
Abstract
Knowledge of the nanoscale changes that take place in individual cells in response to a drug is useful for understanding the drug action. However, due to the lack of adequate techniques, such knowledge was scarce until the advent of atomic force microscopy (AFM), which is a multifunctional tool for investigating cellular behavior with nanometer resolution under near-physiological conditions. In the past decade, researchers have applied AFM to monitor the morphological and mechanical dynamics of individual cells following drug stimulation, yielding considerable novel insight into how the drug molecules affect an individual cell at the nanoscale. In this article we summarize the representative applications of AFM in characterization of drug actions on cell membrane, including topographic imaging, elasticity measurements, molecular interaction quantification, native membrane protein imaging and manipulation, etc. The challenges that are hampering the further development of AFM for studies of cellular activities are aslo discussed.
Similar content being viewed by others

Introduction
The pharmaceutical industry is currently facing unprecedented challenges, primarily due to the increasing cost of developing new drugs, the sharply rising attrition rates and the dissipation of proprietary products1,2,3. Therefore, innovations are urgently needed for promoting the development of the pharmaceutical industry4. Modern drug discovery is primarily based on the search for drug leads against a preselected therapeutic target followed by subsequent testing of the derived drug candidates5. Approximately half of the approved drugs target human membrane proteins on the cell surface6, such as G-protein coupled receptors (GPCRs), ion channels and antigen molecules7. In the process of drug discovery (eg, target validation, lead optimization and preclinical testing), researchers perform complex experiments to demonstrate the efficacy and safety of a candidate drug by using various biochemical approaches (eg, Western blotting, X-ray crystallography and radioimmunoassay). Although valuable, these assays provide averaged information obtained on large ensembles of molecules contained in many cells8, thereby masking the rare events that take place in single cells. Recent studies have shown that even when derived from a seemingly homogenous population, individual cells can exhibit substantial differences in their gene expression, protein levels, phenotypic output and function9. It has become increasingly apparent that obtaining information at the single-cell level is of central importance for revealing the novel regulatory mechanisms of diverse cellular processes10,11. Single-cell approaches also provide opportunities to investigate many important biological processes that cannot be assessed by ensemble experiments, such as mono-allelic gene expression, lineage tracing during cell differentiation and biofilm formation in bacteria12. Therefore, it can be reasonably anticipated that advances in single-cell techniques will help the pharmaceutical industry to partly overcome its current challenges. However, for potential practical application in drug discovery, single-cell techniques that can rapidly and automatically obtain the multiple parameters of individual living cells with high spatial resolution are needed.
The invention of atomic force microscopy (AFM) has provided an excellent means for observing, manipulating and exploring the functional components of single living cells with nanometer resolution13. Compared with the commonly used biochemical methods for assessing drugs, AFM has clear advantages. For example, optical microscopy cannot visualize nanoscale cellular structures and electron microscopy requires that cells be fixed and dried. By contrast, AFM can be used to observe living cells in aqueous solutions with nanometer resolution, so that the dynamic changes of cellular ultra-microstructures on single cells in response to drugs can be monitored by time-lapse AFM14. Additionally, the drawback of Western blotting, polymerase chain reaction and X-ray crystallography is that they require the extraction of target molecules from cells, which destroys the intrinsic structures of the cells. By contrast, AFM can image the integral membrane proteins in their native membrane environment at submolecular resolution15, allowing the direct observation of drug-induced morphological changes in target molecules16,17. Traditional methods (eg, surface plasma resonance, radioimmunoassay) that quantify the binding constant of a drug to its target molecule cannot reveal the molecular dynamics taking place during the association and dissociation processes of individual drug-target interactions18. By contrast, AFM can directly reveal the detailed dynamics of the dissociation process (eg, how many energy barriers are overcome during dissociation) by simply pulling a drug molecule from its target molecule with different pulling rates19,20; therefore, there is no need to model the dynamic processes of drug-target interactions using complex molecular dynamics simulations21. In addition, recent advances (eg, high-speed AFM22, peak force tapping AFM23) in AFM have increased the power of the technique. Currently, AFM can simultaneously obtain multiple physicochemical parameters (such as morphology, elasticity, adhesion, deformation and energy dissipation)23 of biological systems in a relatively short amount of time (approximately several minutes), allowing the nanoscale structures, properties and functions of cells/targets in response to drugs to be correlated.
In the past decade, AFM has emerged as a valuable addition to the pharmaceutical industry's toolkit24 and has been demonstrated to be useful in assisting drug evaluations and assays (eg, drug-target interactions25, target molecules26,27 and drug delivery system characterizations28,29). The applications of AFM in pharmacology have provided novel fundamental insight for understanding drug actions30. The increased knowledge obtained through AFM will be significant for making improvements to the current drug discovery process31. Here, based on the authors' own research on AFM investigations of cell-drug interactions, the principle of AFM imaging and representative quantitative measurements will be first presented, followed by a summary of the typical applications (eg, topographic imaging, elasticity measurements, molecular interaction quantification, native membrane protein imaging and manipulation) of AFM in assessing drug actions. Finally, the current challenges and future directions of this field will be discussed.
AFM imaging and measurements: principle and methods
The main components of an AFM include a piezoelectric tube driver, which is connected to a flexible cantilever with a sharp tip mounted at its end, a laser photodiode detector and feedback electronics, as shown in Figure 1A. High-resolution imaging of the topography of active biological samples (eg, living cells, native molecules) is the most important application of AFM in the life sciences. The two modes of AFM commonly used are contact mode and tapping mode. In contact mode (Figure 1Ai), the tip scans over the sample surface while changes in the cantilever deflection are monitored by the photodiode detector (the detector monitors a beam of laser light reflected from the backside of the cantilever). The cantilever is driven vertically by the piezoelectric tube to maintain a constant deflection according to the feedback electronics, thereby maintaining a constant loading force on the sample. In tapping mode (Figure 1Aii), the cantilever oscillates near its resonance frequency and is scanned over the sample surface. In liquid, the driving frequency of the cantilever is often approximately 10 kHz. The piezoelectric tube drives the cantilever to move vertically to maintain a set constant amplitude according to the feedback electronics. Presently, contact mode is the more widely used imaging mode32. Contact mode is particularly suitable for obtaining high-resolution images from living adherent cell imaging33,34,35 and native protein imaging15,16,17,36,37,38. A notable point regarding cell imaging using contact mode is that the scan force between the tip and cells should be low enough to prevent sample damage. For suspended mammalian living cells, however, tapping mode is adequate35,39,40, because suspended mammalian cells cannot spread on the substrate and have soft surfaces. Tapping mode can significantly diminish the influence of lateral forces during the scanning, so that there is no scraping of the sample. Moreover, tapping mode is suitable for some special applications, such as high-speed scanning41 and multiparametric imaging23. Under near-physiological conditions (eg, cell culture medium, buffered solution), the dynamic changes of cellular ultra-microstructures following the addition of drugs can be visualized to directly assess their influence on individual cells. By immobilizing cell membrane patches onto the supported substrate (eg, mica), the morphological changes of single target molecules in response to the drug molecules can also be investigated.

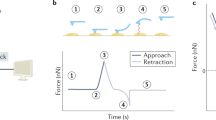
Using AFM to investigate the drug actions on single cells. (A) The main components of an AFM. Contact mode (i) and tapping mode (ii) are the two commonly used imaging modes. (B) Measuring the elastic properties of cells using AFM indenting technique. The cellular Young's modulus is calculated from the approach curve. (i) Optical and SEM images of a sphere probe49. (ii) Schematic diagram. (C) Measuring the binding force of a drug molecule to the target molecule by pulling a drug molecule from its target molecule54. The binding force is calculated from the retract curve which has a specific unbinding peak (denoted by the green arrow).
