
Abstract
In the absence of detailed pedigree records, researchers have attempted to estimate individuals' levels of inbreeding using molecular markers, generally making use of heterozygosity measures based on microsatellite markers. Here we report and validate a method for estimating an individual's inbreeding coefficient, f, using amplified fragment length polymorphism (AFLP) markers. We use simulations to confirm that our measure scales appropriately with f when allele frequencies can be estimated from a subset of outbred individuals. We also present an approach for obtaining satisfactory estimates even in the absence of an independent set of known outbred individuals from which to estimate allele frequencies. We then test our method against empirical data from 179 wild and captive-bred old-field mice, Peromyscus polionotus subgriseus, comprising pedigree-based estimates of f, along with genetic data from 94 AFLP markers and 12 microsatellites. Inbreeding estimates based on both AFLP and microsatellite markers were found to correlate strongly with pedigree-based inbreeding coefficients. Owing to their ease of amplification in any species, AFLP markers may prove to be a valuable new tool for estimating f in natural populations and for examining correlations between heterozygosity and fitness.
Similar content being viewed by others


Introduction
When individuals who are related by descent mate, the resulting offspring have lower fitness compared to the mean fitness level of the population. This phenomenon is known as inbreeding depression (Frankham et al., 2002). A large number of studies demonstrating inbreeding depression in both plants and animals have been published, the most convincing of which are those based on laboratory or captive population (Ralls et al., 1988; Pray and Goodnight, 1995; Hauser and Loeschke, 1996; Lacy et al., 1996; Ballou, 1997). In contrast to laboratory studies, evidence for inbreeding depression in wild populations is difficult to find (Crnokrak and Roff, 1999). While this may be because many organisms have evolved mechanisms to avoid inbreeding, it is also true that measuring inbreeding coefficients (f) in natural populations is notoriously difficult. The most direct approach requires accurate pedigree information extending back at least three to four generations. Such detailed long-term observations are available for only a handful of populations in a few species such as the Mandarte Island song sparrow (Keller, 1998), Darwin's finches (Keller et al., 2007), the great tit (Greenwood et al., 1978), blue tit (Kempenaers et al., 1996), Soay sheep and red deer (Marshall et al., 2002). In general, these are small and often isolated populations where most individuals can be tagged and genetically sampled.
While pedigree-based measures provide the best estimate of f, suitable long-term data are not available for the majority of species. An alternative approach is to exploit the fact that inbreeding decreases heterozygosity. Consequently, f may, in principle, be estimated from heterozygosity at a panel of neutral genetic markers. Indeed, if a very large number of genetic markers are available, heterozygosity may even provide a more reliable estimate of actual inbreeding compared to (probably imperfect) pedigree data stretching back a small number of generations. Early studies employed allozyme markers (Houle, 1989; Pemberton et al., 1991), but more recently microsatellite markers have dominated because of their high variability and their ubiquity in most genomes (Coltman et al., 1998, 1999; Coulson et al., 1999; Amos et al., 2001).
Although microsatellites are informative markers for estimating heterozygosity, there are two important problems. First, there remain many species for which microsatellites have not been developed, and although cross-species amplification often allows the use of markers developed in related species (Menotti-Raymond and O'Brien, 1995; Primmer et al., 1996), such amplification can be prone to higher errors in the form of elevated null allele frequencies. Second, although many studies have revealed correlations between heterozygosity and fitness using only around 10 markers, recent theory stresses the benefits of using larger numbers of markers, both to give improved estimates of genome-wide heterozygosity (and hence f) and to increase the chance of linkage to genes experiencing balancing selection (Balloux et al., 2004; Slate et al., 2004). Some recent studies have used larger numbers of markers (Slate et al., 2004), but for the most part microsatellite development often ceases when around 10 loci have been found, and most studies could not increase marker numbers without further cloning.
One way to increase marker numbers without the necessity for cloning could be to use techniques that detect large numbers of anonymous loci without prior knowledge of the genome. Suitable techniques include DNA fingerprinting (Jeffreys et al., 1985), random amplified polymorphic DNA (RAPDs; Williams et al., 1990), and amplified fragment length polymorphisms (AFLPs; Vos et al., 1995). Of these, AFLP markers appear to offer the best balance between reliability (Jones et al., 1997; Questiau et al., 1999), ease of use and applicability across a wide range of taxa (Rosendahl and Taylor, 1997; Mueller and Wolfenbarger, 1999; Madden et al., 2004; Siemer et al., 2004).
