressive Treg-cell population, indicating that the Treg cells induced by M2-like macrophages can directly mediate an inhibitory effect on tumour immunity.
Conversely, to examine the effect of aTreg cells on macrophage functions in the context of tumour-induced immunosuppression, we added TSNs to the co-culture system to mimic the tumour microenvironment (TME). Interestingly, aTreg cell-affected monocytes expressed significantly higher levels of an M2-specific marker in the TSNs compared with the medium alone, wheresas single TSNs had only a marginal effect on M2 marker induction within the specific time. These results indicated that TSNs have a good promotion effect on M2 macrophage induction by aTreg cells. The reverse process connected the formation of M2 macrophages and aTreg cells to a positive-feedback loop that may contribute to an increase of their number in tumours. Therefore, this biological mechanism can serve as an explanation for the positive correlation between aTreg cells and M2 macrophages in LSCC tissue.
To validate the positive-feedback relationship between M2 macrophages and aTreg cells and its importance in vivo, we targeted the two populations individually in a tumour-bearing mouse model. As a result, depletion of intratumoural M2 macrophages simultaneously attenuated the intratumoural aTreg cell frequency, whereas blockage of aTreg cell tumour trafficking indirectly decreased the percentage of intratumoural M2 macrophages. These data indicate that it is imperative to further investigate combined strategies to interfere with both M2 macrophages and aTreg cells in the TME.
Indeed, targeting M2 macrophages and aTreg cells simultaneously had a superior effect on regulating the two cell populations. Both aTreg cells and M2 macrophages have a key role in subversion of anti-tumour immunity. The former can suppress TAA-T-cell responses by cell contact, while the latter have a poor antigen-presenting capacity and suppress T-cell activation and proliferation by releasing prostaglandins, IL-10, and TGF-β (Kambayashi et al, 1995; Maeda et al, 1995; Loercher et al, 1999; Sakaguchi et al, 2009). In mice, we found that the combined strategy was more efficient than single targeting for inhibition of tumour growth and prolongation of survival. The efficiency is derived from deregulation of antitumour immunity because the proportions of CD4+ and CD8+ T cells and Th1 cells were increased significantly. Furthermore, we found that this strategy had no significant influence on the peripheral immune status, thus indicating greater potential therapeutic implications.
This combined targeting strategy, based on the theory of the M2 macrophage-aTreg cell positive-feedback loop, may offer new approaches to overcome the barriers of immunosuppression in the TME. Various options can be provided for the specific therapeutic program, including blocking tumour trafficking of macrophages/Treg cells using CSF1R/CCR4 antagonists, respectively (Sugiyama et al, 2013; Ries et al, 2014; Kurose et al, 2015), ligation of GITR to down-regulate Foxp3 expression and reduce production of IL-10 by Treg cells (Schaer et al, 2013), and re-educating M2 macrophages to an M1 phenotype using Toll-like receptor 3 ligands or STING agonists (Shime et al, 2012; Downey et al, 2014; Baird et al, 2016). A rational combination of the above methods may exert an unexpected antitumour effect in LSCC treatment.
In summary, our study suggests an important role of the positive-feedback loop between tumour-infiltrating M2 macrophages and aTreg cells in tumour progression. This positive-feedback loop is essential to maintain or promote immunosuppression in the TME and could be a potential therapeutic target against tumour progression.
Change history
21 November 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alhamarneh O, Amarnath SM, Stafford ND, Greenman J (2008) Regulatory T cells: what role do they play in antitumor immunity in patients with head and neck cancer? Head Neck 30: 251–261.
Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay NE, Mosseri V, Laccourreye O, Bruneval P, Fridman WH (2006) Prognostic value of tumor-infiltrating CD4(+) T-cell subpopulations in head and neck cancers. Clin Cancer Res 12: 465–472.
Baird JR, Friedman D, Cottam B, Dubensky TJ, Kanne DB, Bambina S, Bahjat K, Crittenden MR, Gough MJ (2016) Radiotherapy combined with Novel STING-targeting oligonucleotides results in regression of established tumors. Cancer Res 76: 50–61.
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357: 539–545.
Begum S, Westra WH (2008) Basaloid squamous cell carcinoma of the head and neck is a mixed variant that can be further resolved by HPV status. Am J Surg Pathol 32: 1044–1050.
Biswas SK, Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11: 889–896.
Bron L, Jandus C, Andrejevic-Blant S, Speiser DE, Monnier P, Romero P, Rivals JP (2013) Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int J Cancer 132: E85–E93.
Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC (2005) Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol 23: 953–964.
Curiel TJ, Coukos G, Zou LH, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10: 942–949.
DeLeeuw RJ, Kost SE, Kakal JA, Nelson BH (2012) The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res 18: 3022–3029.
Downey CM, Aghaei M, Schwendener RA, Jirik FR (2014) DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2'3'-cGAMP, induces M2 macrophage repolarization. PLoS One 9: e99988.
Ferris RL (2015) Immunology and immunotherapy of head and neck cancer. J Clin Oncol 33: 3293–3304.
Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, Oertli D, Kettelhack C, Terracciano L, Tornillo L (2010) High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer 126: 2635–2643.
Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, Yao J, Jin L, Wang H, Yang Y, Fu YX, Wang FS (2007) Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 132: 2328–2339.
Hiraoka N, Onozato K, Kosuge T, Hirohashi S (2006) Prevalence of FOXP3(+) regulatory T cells increases during the progression of pancreatic ductal adenocarcinorna and its premalignant lesions. Clin Cancer Res 12: 5423–5434.
Jiang W, Sun R, Wei HM, Tian ZG (2005) Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of toll-like receptor 4 expression on macrophages. Proc Natl Acad Sci USA 102: 17077–17082.
Jie H, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, Whiteside TL, Ferris RL (2015) CTLA-4(+) Regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress nk cell cytotoxicity and correlate with poor prognosis. Cancer Res 75: 2200–2210.
Kambayashi T, Alexander HR, Fong M, Strassmann G (1995) Potential involvement of IL-10 in suppressing tumor-associated macrophages. Colon-26-derived prostaglandin E2 inhibits TNF-alpha release via a mechanism involving IL-10. J Immunol 154: 3383–3390.
Khazaie K, von Boehmer H (2006) The impact of CD4+CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin Cancer Biol 16: 124–136.
Kim DW, Min HS, Lee KH, Kim YJ, Oh DY, Jeon YK, Lee SH, Im SA, Chung DH, Kim YT, Kim TY, Bang YJ, Sung SW, Kim JH, Heo DS (2008) High tumour islet macrophage infiltration correlates with improved patient survival but not with EGFR mutations, gene copy number or protein expression in resected non-small cell lung cancer. Br J Cancer 98: 1118–1124.
Kindt N, Descamps G, Seminerio I, Bellier J, Lechien JR, Mat Q, Pottier C, Delvenne P, Journe F, Saussez S (2017) High stromal Foxp3-positive T cell number combined to tumor stage improved prognosis in head and neck squamous cell carcinoma. Oral Oncol 67: 183–191.
Kuang DM, Wu Y, Chen N, Cheng JS, Zhuang SM, Zheng LM (2007) Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 110: 587–595.
Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, Doi T, Suzuki S, Isobe M, Funakoshi T, Kakimi K, Nishikawa H, Udono H, Oka M, Ueda R, Nakayama E (2015) Phase Ia Study of FoxP3+ CD4 treg depletion by infusion of a humanized anti-CCR4 Antibody, KW-0761, in cancer patients. Clin Cancer Res 21: 4327–4336.
Li Q, Prince MEP, Moyer JS (2015) Immunotherapy for head and neck squamous cell carcinoma. Oral Oncol 51: 299–304.
Liu Z, Kim JH, Falo LJ, You Z (2009) Tumor regulatory T cells potently abrogate antitumor immunity. J Immunol 182: 6160–6167.
Loercher AE, Nash MA, Kavanagh JJ, Platsoucas CD, Freedman RS (1999) Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J Immunol 163: 6251–6260.
Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A (1995) TGF-beta enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol 155: 4926–4932.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555.
Mantovani A, Locati M (2013) Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: lessons and open questions. Arterioscl Throm Vasc Biol 33: 1478–1483.
Marcus B, Arenberg D, Lee J, Kleer C, Chepeha DB, Schmalbach CE, Islam M, Paul S, Pan Q, Hanash S, Kuick R, Merajver SD, Teknos TN (2004) Prognostic factors in oral cavity and oropharyngeal squamous cell carcinoma–The impact of tumor-associated macrophages. Cancer 101: 2779–2787.
Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D (2009) Functional Delineation and Differentiation Dynamics of Human CD4(+) T Cells Expressing the FoxP3 Transcription Factor. Immunity 30: 899–911.
Noy R, Pollard JW (2014) Tumor-associated macrophages: from mechanisms to therapy. Immunity 41: 49–61.
Qian BZ, Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141: 39–51.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Rüttinger D (2014) Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25: 846–859.
Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA (2008) Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer 15: 1069–1074.
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, Maeda Y, Hamaguchi M, Ohkura N, Sato E, Nagase H, Nishimura J, Yamamoto H, Takiguchi S, Tanoue T, Suda W, Morita H, Hattori M, Honda K, Mori M, Doki Y, Sakaguchi S (2016) Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 22: 679–684.
Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T (2009) Regulatory T cells: how do they suppress immune responses? Int Immunol 21: 1105–1111.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen YT, Ohtani H, Old LJ, Odunsi K (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102: 18538–18543.
Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, Zhong H, Yang X, Houghton AN, Merghoub T, Wolchok JD (2013) GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res 1: 320–331.
Schuler PJ, Harasymczuk M, Visus C, Deleo A, Trivedi S, Lei Y, Argiris A, Gooding W, Butterfield LH, Whiteside TL, Ferris RL (2014) Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin Cancer Res 20: 2433–2444.
Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, Tahara H, Inoue N, Seya T (2012) Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci USA 109: 2066–2071.
Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, Karbach J, Jäger E, Sakaguchi S (2013) Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA 110: 17945–17950.
Sun W, Li WJ, Fu QL, Wu CY, Lin JZ, Zhu XL, Hou WJ, Wei Y, Wen YH, Wang YJ (2015) Functionally distinct subsets of CD4(+) regulatory T cells in patients with laryngeal squamous cell carcinoma are indicative of immune deregulation and disease progression. Oncol Rep 33: 354–362.
Sun W, Li WJ, Wei FQ, Wong TS, Lei WB, Zhu XL, Li J, Wen WP (2016) Blockade of MCP-1/CCR4 signaling-induced recruitment of activated regulatory cells evokes an antitumor immune response in head and neck squamous cell carcinoma. Oncotarget 7: 37714–37727.
Sun W, Li WJ, Wu CY, Zhong H, Wen WP (2014) CD45RA(-)Foxp3(high) but not CD45RA(+) Foxp3(low) suppressive T regulatory cells increased in the peripheral circulation of patients with head and neck squamous cell carcinoma and correlated with tumor progression. J Exp Clin Canc Res 33: 35.
Wang B, Xu DZ, Yu XJ, Ding T, Rao HL, Zhan YQ, Zheng LM, Li L (2011) Association of intra-tumoral infiltrating macrophages and regulatory t cells is an independent prognostic factor in gastric cancer after radical resection. Ann Surg Oncol 18: 2585–2593.
Wei FQ, Sun W, Wong TS, Gao W, Wen YH, Wei JW, Wei Y, Wen WP (2016) Eliciting cytotoxic T lymphocytes against human laryngeal cancer-derived antigens: evaluation of dendritic cells pulsed with a heat-treated tumor lysate and other antigen-loading strategies for dendritic-cell-based vaccination. J Exp Clin Cancer Res 35: 18.
Wolf D, Wolf AM, Rumpold H, Fiegl H, Zeimet AG, Muller-Holzner E, Deibl M, Gastl G, Gunsilius E, Marth C (2005) The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res 11: 8326–8331.
Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, Wu WZ, Wang L, Tang ZY, Sun HC (2008) High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol 26: 2707–2716.
Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6: 295–307.
Acknowledgements
This work was supported by grants from the Natural Science Foundation of Guangdong Province (2016A030310153, 2014A030313031, and 2016A030313257), the Science and Technology Planning Project of Guangdong Province (2014A020212141), the Natural Science Foundation of China (81602365, 81670902, and 81470674), the Medical Scientific Research Foundation of Guangdong Province (A2017216), and the Guangzhou Key Laboratory of Otorhinolaryngology Head and Neck Surgery (201605030003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Sun, W., Wei, FQ., Li, WJ. et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br J Cancer 117, 1631–1643 (2017). https://doi.org/10.1038/bjc.2017.329
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.329
Keywords
This article is cited by
-
The paradoxical role of cytokines and chemokines at the tumor microenvironment: a comprehensive review
European Journal of Medical Research (2024)
-
Selective targeting or reprogramming of intra-tumoral Tregs
Medical Oncology (2024)
-
Targeting tumor-associated macrophages for cancer treatment
Cell & Bioscience (2022)
-
Metabolic guidance and stress in tumors modulate antigen-presenting cells
Oncogenesis (2022)
-
Immunologically “cold” triple negative breast cancers engraft at a higher rate in patient derived xenografts
npj Breast Cancer (2022)

{kind=link}
{kind=link}