The elastic properties of samples are measured by controlling the AFM tip to indent the samples, as shown in Figure 1B. For measuring the elastic properties of cells, the tip performs approach-retract cycles on the cell surface. During the process, the deflection of the cantilever and the movement of the piezo tube are recorded to produce so-called force curves. Figure 1B shows a schematic diagram of a force curve obtained during the approach-retract cycles. A force curve contains two components: an approach curve and a retract curve. The approach curve is used to calculate the cellular Young's modulus42 because the retract curve is often influenced by tip-sample adhesions, making it difficult to discern the contact point of the latter. By contrast, the contact point of the approach curve can be clearly visualized (denoted in Figure 1B). The cantilever deflection is constant prior to when the tip contacts the cell, and when the tip begins to indent the cell, the shape of the approach curve becomes bent. In practice, the approach curve is first converted into an indentation curve according to the contact point, and then the Hertz-Sneddon model is applied to calculate the cell Young's modulus43. The Hertz-Sneddon model is based on the following expressions44:




where υtip and υcell are the Poisson ratios of the tip and cell, respectively, F is the loading force of tip, δ is the indentation depth, E is the effective Young's modulus, Etip is the Young's modulus of tip, Ecell is the Young's modulus of cell, θ is the half-opening angle of the tip, R is the effective radius, Rtip is the radius of tip and Rcell is the radius of cell. Because the Young's modulus of living cells is much smaller than that of the tip (Ecell<<Etip), formula (3) can be rewritten as:

Based on this approximation, formulas (1) and (2) can be recast as:


Living cells are often considered to be incompressible materials; therefore, the value of υcell is 0.5. According to Hooke's law (F=kx, where k is the spring constant of the cantilever), the loading force F can be calculated from the cantilever deflection x. After obtaining the loading force F and the indentation depth δ, the cellular Young's modulus Ecell can be calculated according to formulas (6) and (7). It should be noted that the Hertz-Sneddon model is based on a number of assumptions applied to the specimen to be indented, including homogeneity, isotropicity, linear elastic material properties, axisymmetry and infinitesimal deformation of the sample, as well as infinite sample thickness and a smooth sample surface43,45. Studies have shown that the Hertz-Sneddon model is appropriate as long as the indentation into the specimen is no more than 10% of the specimen thickness46. However, the indentations should be larger than 400 nm to avoid errors due to the uncertain determination of the contact point, which leads to measurements often being performed on the central region of the cell47. The Hertz-Sneddon model does not consider the forces between the contact surfaces (eg, electrostatic forces, adhesion forces and friction forces)42 and is therefore applicable when the forces between the contact surfaces are negligible compared with the maximum load. By contrast, the Johnson-Kendall-Roberts (JKR) model and Derjaguin-Muller-Toporov (DMT) model take into account the adhesion forces between the contact surfaces. The JKR model considers the adhesion forces inside the contact area whereas the DMT model considers the forces outside the contact area48. Researchers have shown that the JKR model can be applied in the case of large tips and soft samples with a large adhesion, whereas the DMT model is applicable in the case of small tips and stiff samples with a small adhesion48. The conventional cone tip is sharp and thus is prone to causing cell damage. To avoid this problem, a sphere can be glued to the cantilever (Figure 1Bi) and the spherical tip can be used to measure the cellular Young's modulus. A simple method to prepare a spherical tip is to attach a sphere to a tipless cantilever with epoxy resin glue via AFM manipulation under the guidance of optical microscopy49. Cell mechanics is closely related to cell states and can serve as a novel biomarker50. Therefore, an important application of AFM elasticity measurements in drug assays is the monitoring of the elastic changes of a single cell after the stimulation of drugs51,52,53 to assess the drug actions from the viewpoint of cell mechanics.
By linking drug molecules to the tip, AFM can probe the target molecules on the cell surface. This technique is called single-molecule force spectroscopy (SMFS)54, as shown in Figure 1C. By performing approach-retract cycles on the cell surface using drug-tethered tips, force curves can be recorded. If a drug molecule binds to a target molecule during the contact between the tip and cell, the drug-target complex is then pulled by the AFM tip during the retract process, leading to a non-linear unbinding peak on the retract curve (indicated by the green arrow in Figure 1C). The non-linear shape of the peak is caused by the stretching of flexible linker molecules, which have non-linear elasticity19. The magnitude of the peak is equal to the binding force between a target molecule and a drug molecule (Figure 1C). There are several methods for linking molecules onto AFM tips55,56, including physisorption, covalent coupling with organosilanes or ethanolamine hydrochloride and the functionalization of gold-coated tips. Using indolent polyethylene glycol (PEG) molecules to tether drug molecules onto the AFM tip is the most common strategy due to its many advantages, such as allowing the drug molecules to freely reorient with respect to the target molecules on the cell surface, preventing the danger of drug molecules being compressed when the tip contacts the cell surface, and enabling the clear identification of the specific binding according to the unbinding peak on the retract curve54. Traditional methods for characterizing the binding constant of drug molecules cannot reveal the dynamics taking place during the dissociation of a drug-target complex. However, because SMFS can reveal the dynamics taking place during the process of pulling drug molecules from targets cells, it enables better understanding of the drugs and is useful for drug design. We can thus conclude that AFM is a powerful method that can be used to obtain multiple physicochemical parameters, such as morphology, elasticity, and drug-target interactions, on single cells and native proteins under the stimulation of drugs. In the following sections, we will summarize the recent progress of AFM as applied to the monitoring of drug actions on membranes.
Dynamic changes of cellular ultra-microstructure and elasticity in response to drugs
The morphology of a cell is closely related to its functions. When a cell becomes pathological, its morphology changes. For example, the shape of normal red blood cells is an oval biconcave disk, whereas those of anemia patients become sickle-shaped57. Additionally, the morphology of a normal cell changes when it transforms into a cancerous one58. For example, the nuclear volume of cancer cells is often larger than those of normal cells and the nuclear contour of cancer cells is often irregular59. There is also difference in the cell surface brush between normal cells and cancer cells60. In fact, because of the significant morphological differences between normal cells and cancer cells, the primary method for diagnosing cancers in the clinic is based on the observation of stained pathological tissue samples58. Due to its 200 nm limitation in resolution, the traditional observation of cellular morphology using optical microscopy cannot reveal cellular nanostructures. Thus, the detailed morphological changes of cells after the stimulation of drugs cannot be explored with optical microscopy.
AFM opens the door to in situ investigations of the nanoscale morphological changes in single cells after drug stimulation. Antimicrobial peptides are a promising class of antimicrobials that have demonstrated activity against antibiotic-resistant bacteria, parasites, viruses and fungi14. In 2010, Fantner et al14 used AFM to image the dynamic nanoscale changes on single living bacterial cells in response to CM15 (an antimicrobial peptide drug), revealing that the cell surface changed from smooth to corrugated after the stimulation of CM15. Glyphosate is an extensively used herbicide that has been shown to increase the risk of cancer and quercetin is a well-known antioxidant61. In 2012, Heu et al61 investigated the effect of glyphosate and quercetin on single living keratinocyte cells by AFM peak force tapping mode, showing that cells develop a filamentous cytoskeleton network after the stimulation of glyphosate, whereas the filamentous structures disappeared after the subsequent addition of quercetin. Daunorubicin (Dau) is a drug used to treat several types of cancers, such as leukemia and neuroblastomas62. Dau intercalates in DNA to cause a local unwinding of the DNA helix. In 2013, Alonso-Sarduy et al62 applied time-lapse AFM to investigate the dynamic changes of DNA molecules in response to Dau in a physiological medium. The results clearly showed conformational changes in the DNA molecules after the addition of Dau. These studies have provided novel insights that are useful for understanding the drug actions and to assess drugs from the viewpoint of cellular nanostructures (eg, cell surface, cytoskeleton, DNA).