In this study we first predict the theoretical relationship between band absence at dominant markers and inbreeding coefficient. This relationship is then empirically investigated using AFLP markers to estimate levels of inbreeding with a dataset of 179 wild and captive-bred old-field mice, P. polionotus subgriseus, with known pedigree-based inbreeding coefficients. In addition, the utility of AFLP markers at estimating levels of inbreeding is compared with that of microsatellite markers.
Materials and methods
Theoretical expectations and simulations
An extensive literature exists on alternative methods to estimate relatedness between pairs of individuals using genetic markers (Queller and Goodnight, 1989; Ritland, 1996; Lynch and Ritland, 1999; Wang, 2002). Applied to codominant data such as microsatellites, these methods appear to perform well and suffer relatively little bias (Wang, 2004). Dominant loci such as AFLP markers present greater problems because while band absence at a locus can be inferred to represent homozygosity for the null allele, presence can indicate either homozygosity for the present allele or a heterozygous individual carrying both a present and an absent allele. Several methods have been proposed to deal with such data (Lynch and Milligan, 1994; Hardy, 2003; Wang, 2004; Ritland, 2005), but these tend to suffer from bias in populations where inbreeding occurs (Wang, 2004). However, when the required quantity is the inbreeding coefficient itself, a simpler approach may be possible.
An individual's inbreeding coefficient can be defined as the probability that two alleles at a locus are identical by descent. More inbred individuals therefore exhibit increased homozygosity. At a dominant AFLP locus, absence of a band is deemed to indicate homozygosity for the null allele. Therefore, as homozygosity is increased through inbreeding, the number of bands carried by an individual decreases and, consequently, the number of absent phenotypes increases. This idea has in the past been applied to DNA fingerprint data (Kuhnlein et al., 1990; Stephens et al., 1992).
The expected number of null phenotypes, Pexp, carried by an individual born to unrelated parents and scored at n AFLP bands is given by:

where pi is the frequency of the null allele at locus i in the population. In an inbred individual with inbreeding coefficient f, this increases to:

Hence, on average an individual with inbreeding coefficient f will carry  more null phenotypes than an equivalent non-inbred individual, allowing an individual's AFLP-based inbreeding coefficient, fAFLP to be estimated as:
more null phenotypes than an equivalent non-inbred individual, allowing an individual's AFLP-based inbreeding coefficient, fAFLP to be estimated as:

where Pobs is the observed number of null phenotypes in the individual. This is directly equivalent to the estimator proposed by Ritland and Travis (Ritland and Travis, 2004) for codominant markers:

where δij=1 if the locus is homozygous (allele i=allele j) and pi is the population frequency of allele i.
To explore the behaviour of the estimator fAFLP, we used stochastic simulations based on either 100 loci, a similar number to that used in this study, or 1000 loci, the maximum number likely to be employed in a large study. Null allele frequencies were sampled from a flat distribution in the range 0.05–0.95. For each value of f (f was varied from 0 to 0.5 in steps of 0.05), 100 genotypes were simulated and fAFLP calculated, yielding a mean and standard deviation.
In our study, we are fortunate to have an external group of known outbred mice from which to obtain reasonably unbiased estimates of pi. When such a group is not available, it becomes impossible to estimate simultaneously both the allele frequency at a locus and f. In particular, one cannot distinguish between groups of individuals with different mean f but the same variance. One attempt to circumvent this problem has been to assume that allele frequencies follow a β distribution and to estimate the parameters of this distribution rather than individual allele frequencies. Unfortunately, although this works well for estimating Fst, its accuracy is very poor when applied to estimate f (Holsinger et al., 2002). The proportion of inbred individuals in wild populations is in general very low (Marshall et al., 2002). Therefore, we follow an alternative approach that makes the reasonable assumption that appreciable numbers of outbred individuals are present. Thus, when the variance in fAFLP is low, indicating homogeneous f, the sample is inferred to be outbred.