Studies in the past decade have demonstrated that cell mechanics play an important role in maintaining cellular physiological activities53,63. Cells sense external mechanical stimuli and convert them into intracellular chemical signals via proteins on the cell surface to regulate cellular physiological activities64. For example, in the process of cancer metastasis, circulating tumor cells (CTCs) must undergo large elastic deformations to penetrate endothelial cell-cell junctions as well as overcome the effect of fluid shear to adhere to the vascular endothelium of distant organs65. Researchers have shown that pathological changes can cause changes in cellular mechanical properties (such as changes in cell deformability and altered cytoadherence characteristics), which then result in changes in cellular physiological properties (such as protein expression and cell functions) and finally lead to the occurrence of cancer50. In 2012, Plodinec et al66 comprehensively investigated the stiffness of three types of biopsy samples (normal breast tissues, benign breast tissues and malignant breast tissues), and the results showed that the stiffness profiles of breast tissues changed remarkably during the development of breast cancer, providing quantitative indicators in the clinical diagnostics of cancer with translational significance. In recent years, there has been an increasing consensus that cell mechanics can serve as an effective label-free biomarker for the indication of cell states67,68.
Investigating drug-cell actions from the viewpoint of cell mechanics provides a novel way to evaluate drugs. The cytoskeleton is crucial for maintaining the cell shape and therefore some drugs, such as cytochalasin and taxol, influence the cytoskeleton to inhibit cell migration. In 2000, Rotsch et al51 investigated the effect of various drugs (cytochalasin, taxol, colchicine, and latrunculin) on fibroblasts by measuring the cell elasticity before and after the stimulation of drugs. The results showed that drugs (cytochalasin, latrunculin) that disaggregated the actin filaments could cause a distinct decrease in cell elastic modulus whereas drugs (taxol, colchicine) that stabilized microtubules could not significantly influence the cell elasticity. In 2007, Lam et al52 investigated the mechanical properties of leukemia cells (isolated from the blood of patients with diagnosed acute leukemia) in response to chemotherapy drugs (dexamethasone, daunorubicin), and the results showed that leukemia cell stiffness increased by nearly two orders of magnitude after exposure to dexamethasone or daunorubicin. N-methyl-D-aspartate (NMDA) specifically binds to the NMDA receptors in the cytomembrane that opens the ligand gated ion channel to facilitate Ca2+ influx into the cell53. In 2014, Fang et al53 investigated the effect of NMDA on SH-SY5Y human neuroblastoma cells from the viewpoint of cell elasticity, and the results showed that the cell Young's modulus significantly increased after the treatment of NMDA. These studies provide novel insight into the effects of drugs and also facilitate the development of new therapeutic strategies for combating diseases such as cancer. One important issue regarding measuring the elasticity of living cells using AFM is whether the consecutive probing by the tip itself causes the observed changes in cellular elasticity. Researchers have shown that prolonged poking (200 s) of the cell can lead to the remodeling of the cell cytoskeleton, and thus the probing time should be limited during the measurements to avoid this effect69. In practice, when we investigate the effect of external stimuli such as drugs on the cellular elasticity, control experiments (eg, monitoring changes in the cellular elasticity without the addition of drugs)51,53 are often required to determine the influence of tip poking on the cellular elasticity.
Rituximab is a monoclonal antibody that targets the CD20 antigen on the surface of B-cell non-Hodgkin lymphoma cells. One of the primary functions of rituximab is that it can activate the classical complement pathway to lyse the cancer cells; this effect is called complement-mediated cytotoxicity (CMC). We have investigated the dynamic changes of cellular ultra-microstructures and their mechanical properties during the CMC process70, as shown in Figure 2. A schematic diagram of the CMC process is illustrated in Figure 2A. The binding of rituximab to CD20 on a lymphoma cancer cell can activate the C1 complex, which then triggers the classical pathway by the cascade interactions of complement proteins (such as C2, C4, C3). The terminal product of CMC is the membrane attack complex (MAC) formed on the cell surface. Small molecules (eg, water, ion) can enter into the cells freely, which causes them to swell and burst. Traditional methods for investigating the CMC effect are based on optical microscopy, which cannot resolve the detailed events taking place on the cell surface during CMC. We therefore used AFM to image the nanoscale morphology of lymphoma cells that exhibited the CMC effect, as shown in Figure 2B–2J. In the early stage of CMC, some micro-holes appeared on the cell surface (denoted by the arrows in Figure 2C), which were related to the formation of MAC on the cell surface due to the CMC effect. In the middle stage, we observed that the size of the holes increased (denoted by the arrows in Figure 2F) and that cytoplasm had escaped from the cell (denoted by the arrow in Figure 2E). In the final stage, the cells collapsed, leaving behind only cell membrane debris (Figure 2H–2J). Collectively, the AFM images clearly showed the dynamic processes of CMC from the nanoscale cellular topography. Additionally, changes in the cellular mechanical properties during CMC were also measured. The cells were PI-stained to identify the CMC effect (Figure 2K). Under the guidance of fluorescence, the AFM tip was moved toward the cells exhibiting the CMC effect (Figure 2I). The Young's modulus of lymphoma Raji cells was measured at three intervals (before treatment, after treatment without CMC, and after treatment with CMC). The results (Figure 2M) revealed that during CMC the cellular Young's modulus first decreased and then significantly increased, improving our understanding of rituximab's CMC effect. The studies also indicated that the efficacy of drugs could be assessed by monitoring the mechanical properties of the cells. Both experimental50,66 and theoretical71 studies have shown that cell mechanical properties are closely related to the uncontrolled division and migration of tumor cells; therefore, we can consider designing drugs that alter the mechanical properties of tumor cells. These drugs would likely not kill cancer cells, but may effectively hinder the propagation of the neoplasm72.

Dynamic changes of cellular nanostructures and mechanical properties during CMC investigated by AFM70. (A) Schematic diagram of CMC. (B–J) AFM images of lymphoma Raji cells after the treatment of Rituximab and human serum. (K) Fluorescence image of a PI-stained Raji cell. (L) Under the guidance of fluorescence, AFM tip was moved to the cell. (M) Dynamic changes of cellular Young's modulus during CMC. (B–M) were reprinted with permission from Ref70. Copyright 2014 IEEE.
In situ quantification of individual drug-target interactions
Drug-target interactions (such as the binding force) are closely related to the overall drug efficacy; therefore, investigating drug-target interactions is important for better understanding the drug action. Traditional methods for characterizing drug-target interactions require many purified target molecules that are isolated from cells. Studies have shown that the cell membrane plays an essential role in determining the functions of the membrane proteins73. Therefore, the results obtained from purified proteins cannot faithfully reflect the real properties of the same molecules in vivo74. The pioneering work of Gaub et al showed that AFM could be used to measure the binding force of individual receptor-ligand pairs75 and to investigate the unfolding dynamics of single molecules76. Following his work, similar studies on different types of purified molecules emerged77,78, providing a better understanding of the molecular interactions. In 2000, Benoit et al79 investigated cell-cell adhesion interactions by AFM force spectroscopy, demonstrating the potential of AFM to measure individual molecular interactions in situ. With AFM force spectroscopy, the binding force of individual drug-target pairs can be directly quantified by linking drug molecules to the AFM tip. Compared with traditional methods, AFM provides a novel approach to investigate the drug-target interactions and provides unique data that are useful for the improvement of drug design. Sodium-glucose co-transporter (SGLT) plays an important role in intestinal glucose adsorption and in renal glucose reabsorption in many organisms. SGLT is the target for a series of drugs used in the treatment of diabetes, such as canagliflozin80 and ipragliflozin81. In 2006, Puntheeranurak et al82 applied AFM force spectroscopy to measure the binding force of SGLT on living CHO cells by linking its antibody to an AFM tip. The results demonstrated the potential of AFM to study the presence and dynamics of plasma membrane transporters in intact cells at the single-molecule level. Vascular endothelial growth factor receptors (VEGFRs) are important targets for cancer treatment because of their crucial roles in vascular development, angiogenesis and lymphangiogenesis83. In 2007, Lee et al84 combined AFM imaging and molecular recognition to investigate the location and binding kinetics of VEGFRs on human umbilical vein endothelial cells (HUVEC) by linking monoclonal anti-VEGFR2 antibody to an AFM tip. The results demonstrated that AFM can provide spatiotemporal visualization of cell surface dynamics that regulate receptor-mediated behaviors. So far, researchers have applied AFM force spectroscopy to investigate a wide range of drug-target interactions on a series of cells, such as Herceptin-EGFR on HEK 293 cells85, quantum dot-HeLa cell interactions86, hormone-drug on HeLa cells87, transporter-antibody on erythrocyte88, and aptamer-glycoprotein on U251 cells89. Although these studies improve our understanding of drug-cell interactions at the single-molecule level, they were performed on cell lines cultured in vitro. In fact, the conditions used in research laboratories (eg, tumor cells are often selected based on their ability to grow rapidly in vitro and occasionally their relative sensitivity to therapy) vary significantly from those used in the real-world clinical environment90. Therefore, the results obtained using cell lines may not accurately reflect realistic drug-cell interactions.