Given a set of samples with AFLP genotypes but unknown f, we attempt to find the best match between empirically derived f-values and equivalent values derived by simulation. Starting with the assumption that at least 50% of individuals are outbred, f=0, we explore all possible combinations of individuals with f=0, f=0.1, f=0.25 and f=0.5, in steps of two individuals, from all with f=0 to 50% with f=0 and 50% with f=0.5. For each combination we test the hypothesis that this is the correct combination of f-values, as the observed band counts overestimate the frequency of null alleles by a quantity that depends on the mean f-value. Consequently, we begin by calculating mean f over all individuals and use this to adjust the raw frequencies of null alleles using Equation (1) above. We then use these adjusted frequencies to generate 10 000 simulated genotypes in input proportions. Finally, we calculate fAFLP for both the empirical and the simulated genotypes and test the how similar the resulting frequencies are using a χ2 test. Repeating over all combinations of input f-values, we search for the combination that yields the lowest χ2 value.
To test the effectiveness of our frequency distribution matching, we generated a range of simulated datasets based on 200 individuals typed for 100 AFLP bands with the frequency of null phenotypes chosen at random from a flat distribution with bounds 0.05 and 0.95. We examined five combinations of f-values:
-
a)
200 individuals with f=0
-
b)
150 individuals with f=0, 50 individuals with f=0.1
-
c)
150 individuals with f=0, 50 individuals with f=0.5
-
d)
100 individuals with f=0, 50 individuals with f=0.1, 50 individuals with f=0.5
-
e)
100 individuals with f=0, 50 individuals with f=0.25, 50 individuals with f=0.5.
This frequency matching procedure was repeated 100 times for each scenario to yield a mean and standard deviation for the estimated mean f-value (fAFLPFM) of each f-class. For comparison we also calculated fAFLP values using both the input band frequencies (fAFLPI) and frequencies based on the unadjusted band counts (fAFLPR).
Empirical data
Old-field mice, P. polionotus subgriseus, from Ocala National Forest, Marion County, Florida, USA were trapped in 1998. Thirty-five mice were collected, of which 26 were randomly selected to found the experimental stocks at Brookfield Zoo (Brookfield, IL, USA). Mice were paired to produce offspring with a range of inbreeding coefficients (0–0.453) over five generations of laboratory breeding and the resulting pedigree was recorded. The breeding design was not regular and pairings were arranged to produce a mixture of inbred and non-inbred (or weakly inbred) mice each generation. Breeding protocols followed those reported in earlier studies of this species (Lacy et al., 1996; Lacy and Ballou, 1998).
DNA was extracted from tissue samples of all 35 of the original wild mice and 144 captive-bred mice (representing animals with the full range of inbreeding coefficients) by Proteinase K digestion using an adapted Chelex 100 protocol (Walsh et al., 1991) and the DNA purified using a standard phenol-chloroform procedure (Sambrook et al., 1989). The samples were genotyped using eight AFLP primer combinations (TaqI-CAC with EcoRI-ACA, TaqI-CCA with EcoRI-ACA, TaqI-CGA with EcoRI-ACA, TaqI-CTG with EcoRI-ACA, TaqI-CAG with EcoRI-ACA, TaqI-CAC with EcoRI-AAC, TaqI-CAC with EcoRI-ATG, TaqI-CAC with EcoRI-AGC). The AFLP protocol was similar to that used in Vos et al. (1995) and the primer sequences and reaction conditions are described in Madden et al. (2004). Both AFLP and microsatellite PCR products were resolved by electrophoresis through 6% acrylamide gels, visualised by autoradiography and scored by eye. Ninety-four AFLP loci were polymorphic and could be scored unambiguously.