To better understand drug-cell interactions, direct investigations of pathological cells from clinical patients are required. Experiments performed on patient cells can provide novel insights with translational medical significance. To this end, we have investigated drug-target interactions directly on cells from clinical lymphoma patients91,92,93, as shown in Figure 3. The binding of rituximab to the CD20s on lymphoma cancer cells can lead to cell lysis via three mechanisms, including direct induced apoptosis, antibody-dependent cellular cytotoxicity (ADCC) and CMC (Figure 3A). To investigate the CD20-rituximab interactions directly on cancer cells from lymphoma patients, the cancer cells from the clinical biopsy samples should first be identified because in biopsy samples (such as bone marrow), cancer cells and normal cells are mixed together. Receptor tyrosine-like orphan receptor 1 (ROR1) is a specific cell surface marker that is highly expressed on B lymphoma cancer cells, but not on normal cells94,95. Therefore, we used ROR1 to identify the cancer cells in the bone marrow samples prepared from B-cell lymphoma patients. Then, the CD20s on the cancer cells could be probed using rituximab-tethered tips (Figure 3A). Under the guidance of ROR1 fluorescence recognition, the AFM tip was positioned on the cancer cell (denoted by the black arrow in Figure 3B). The obtained typical force curves contained a specific unbinding peak in the retract curve (denoted by the green arrow in Figure 3C), which was caused by the stretching of PEG spacer molecules. By obtaining thousands of force curves at different points on the cell surface, a histogram of the binding force was constructed (Figure 3D). Additionally, by obtaining 16×16 force curves on the local cell surface (500 nm×500 nm), gray maps that reflected the nanoscale distribution of CD20s were constructed (Figure 3E). For contrast, few gray pixels in the gray maps were obtained on normal cells (red blood cells) (Figure 3F) that did not express CD20s. To investigate the role of CD20-rituximab interactions in the rituximab clinical therapy, biopsy samples from three lymphoma patients were tested. The results showed that for the three patients, there was no significant difference in the binding force of CD20 on cancer cells (Figure 3G); however, there was significant difference in the distribution density of CD20 on cancer cells (Figure 3H). From the results obtained from combining AFM together with the clinical treatment information (Figure 3I), we were able to conclude that the distribution density of CD20 on the lymphoma cancer cells had a direct impact on the clinical efficacies of rituximab. Current biochemical research is typically performed on cells grown in vitro, which leads to a large gap between the laboratory results and clinical requirements54. The results shown in Figure 3 represent a new method to deepen the relationship between clinical treatment and laboratory research and also demonstrate the potential of using AFM force spectroscopy to evaluate drug efficacies from the perspective of clinical medicine.

Investigating the CD20-rituximab interactions on cancer cells in the biopsy samples prepared from clinical lymphoma patients by AFM force spectroscopy91,92,93. (A) Schematic diagram of rituximab's mechanisms and probing the CD20-rituximab interactions on lymphoma cancer cells. (B) Under the guidance of ROR1 fluorescence recognition, AFM tip carrying rituximabs was moved to cancer cell. (C) Typical force curve obtained on cancer cell with specific unbinding peak. (D) Histogram of binding force. CD20 distribution map obtained on cancer cell (E) and normal cell (F). Histogram of binding force (G) and distribution density (H) of CD20s on the cancer cells from three lymphoma patients. (i) Clinical treatment information of the tree patients. (B) was reprinted with permission from Ref91. Copyright 2013 John Wiley & Sons, Ltd. (C–F) were reprinted with permission from Ref92. Copyright 2013 Elsevier Inc. (G–I) were reprinted with permission from Ref93. Copyright 2014 Royal Microscopical Society.
High-resolution imaging and manipulation of individual native target molecules
Determining the structure of target molecules is crucial for understanding their interactions with drug molecules26. Most of the targets are membrane proteins. The amphiphilic nature of membrane proteins makes the application of traditional structural biology methods (eg, nuclear magnetic resonance spectroscopy, electron microscopy and X-ray crystallography) toward their study challenging, especially with regards to protein expression, protein purification, and crystallization96. AFM provides an alternative way to investigate the nanoscale morphology and mechanical properties of individual native membrane proteins. By reconstituting purified membrane proteins into a lipid bilayer to mimic their native environment, AFM can observe single native membrane proteins at work, which can thus provide different and complementary information to that obtained from traditional structural biology methods38. Gap junction channels mediate communication between adjacent cells97. In 2002, Muller et al97 imaged the conformational changes of single native junction channels in response to ions in buffer solution by adsorbing the purified gap junction fractions to mica. The results showed that calcium ions could induce closing of the channels, as shown in Figure 4A and 4B. Potassium channels are tetrametric membrane proteins that facilitate the permeation of potassium ions through the membrane with high specificity and high-throughput rates16. In 2011, Mari et al16 visualized the conformational changes of a single MlokiK1 potassium channel mediated by ligands. The AFM images clearly showed that the binding of cAMP molecules could lead to the opening of the potassium channels, whereas the potassium channels closed in the absence of cAMP, as shown in Figure 4C and 4D. It has been established that traditional structural biology methods such as X-ray crystallography and electron microscopy can only provide the static structure of a membrane protein96,98. By contrast, AFM can image the dynamic conformational changes of single native membrane proteins in response to external stimuli such as drugs, providing direct evidence of the drug actions from the in situ single molecules.

Imaging and manipulating single native membrane proteins by high-resolution AFM and force spectroscopy16,97,99. (A, B) Imaging Ca2+-induced conformation change of the extracellular connexon surface of gap junction channels. (A) In Ca2+-free buffer solution. (B) In the presence of 0.5 mmol/L CaCl2. The insets are the average of the raw data. (C, D) Visualizing ligand-induced conformation change of the potassium channel. (C) In the presence of cAMP. (D) In the absence of cAMP. The insets are the average (top) and SD map (bottom) of raw data. (E, F) Single-molecule force spectroscopy of membrane protein NhaA. (E) Schematic diagram. (F) Force curve revealing the unfolding of a single NhaA molecule. (A, B) were reprinted with permission from Ref97. Copyright 2002 European Molecular Biology Organization. (C, D) were reprinted with permission from Ref16. Copyright 2011 National Academy of Sciences. (E, F) were reprinted with permission from Ref99. Copyright 2006 Elsevier Ltd.
In addition to imaging the two-dimensional morphology, AFM can also reveal the unfolding dynamics of a single membrane protein by pulling one of its ends, as shown in Figure 4E. In 2006, Kedrov et al99 investigated the unfolding dynamics of individual antiporter NhaAs that were reconstituted into the lipid bilayers by obtaining force curves on the protein. The obtained force curves shown in Figure 4F clearly revealed serial serrated peaks, each of which corresponded to the unfolding of one domain of the membrane protein. By applying AFM force spectroscopy, the unfolding dynamics of various types of membrane proteins have been investigated100,101,102. In a recent study, high-speed force spectroscopy was developed to unfold single proteins at velocities previously only reached by simulations. This finding has enabled direct comparison of experimental results to those of simulations, which is of considerable significance in studies of molecular mechanical dynamics103. These studies have demonstrated the ability of AFM to investigate the dynamic morphology and unfolding process of single native target molecules, which is of potential significance for pharmacology and drug development. AFM can be used to directly visualize drug effects on single target molecules as well as to obtain the unfolding dynamics of a single target molecule. Current molecular simulations are based on 3-D structural data obtained by traditional structural biology methods104,105. The information obtained by AFM will provide complementary data, which may in turn improve the computer simulations of drug-target interactions.
Challenge and outlook
AFM provides a novel powerful instrument for researchers to investigate drug-target interactions at the single-cell and single-molecule levels under near-physiological conditions. It allows us to visually and physically probe drug effects at the nanoscale, which are inaccessible by traditional biochemical methods. Measuring the physical properties of active biological samples (such as living cells and native membrane proteins) probed by AFM is advantageous in that it does not require costly labeling or sample preparation106. Single-cell assays based on AFM offer new opportunities for analyzing the heterogeneity of individual cells, which is masked by traditional ensemble experiments107. The typical applications summarized here of AFM in the past decade (topography imaging, elasticity measurements, molecular interaction quantification, native membrane protein imaging and manipulation) for monitoring the physiological activities on single cells and single molecules after the stimulation of drugs have provided considerable novel insight into our understanding of drug actions, demonstrating the valuable role of AFM for pharmacology and drug development. However, there are still several challenges that need to be addressed to improve the current AFM-based research.
The first challenge is how to improve the spatial resolution and reliability of AFM imaging and force spectroscopy probing of living cells. Although AFM imaging and force spectroscopy can directly image and reveal the unfolding dynamics of single native membrane proteins, the current research has been performed on reconstituted lipid bilayers. Clearly, the mechanistic results derived from an in vitro simulacrum must be addressed at the cellular level108. AFM has achieved great success in studies of microbial cells (such as bacteria and fungi) because of the well-defined and rigid nature of the microbial cell envelope107,109. However, AFM imaging of mammalian cells remains limited to resolutions in the 50–100 nm range32 due to the soft, dynamic and highly heterogeneous nature of the cell surface, meaning that single proteins on living cells cannot be observed. For mammalian suspended cells in particular, the prerequisite of performing AFM experiments is to immobilize them onto the substrate. Current immobilization methods such as the micro-well array35 and micro-pillar array39,40 methods are effective for trapping the whole cell. However, they cannot eliminate the dynamic movement of the cell membrane, which prevents the acquisition of higher resolution images of local areas on living mammalian cells. Methods that can inhibit the dynamics of the cell surface are still lacking. Moreover, the vast majority of interactions detected during AFM force spectroscopy experiments are unspecific110 because of the high complexity of the cell surface (eg, a cell membrane contains hundreds of lipids and a plethora of proteins, and also exhibits raft-based heterogeneity111). When performing force spectroscopy on living cells, the AFM probe can be contaminated after recording only a few force curves110. Therefore, current AFM methods (such as simultaneous topography and recognition imaging112,113) are mainly adapted for chemically fixed cells, which allow us to obtain data with a higher signal-noise ratio. Studies that convincingly demonstrate the reliability of force spectroscopy experiments on living mammalian cells are still needed.
The second challenge stems from the low throughput of AFM experiments and the intensive labor required to perform them. Currently, AFM experiments are not automatic and must be performed manually. Under the guidance of optical microscopy (or fluorescence), an operator moves the AFM tip to the cell of interest to perform measurements (such as morphology imaging and force spectroscopy). After the measurements are finished, the AFM tip is directed toward another cell. This process leads to a very low throughput (the time of handling one cell is often several minutes)67. To enable practical applications such as drug screening and evaluation, the throughput of AFM should be considerably improved and thus automation strategies may be required. Current AFM measurement experiments (such as elasticity measurements and molecular interactions measurements) also require the off-line processing of force curves, which is time-consuming (typically thousands of force curves need to be analyzed). Recently, the advent of a novel AFM mode called peak force tapping mode23 has enabled analysis of the obtained force curves in real-time and directly provides the multiparametric images (such as the Young's modulus and adhesion force). This development has greatly improved the efficiency of AFM force measurements. We have presented a strategy for automatically performing AFM measurements by cell recognition via an image processing method114, and the results demonstrated that it could significantly reduce the measurement time. However, there are still several issues that need to be addressed to achieve fully automated AFM measurements, such as the integration of cell delivery techniques, automation of the system calibration and visualization of the detection results. Regarding cell delivery, microfluidic techniques115 are of potential significance, whereas for data visualization, the 2-D scatter plots and gating approaches widely used in flow cytometry67 will be useful. If fully automated AFM measurements, such as automatic monitoring of the changes in cellular elastic properties, can be achieved, then they can be applied to drug research. For example, we can imagine employing AFM to screen out drugs that can bind to cytoskeleton proteins and change the elastic properties of cells, which is of significance in promoting the development of the pharmaceutical industry and clinical disease therapy.
The third challenge is to improve the time resolution of AFM measurements. Currently, the time needed to acquire a high-resolution image (often several minutes) is much greater than the time scale at which dynamic processes usually occur in biology32 (eg, bacteria respond to antibiotics on the time scale of seconds116). Although high-speed scanning AFM has reduced the acquisition time for an image to less than one second22,41, it is primarily limited to certain samples (such as rigid microbial cells14 and isolated molecules adsorbed on substrate41) and the scan size is often small (less than one micrometer). To make high-speed AFM applicable to living mammalian cells, which are large and have rough surfaces, several technical and conceptual obstacles need to be overcome117. Moreover, so far AFM cannot access the interior of cells. A recent study has combined AFM with confocal fluorescence microscopy to measure in situ the elasticity of the cell nucleus directly on living cells via a needle tip (the tip can penetrate the cell membrane)118, demonstrating the potential of AFM for intracellular mechanics studies. However, this penetration method is in fact invasive; therefore, the influence of the manipulation process on the biological activity of cells needs to be clarified (eg, whether the cell is still alive and whether it can be used for subsequent experiments). In addition, some applications (eg, force spectroscopy for probing molecular interactions or cell-cell adhesions) require particular modifications of the AFM tip, such as linking specific molecules (eg, antibodies, ligands, viruses) or a living cell to the AFM tip119. However, the currently available protocols are labor-intensive, time-consuming and complex, and the lifetime of the functionalized tip is often short. Therefore, developing simple and standardized protocols for tip functionalization32 will certainly contribute to advances in AFM force spectroscopy experiments.
In summary, the results achieved by applying AFM to the monitoring of drug actions on membranes at the nanoscale in recent decades have provided new opportunities for drug development. However, there are still challenges, as described above, which are hampering the further development of AFM. Overcoming these challenges will require the efforts of researchers from different disciplines (eg, biology, engineering, physics, automation). Nonetheless, it is gratifying to see that in recent years important progress has been made to enhance the functions of AFM, such as high-speed imaging (to improve the scan speed)41, multiparametric mode (to simultaneously obtain multiple parameters)23, high-speed force spectroscopy (to improve the speed of obtaining force curves)103, nanofluidics AFM (to allow the injection of chemical molecules or drugs into a single cell)120, tomographic contact resonance AFM (to generate cross-sectional stiff images of samples)121 and fast nanomechanical spectroscopy (to improve the speed of mapping the mechanical properties)122. Moreover, AFM can also be combined with other techniques to obtain complementary information, and this will be an important aspect for future AFM studies. For example, the spatial resolution of optical microscopy can now reach the nanometer level via fluorescence123. Under the guidance of ultra-resolution optical (fluorescence) microscopy, AFM may be used to probe individual structures such as cellular machinery on the surface of a living cell (eg, to measure their mechanical properties). Patch-clamp is a commonly used technique in studies of ion channels. AFM has been successfully combined with patch-clamp to simultaneously obtain the mechanical dynamics and current responses of the ion channels on a living cell124, providing novel insights into the mechanosensitive ion channels. Scanning ion conductance microscopy has been shown to be effective in imaging the surface of living cells, as it has a noncontact character125. This technique has inspired us to improve the spatial resolution of imaging living cells by developing noncontact AFM scanning mode in liquids. Collectively, the exciting results obtained with AFM indicate that its further applications in the life sciences will bring about yet more changes in related fields such as pharmaceutical industry and clinical medicine.