To enable a comparison between AFLP markers and the more widely used microsatellite markers, all 179 samples were also genotyped at 12 microsatellite loci: Pml1, Pml2, Pml4, Pml6, Pml7, Pml10, Pml11 (Chirhart et al., 2000), Plgt58, Plgt62, Plgt66 (Schmidt, 1999), Po3-68 and Po97 (Prince et al., 2002). Amplification was carried out in 10 μl volumes containing 0.5 μl of diluted template DNA, 1 × buffer (100 mM Tris-HCl pH 8.0, 50 mM KCl, 1.5 mM MgCl2, 0.01% Tween 20, 0.01% gelatine, 0.01% IGepal), 0.5 mM additional MgCl2, 0.2 mM each of dATP, dTTP and dGTP, 0.05 mM dCTP, 400 nM of each primer, 0.25 U Taq polymerase, and 0.1 μCi [α32P]-dCTP. The following PCR program was used: 3 min denaturing at 94 °C; followed by 35 cycles of 30 s at 94 °C, 30 s at the annealing temperature (51 °C to 63 °C depending on the locus), 25 s at 72 °C; ending with a 20 min final elongation stage at 72 °C. All microsatellite loci were found to be in Hardy–Weinburg equilibrium in the wild mice (GENEPOP 3.3; Raymond and Rousset, 1995).
Assuming that the founding wild mice are non-inbred and unrelated to one another, the known pedigree was used to calculate f, the probability that two homologous alleles in an individual are identical by descent (Wright, 1922), for each of the 145 captive-bred mice. The resulting inbreeding coefficients ranged from 0 to 0.453, with a mean of 0.183. Relative levels of inbreeding of each of the wild and captive-bred mice were also estimated from microsatellite genotypes by calculating microsatellite heterozygosity, Hμsat, calculated as the total number of loci at which a particular individual is heterozygous, divided by the number of loci at which it was genotyped. Estimators of inbreeding that require knowledge of allele frequencies, such as internal relatedness (Amos et al., 2001), were not used as the microsatellites have high allelic diversities and allele frequency estimates based on only 35 individuals are rather inaccurate. A third measure of inbreeding estimated from AFLP genotypes, the AFLP-based inbreeding coefficient, was calculated using Equation (3), with Pexp and pi estimated from the 35 wild caught individuals.
Results
Simulations show that the AFLP-based inbreeding estimator, fAFLP, varies linearly with inbreeding coefficient (Figure 1). Neither the gradient of the relationship nor the y-intercept differs significantly from 1 and 0 respectively, indicating that the average values of our estimator agree closely with the inputted values of f. As expected, using more loci improves the performance of the estimator (Figure 1).

Expected theoretical relationship between inbreeding coefficient and amplified fragment length polymorphism (AFLP)-based inbreeding coefficient, fAFLP, obtained from genotypes with simulated allele frequencies using (a) 100 AFLP loci and (b) 1000 AFLP loci. Vertical lines represent standard deviations.
We next sought to estimate fAFLP in simulated samples of animals where the allele frequencies have to be estimated from the data themselves, without an extrinsic sample of outbred individuals. Summary results for the frequency matching method (fAFLPFM) are presented in Table 1 with, for comparison, equivalent estimates based on the input allele frequencies (fAFLPI) and on unadjusted allele frequencies based on raw allele counts (fAFLPR). For each of the five scenarios, the three methods of inferring f and three classes of f, values are presented for the average estimate of f and the associated standard deviation calculated across 100 replicates. As expected, fAFLPI recovers the input distributions with high fidelity, while fAFLPR increasingly underestimates f as mean f increases. In comparison, fAFLPFM appears to be quite effective, even though the variance of the estimates is almost double that of fAFLPI. fAFLPFM does show an upward bias when inbred individuals are scarce/absent, presumably because any chance deviations from the perfect inference of f=0 are only allowed in the direction of finding some (rather than negative numbers of) inbred individuals. This bias could perhaps be addressed by allowing hypothetical negative deviations in mean f, or, more pragmatically, by requiring that any inferred presence of inbred individuals is allowed only if it yields a statistically significant improvement in fit over the null condition of all individuals having f=0. As soon as appreciable numbers of inbred individuals are generated, this bias is eroded and the fit between the input conditions and the inferred frequencies becomes good.