References
Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 2010; 9: 203–14.
Pammolli F, Magazzini L, Riccaboni M . The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov 2011; 10: 428–38.
Khanna I . Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov Today 2012; 17: 1088–102.
Bennani YL . Drug discovery in the next decade: innovation needed ASAP. Drug Discov Today 2011; 16: 779–92.
Zheng CJ, Han LY, Yap CW, Ji ZL, Cao ZW, Chen YZ . Therapeutic targets: progress of their exploration and investigation of their characteristics. Pharmacol Rev 2006; 58: 259–79.
Baker M . Making membrane proteins for structures: a trillion tiny tweaks. Nat Methods 2010; 7: 429–33.
Overington JP, Al-Lazikani B, Hopkins AL . How many drug targets are there? Nat Rev Drug Discov 2006; 5: 993–6.
Dufrene YF . Atomic force microscopy of fungal cell walls: an update. Yeast 2010; 27: 465–71.
Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013; 498: 236–40.
Pelkmans L . Using cell-to-cell variability — a new era in molecular biology. Science 2012; 336: 425–6.
Guillaume-Gentil O, Potthoff E, Ossola D, Franz CM, Zambelli T, Vorholt JA . Force-controlled manipulation of single cells: from AFM to fluidFM. Trends Biotechnol 2014; 32: 381–8.
Saliba AE, Westermann AJ, Gorski SA, Vogel J . Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 2014; 42: 8845–60.
Muller DJ, Dufrene YF . Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat Nanotechnol 2008; 3: 261–9.
Fantner GE, Barbero RJ, Gray DS, Belcher AM . Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat Nanotechnol 2010; 5: 280–5.
Liu LN, Scheuring S . Investigation of photosynthetic membrane structure using atomic force microscopy. Trends Plant Sci 2013; 18: 277–86.
Mari SA, Pessoa J, Altieri S, Hensen U, Thomas L, Morais-Cabral JH, et al. Gating of the Mlotik1 potassium channel involves large rearrangements of the cyclic nucleotide-binding domains. Proc Natl Acad Sci U S A 2011; 108: 20802–7.
Kowal J, Chami M, Baumgartner P, Arheit M, Chiu PL, Rangl M, et al. Ligand-induced structural changes in the cyclic nucleotide-modulated potassium channel Mlok1. Nat Commun 2014; 5: 3106.
Madeira A, Ohman E, Nilsson A, Sjogren B, Andren PE, Svenningsson P . Coupling surface plasmon resonance to mass spectroscopy to discover novel protein-protein interactions. Nat Protoc 2009; 4: 1023–37.
Hinterdorfer P, Dufrene Y . Detection and localization of single molecule recognition events using atomic force microscopy. Nat Methods 2006; 3: 347–55.
Zhang X, Shi X, Xu L, Yuan J, Fang X . Atomic force microscopy study of the effect of HER 2 antibody on EGF mediated ErbB ligand-receptor interaction. Nanomedicine 2013; 9: 627–35.
Katan AJ, Dekker C . High-speed AFM reveals the dynamics of single biomolecules at the nanometer scale. Cell 2011; 147: 979–82.
Ando T . High-speed atomic force microscopy coming of age. Nanotechnology 2012; 23: 062001.
Dufrene YF, Martinez-Martin D, Medalsy I, Alsteens D, Muller DJ . Multiparametric imaging of biological systems by force-distance curve-based AFM. Nat Methods 2013; 10: 847–54.
Edwardson JM, Henderson RM . Atomic force microscopy and drug discovery. Drug Discov Today 2004; 9: 64–71.
Vliet KJV, Hinterdorfer P . Probing drug-cell interactions. Nano Today 2006; 1: 18–25.
Muller DJ, Wu N, Palczewski K . Vertebrate membrane proteins: structure, function, and insights from biophysical approaches. Pharmacol Rev 2008; 60: 43–78.
Whited AM, Park PS . Atomic force microscopy: a multifaceted tool to study membrane proteins and their interactions with ligands. Biochim Biophys Acta 2014; 1838: 56–68.
Spyratou E, Mourelatou EA, Makropoulou M, Demetzos C . Atomic force microscopy: a tool to study the structure, dynamics and stability of liposomal drug delivery systems. Expert Opin Drug Deliv 2009; 6: 305–17.
Lamprecht C, Hinterdorfer P, Ebner A . Applications of biosensing atomic force microscopy in monitoring drug and nanoparticle delivery. Expert Opin Drug Deliv 2014; 11: 1237–53.
Pillet F, Chopinet L, Formosa C, Dague E . Atomic force microscopy and pharmacology: from microbiology to cancerology. Biochim Biophys Acta 2014; 1840: 1028–50.
Jain KK . The role of nanobiotechnology in drug discovery. Drug Discov Today 2005; 10: 1435–42.
Muller DJ, Dufrene YF . Atomic force microscopy: a nanoscopic window on the cell surface. Trends Cell Biol 2011; 21: 461–9.
Kirmse R, Otto H, Ludwig T . Interdependency of cell adhesion, force generation and extracellular proteolysis in matrix remodeling. J Cell Sci 2011; 124: 1857–66.
Hecht E, Thompson K, Frick M, Wittekindt OH, Dietl P, Mizaikoff B, et al. Combined atomic force microscopy-fluorescence microscopy: analyzing exocytosis in alveolar type II cells. Anal Chem 2012; 84: 5716–22.
Li M, Liu L, Xi N, Wang Y, Dong Z, Xiao X, et al. Atomic force microscopy imaging of live mammalian cells. Sci China Life Sci 2013; 56: 811–7.
Engel A, Gaub HE . Structure and mechanics of membrane proteins. Annu Rev Biochem 2008; 77: 127–48.
Bippes CA, Muller DJ . High-resolution atomic force microscopy and spectroscopy of native membrane proteins. Rep Prog Phys 2011; 74: 086601.
Fotiadis D . Atomic force microscopy for the study of membrane proteins. Curr Opin Biotechnol 2012; 23: 510–5.
Li M, Liu L, Xi N, Wang Y, Dong Z, Tabata O, et al. Imaging and measuring the rituximab-induced changes of mechanical properties in B-lymphoma cells using atomic force microscopy. Biochem Biophys Res Commun 2011; 404: 689–94.
Li M, Liu L, Xi N, Wang Y, Dong Z, Xiao X, et al. Drug-induced changes of topography and elasticity in living B lymphoma cells based on atomic force microscopy. Acta Phys Chim Sin 2012; 28: 1502–8.
Kodera N, Yamamoto D, Ishikawa R, Ando T . Video imaging of walking myosin V by high-speed atomic force microscopy. Nature 2010; 468: 72–6.
Kasas S, Longo G, Dietler G . Mechanical properties of biological specimens explored by atomic force microscopy. J Phys D Appl Phys 2013; 46: 133001.
Li M, Liu L, Xi N, Wang Y . Research progress in quantifying the mechanical properties of single living cells using atomic force microscopy. Chin Sci Bull 2014; 59: 4020–9.
Lekka M, Pogoda K, Gostek J, Klymenko O, Prauzner-Bechcicki S, Wiltowska-Zuber J, et al. Cancer cell recognition-mechanical phenotype. Micron 2012; 43: 1259–66.
Costa KD . Single-cell elastography: probing for disease with the atomic force microscope. Dis Markers 2004; 19: 139–54.
Stolz M, Raiteri R, Daniels AU, VanLandingham MR, Baschong W, Aebi U . Dynamic elastic modulus of porcine articular cartilage determined at two different levels of tissue organization by indentation-type atomic force microscopy. Biophys J 2004; 86: 3269–83.
Gavara N, Chadwick RS . Determination of the elastic moduli of thin samples and adherent cells using conical atomic force microscope tips. Nat Nanotechnol 2012; 7: 733–6.
Butt HJ, Cappella B, Kappl M . Force measurements with the atomic force microscope: technique, interpretation and applications. Surf Sci Rep 2005; 59: 1–152.
Li M, Liu L, Xi N, Wang Y, Dong Z, Xiao X, et al. Atomic force microscopy imaging and mechanical properties measurement of red blood cells and aggressive cancer cells. Sci China Life Sci 2012; 55: 968–73.
Suresh S . Biomechanics and biophysics of cancer cells. Acta Biomater 2007; 3: 413–38.
Rotsch C, Radmacher M . Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J 2000; 78: 520–35.
Lam WA, Rosenbluth MJ, Fletcher DA . Chemotherapy exposure increases leukemia cell stiffness. Blood 2007; 109: 3505–8.
Fang Y, Lu CY, Lui CNP, Zou Y, Fung CKM, Li HW, et al. Investigating dynamic structural and mechanical changes of neuroblastoma cells associated with glutamate-mediated neurodegeneration. Sci Rep 2014; 4: 7074.
Li M, Liu L, Xi N, Wang Y . Progress in measuring biophysical properties of membrane proteins with AFM single-molecule force spectroscopy. Chin Sci Bull 2014; 59: 2717–25.
Barattin R, Voyer N . Chemical modification of AFM tips for the study of molecular recognition events. Chem Commun (Camb) 2008; (13): 1513–32.
Ebner A, Wildling L, Zhu R, Rankl C, Haselgrubler T, Hinterdorfer P, et al. Functionalization of probe tips and supports for single-molecule recognition force microscopy. Top Curr Chem 2008; 285: 29–76.
Darghouth D, Koehl B, Madalinski G, Heilier JF, Bovee P, Xu Y, et al. Pathophysiology of sickle cell disease is mirrored by the red blood cell metabolome. Blood 2011; 117: e57–66.
Suresh S . Elastic clues in cancer detection. Nat Nanotechnol 2007; 2: 748–9.
Chow KH, Factor RE, Ullman KS . The nuclear envelope environment and its cancer connections. Nat Rev Cancer 2012; 12: 196–209.
Iyer S, Gaikwad RM, Subba-Rao V, Woodworth CD, Sokolov I . Atomic force microscopy detects differences in the surface brush of normal and cancerous cells. Nat Nanotechnol 2009; 4: 389–93.
Heu C, Berquand A, Elie-Caille C, Nicod L . Glyphosate-induced stiffening of HaCaT keratinocytes, a Peak Force Tapping study on living cells. J Struct Biol 2012; 178: 1–7.
Alonso-Sarduy L, Longo G, Dietler G, Kasas S . Time-lapse AFM imaging of DNA conformational changes induced by daunorubicin. Nano Lett 2013; 13: 5679–84.
Fletcher DA, Mullins RD . Cell mechanics and the cytoskeleton. Nature 2010; 463: 485–92.
Janmey PA, McCuloch CA . Cell mechanics: integrating cell responses to mechanical stimuli. Ann Rev Biomed Eng 2007; 9: 1–34.
Wirtz D, Konstantopoulos K, Searson PC . The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer 2011; 11: 512–22.
Plodinec M, Loparic M, Monnier CA, Obermann EC, Zanetti-Dallenbach R, Oertle P, et al. The nanomechanical signature of breast cancer. Nat Nanotechnol 2012; 7: 757–65.
Carlo DD . A mechanical biomarker of cell state in medicine. J Lab Autom 2012; 17: 32–42.
Mao X, Huang TJ . Exploiting mechanical biomarkers in microfluidics. Lab Chip 2012; 12: 4006–9.
Lekka M, Laidler P . Applicability of AFM in cancer detection. Nat Nanotechnol 2009; 4: 72–3.
Li M, Liu L, Xi N, Wang Y, Xiao X, Zhang W . Quantitative analysis of drug-induced complement-mediated cytotoxic effect on single tumor cells using atomic force microscopy and fluorescence microscopy. IEEE Trans Nanobiosci 2015; 14: 84–94.
Katira P, Zaman MH, Bonnecaze RT . How changes in cell mechanical properties induce cancerous behavior. Phys Rev Lett 2012; 108: 028103.
Fritsch A, Hockel M, Kiessling T, Nnetu KD, Wetzel F, Zink M, et al. Are biomechanical changes necessary for tumour progression? Nat Phys 2010; 6: 730–2.
Phillips R, Ursell T, Wiggins P, Sens P . Emerging roles for lipids in shaping membrane-protein function. Nature 2009; 459: 379–85.
Oddershede LB . Force probing of individual molecules inside the living cell is now a reality. Nat Chem Biol 2012; 8: 879–86.
Florin EL, Moy VT, Gaub HE . Adhesion forces between individual ligand-receptor pairs. Science 1994; 264: 415–7.
Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE . Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 1997; 276: 1109–12.
Hinterdorfer P, Baumgartner W, Gruber HJ, Schilcher K, Schindler H . Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc Natl Acad Sci U S A 1996; 93: 3477–81.
Fritz J, Katopodis AG, Kolbinger F, Anselmetti D . Force-mediated kinetics of single P-selectin/ligand complexes observed by atomic force microscopy. Proc Natl Acad Sci U S A 1998; 95: 12283–8.
Benoit M, Garbriel D, Gerisch G, Gaub HE . Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat Cell Biol 2000; 2: 313–7.
Sha S, Devineni D, Ghosh A, Polidori D, Chien S, Wexler D, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab 2011; 13: 669–72.
Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol 2014; 727: 66–74.
Puntheeranurak T, Wildling L, Gruber HJ, Kinne RKH, Hinderdorfer P . Ligands on the string: single-molecule AFM studies on the interaction of antibodies and substrates with the Na+-glucose co-transporter SGLT1 in living cells. J Cell Sci 2006; 119: 2960–7.
Holmes K, Roberts OL, Thomas AM, Cross MJ . Vascular endothelial growth factor receptor-2: structure, function, intracellular signaling and therapeutic inhibition. Cell Signal 2007; 19: 2003–12.
Lee S, Mandic J, Vliet KJV . Chemomechanical mapping of ligand-receptor binding kinetics on cells. Proc Natl Acad Sci U S A 2007; 104: 9609–14.
Shi X, Xu L, Yu J, Fang X . Study of inhibition effect of Herceptin on interaction between Heregulin and ErbB receptors HER3/HER2 by single-molecule force spectroscopy. Exp Cell Res 2009; 315: 2847–55.
Shan Y, Hao X, Shang X, Cai M, Jiang J, Tang Z, et al. Recording force events of single quantum-dot endocytosis. Chem Commun 2011; 47: 3377–9.
Zhang J, Wu G, Song C, Li Y, Qiao H, Zhu P, et al. Single molecular recognition force spectroscopy study of a luteinizing hormone-releasing hormone analogue as a carcinoma target drug. J Phys Chem B 2012; 116: 13331–7.
Cai M, Zhao W, Shang X, Jiang J, Ji H, Tang Z, et al. Direct evidence of lipid rafts by in situ atomic force microscopy. Small 2012; 8: 1243–50.
Li Y, Qiao H, Yan W, Zhang J, Xing C, Wang H, et al. Molecular recognition force spectroscopy study of the dynamic interaction between aptamer GBI-10 and extracellular matrix protein tenascin-C on human glioblastoma cell. J Mol Recognit 2013; 26: 46–50.
Weiner GJ . Rituximab: mechanism of action. Semin Hematol 2010; 47: 115–23.
Li M, Xiao X, Liu L, Xi N, Wang Y, Dong Z, et al. Atomic force microscopy study of the antigen-antibody binding force on patient cancer cells based on ROR1 fluorescence recognition. J Mol Recognit 2013; 26: 432–8.