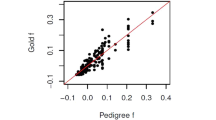
We next examined the correlation between pedigree-based f and estimators based on real AFLP and microsatellite markers genotyped in 179 wild and captive-bred mice. Figures 2a and b show that strong correlations exist for both AFLP markers and microsatellite loci (fAFLP: r2=0.30, P=2 × 10−15; Hμsat: r2=0.39, P<2 × 10−16). There was no significant difference in the amount of variance in f explained by fAFLP or Hμsat (ANOVA, P=0.3). The regression between f and fAFLP had a gradient of 1.44±0.16 and an intercept of 0.085±0.035. These are significantly different from the theoretical expectations of 1 (P=0.02) and 0 (P=0.01), respectively, perhaps indicating the action of selection or some level of non-independence between some bands. When the inbreeding estimators for both wild and captive-bred mice are compared with one another, fAFLP correlates significantly with Hμsat (r2=0.11, n=179, P=8 × 10−6), indicating that these estimators carry significant amounts of common information. However, no such relationship exists between the estimators when only the wild mice are used in this analysis (r2=0.00, n=35, P=0.97). In a multiple regression of f with fAFLP and Hμsat using all 179 wild and captive-bred mice, 53% of the variation in f is explained (r2=0.53, P=5 × 10−11 and <2 × 10−16, respectively), indicating that the two estimators provide independent rather than overlapping information about f.

Pedigree-based inbreeding coefficient vs (a) amplified fragment length polymorphism (AFLP)-based inbreeding coefficient and (b) microsatellite heterozygosity. Pedigree-based pairwise relatedness vs (c) relatedness estimated using AFLP markers and (d) relatedness estimated using microsatellite markers.
In order to investigate how both AFLP-based inbreeding coefficient and microsatellite heterozygosity correlate with inbreeding at lower levels of inbreeding, linear regressions of f against fAFLP and Hμsat were repeated but using progressively fewer inbred individuals. The results of this analysis are summarised in Table 2. In our dataset, the correlations between f and fAFLP and Hμsat were not significant when only individuals with f<0.05 were used in the analysis. At these lower levels of inbreeding (0.05<f<0.15), the correlation between f and fAFLP were stronger than those between f and Hμsat.
Applying our frequency matching analysis to the total mouse dataset (including both wild-caught and captive-bred individuals) we obtain a very satisfactory outcome, if anything, performing better than using allele frequencies calculated from our outbred group. The resulting graph is essentially the same as Figure 2a but with a shifted intercept. This is because the rank order of fAFLP values depends on only the number of null phenotypes in each individual, and therefore does not change. Similarly, in our approach, any refinement of the allele frequencies acts across all loci equally, such that alternative solutions act only to shift the intercept, and have little or no impact on the slope. Bearing this is mind, we can compare the use of raw allele frequencies (intercept=−0.258±0.03 s.e.), with the use of the wild-caught animals to estimate allele frequencies (intercept=0.089±0.03 s.e.) and with our new frequency matching method (intercept=−0.01±0.04 s.e.). In all cases the slope is very similar, at ∼1.4, and steeper than the unity slope expected, but the frequency matching method yields the closest solution to the ideal of the intercept being zero.
Genetic markers can also be used to estimate relatedness between individuals. With reference to a certain base population, the relatedness between two individuals is the probability of sharing alleles that are identical by descent, and can be estimated from genetic data using any of several methods (Queller and Goodnight, 1989; Lynch and Ritland, 1999). Assuming unrelated founders, the pedigree was used to calculate the ‘coefficient of relationship’ or theoretical additive genetic correlation, r (Crow and Kimura, 1970), between all pairs of the 179 study mice. Measures of pairwise relatedness were also calculated based on allele sharing at either microsatellite (rμsat) or AFLP (rAFLP) loci. For microsatellites, we again chose a method that avoids the need to estimate allele frequencies, calculating relatedness as the total number of identical alleles between a pair of individuals (0, 1 or 2 per locus) divided by twice the number of loci considered (Blouin et al., 1996; Ellegren, 1999). For AFLP genotypes, we again opted for simplicity, calculating relatedness values as the number of identical states (presence or absence of the band) between a pair of individuals divided by the number of loci used.