Li M, Xiao X, Liu L, Xi N, Wang Y, Dong Z, et al. Nanoscale mapping and organization analysis of target proteins on cancer cells from B-cell lymphoma patients. Exp Cell Res 2013; 319: 2812–21.
Li M, Xiao X, Zhang W, Liu L, Xi N, Wang Y . Nanoscale distribution of CD20 on B-cell lymphoma tumour cells and its potential role in the clinical efficacy of rituximab. J Microsc 2014; 254: 19–30.
Daneshmanesh AH, Mikaelsson E, Jeddi-Tehrani M, Bayat AA, Ghods R, Ostadkarampour M, et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukamia and may serve as a putative target for therapy. Int J Cancer 2008; 123: 1190–5.
Uhrmacher S, Schmidt C, Erdfelder F, Poll-Wolbeck SJ, Gehrke I, Hallek M, et al. Use of the receptor tyrosine kinase-like orphan receptor 1 (ROR1) as a diagnostic tool in chronic lymphocytic leukemia (CLL). Leuk Res 2011; 35: 1360–6.
Robertson JWF, Kasianowicz JJ, Banerjee S . Analytical approaches for studying transporters, channels and porins. Chem Rev 2012; 112: 6227–49.
Muller DJ, Hand GM, Engel A, Sosinsky GE . Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J 2002; 21: 3598–607.
Ma D, Lu P, Yan C, Fan C, Yin P, Wang J, et al. Structure and mechanism of a glutamate-GABA antiporter. Nature 2012; 483: 632–6.
Kedrov A, Ziegler C, Muller DJ . Differentiating ligand and inhibitor interactions of a single antiporter. J Mol Biol 2006; 362: 925–32.
Kedrov A, Janovjak H, Sapra KT, Muller DJ . Deciphering molecular interactions of native membrane proteins by single-molecule force spectroscopy. Annu Rev Biophys Biomol Struct 2007; 36: 233–60.
Damaghi M, Bippes C, Koster S, Yildiz O, Mari SA, Kuhlbrandlt W, et al. pH-dependent interactions guide the folding and gate the transmembrane pore of the beta-barrel membrane protein OmpG. J Mol Biol 2010; 397: 878–82.
Bujalowski PJ, Oberhauser AF . Tracking unfolding and refolding reactions of single proteins using atomic force microscopy methods. Methods 2013; 60: 151–60.
Rico F, Gonzalez L, Casuso I, Puig-Vidal M, Scheuring S . High-speed force spectroscopy unfolds titin at the velocity of molecular dynamics simulations. Science 2013; 342: 741–3.
Yang H, Yu Y, Li WG, Yu F, Cao H, Xu TL, et al. Inherent dynamics of the acid-sensing ion channel 1 correlates with the gating mechanism. PLoS Biol 2009; 7: e1000151.
Zhang Q, Zhou P, Chen Z, Li M, Jiang H, Gao Z, et al. Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proc Natl Acad Sci U S A 2013; 110: 20093–8.
Gossett DR, Tse HTK, Lee SA, Ying Y, Lindgren AG, Yang OO, et al. Hydrodynamic stretching of single cells for large population mechanical phenotyping: Proc Natl Acad Sci U S A 2012; 109: 7630–5.
Dufrene YF . Atomic force microscopy in microbiology: new structural and functional insights into the microbial cell surface. MBio 2014; 5: e01363–14.
Taatjes DJ, Quinn AS, Rand JH, Jena BP . Atomic force microscopy: high resolution dynamic imaging of cellular and molecular structure in health and disease. J Cell Physiol 2013; 228: 1949–55.
Dorobantu LS, Goss GG, Burrell RE . Atomic force microscopy: a nanoscopic view of microbial cell surfaces. Micron 2012; 43: 1312–22.
Dufrene YF, Evans E, Engel A, Helenius J, Gaub HE, Muller DJ . Five challenges to bringing single-molecule force spectroscopy into living cells. Nat Methods 2011; 8: 123–7.
Simons K, Gerl MJ . Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol 2010; 11: 688–99.
Duman M, Pfleger M, Zhu R, Rankl C, Chtcheglova LA, Neundlinger I, et al. Improved localization of cellular membrane receptors using combined fluorescence microscopy and simultaneous topography and recognition imaging. Nanotechnology 2010; 21: 115504.
Zhang J, Chtcheglova LA, Zhu R, Hinterdorfer P, Zhang B, Tang J . Nanoscale organizatioin of human GnRH-R on human bladder cancer cells. Anal Chem 2014; 86: 2458–64.
Wang Z, Liu L, Wang Y, Xi N, Dong Z, Li M, et al. A fully automated system for measuring cellular mechanical properties. J Lab Autom 2012; 17: 443–8.
Zheng Y, Shojaei-Baghini E, Azad A, Wang C, Sun Y . High-throughput biophysical measurement of human red blood cells. Lab Chip 2012; 12: 2560–7.
Longo G, Aloso-Sarduy L, Rio LM, Bizzini A, Trampuz A, Notz J, et al. Rapid detection of bacterial resistance to antibiotics using AFM cantilevers as nanomechanical sensors. Nat Nanotechnol 2013; 8: 522–6.
Casuso I, Rico F, Scheuring S . High-speed atomic force microccopy: structure and dynamics of single proteins. Curr Opin Chem Biol 2011; 15: 704–9.
Liu H, Wen J, Xiao Y, Liu J, Hopyan S, Radisic M, et al. In situ mechanical characterization of the cell nucleus by atomic force microscopy. ACS Nano 2014; 8: 3821–8.
Muller DJ, Helenius J, Alsteens D, Dufrene YF . Force probing surfaces of living cells to molecular resolution. Nat Chem Biol 2009; 5: 383–90.
Meister A, Gabi M, Behr P, Studer P, Voros J, Niedermann P, et al. FluidFM: combining atomic force microscopy and nanofluidics in a universal liquid delivery system for single cell applications and beyond. Nano Lett 2009; 9: 2501–7.
Stan G, Solares SD, Pittenger B, Erina N, Su C . Nanoscale mechanics by tomographic contact resonance atomic force microscopy. Nanoscale 2014; 6: 962–9.
Herruzo ET, Perrino AP, Garcia R . Fast nanomechanical spectroscopy of soft matter. Nat Commun 2014; 5: 3126.
Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006; 313: 1642–5.
Zhang C, Li P, Liu L, Wang Y, Gao Z, Li G . Development of mechanostimulated patch-clamp system for cellular physiological study. IEEE-ASME Trans Mech 2014; 19: 1138–47.
Rheinhaender J, Geisse NA, Proksch R, Schaffer TE . Comparison of scanning ion conductance microscopy with atomic force microscopy for cell imaging. Langmuir 2011; 27: 697–704.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (61175103, 61375107, 61327014, and 61433017), the Research Fund of the State Key Laboratory of Robotics (No 2014-Z07) and CAS FEA International Partnership Program for Creative Research Teams.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, M., Liu, Lq., Xi, N. et al. Nanoscale monitoring of drug actions on cell membrane using atomic force microscopy. Acta Pharmacol Sin 36, 769–782 (2015). https://doi.org/10.1038/aps.2015.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.28
Keywords
This article is cited by
-
Measuring the elastic modulus of soft culture surfaces and three-dimensional hydrogels using atomic force microscopy
Nature Protocols (2021)
-
Atomic force microscopy for revealing micro/nanoscale mechanics in tumor metastasis: from single cells to microenvironmental cues
Acta Pharmacologica Sinica (2021)
-
Applications of Atomic Force Microscopy in Exploring Drug Actions in Lymphoma-Targeted Therapy at the Nanoscale
BioNanoScience (2016)
-
Atomic Force Microscopy Probing of Cancer Cells and Tumor Microenvironment Components
BioNanoScience (2016)
-
Atomic force microscopy based investigations of anti-inflammatory effects in lipopolysaccharide-stimulated macrophages
Analytical and Bioanalytical Chemistry (2016)