To test the reliability of our genetic relatedness estimators we examined how well they correlated both with pedigree-based coefficient of relationship and with each other. In each comparison, the significance of the correlation between the pairwise relatedness matrices was assessed using a Mantel test, implemented within the software zt (Bonnet and Van de Peer, 2002) with 100 000 randomisations. AFLP and microsatellite-based estimators of relatedness (rAFLP and rμsat) were found to correlate significantly with pedigree-based relatedness, r (r=0.391 and 0.550, respectively, n=179, P=1 × 10−5, Figures 2c and d). The pairwise relatedness matrices rAFLP and rμsat are also significantly correlated with each other (r=0.294, P=4 × 10−5) indicating shared information. A significant correlation exists even when rAFLP and rμsat for only the 35 wild mice are compared (r=0.199, P=4 × 10−5). Relatedness matrices rAFLP and rμsat estimate the relatedness between the wild mice using different types of markers. As they are estimating the same parameter, a significant correlation between the two relatedness matrices indicates that there is an overlap of information between the two estimators, which probably arises from there being detectable levels of relatedness between the wild caught mice.
Effect of locus number
An important question facing any study that attempts to use genetic markers to estimate either f or relatedness is how many markers are required. To investigate how the strengths of the relationships between pedigree-based and genetic measures of f and relatedness vary with the number of loci used, separate locus-dropping simulations were conducted for AFLP and microsatellite loci. In each case, n loci were chosen at random from the available 12-microsatellite/94 AFLP loci. From the genotypes at these n loci, fAFLP, Hμsat and the pairwise relatedness matrices rAFLP and rμsat were calculated for all 179 study mice. For each value of n, the process was repeated with different combinations of the n loci either 100 times or the number of possible unique combinations of loci, whichever was smaller. For microsatellites, n was increased from 2 to 11, and for the AFLPs from 2 to 93, in steps of one. Separate linear regressions of f against fAFLP and Hμsat were carried out for each replicate set, yielding a mean and the standard deviation of r2 (Figures 3a and b). In our dataset we find that 94 AFLP markers are approximately equivalent to 12 microsatellite markers for estimating inbreeding. Finally, we conducted locus-dropping simulations to explore the effect of locus number on AFLP and microsatellite-based relatedness estimates (Figures 3c and d). Here, as few as three to four microsatellite loci appear to perform as well as 90 AFLP loci.

Effect of locus number on the relationship between pedigree-based inbreeding coefficients and (a) amplified fragment length polymorphism (AFLP)-based inbreeding coefficient, (b) microsatellite heterozygosity. Effect of locus number on the relationship between pedigree-based pairwise relatedness and (c) relatedness estimated using AFLP markers, (d) relatedness estimated using microsatellite markers. Vertical lines represent standard deviations.
Discussion
The primary objective of this study was to establish whether AFLP data could be used to estimate levels of inbreeding. A theoretical model shows that AFLP-based inbreeding coefficients should correlate with inbreeding coefficient. This conclusion was supported by empirical data in which AFLP-based inbreeding coefficients were found to correlate strongly with f. Empirical data were also used to compare the performance of AFLP and microsatellite at estimating inbreeding. In this respect, in our dataset 94 AFLP markers were equivalent to 12 microsatellite markers. We also propose a method for estimating allele frequencies and f values from AFLP markers among a group of organisms with unknown f values as long as an appreciable proportion of them are outbred. We show this method works well both on simulated data and on our mouse dataset.
When allele frequencies can be determined extrinsically from a large sample known outbred individuals, f values can be estimated from AFLP data using a simple equation based on the excess of null phenotypes. We confirm this using stochastic simulations. However, in most real scenarios the requirement is to estimate both allele frequencies and f values from the same dataset. A general solution is not possible due to insufficient degrees of freedom, and a partial solution based on estimating the overall distribution of allele frequencies rather than for each locus separately reveals an unusable level of bias (Holsinger et al., 2002). We also find bias in two different approaches we trial, one where progressively more and more individuals are treated as being inbred, and one that matches the real data to simulated distributions. However, these biases are complementary and by combining the two methods and at the same time assuming that at least half the individuals are outbred (typically levels of inbreeding are far lower in wild populations), we produce an algorithm that seems effective at estimating the underlying allele frequencies and hence the f values of individuals concerned. Applied to our mouse dataset, this method is strikingly successful, recovering estimates of f that are arguably better than those obtained using allele frequencies from our small set of outbred individuals. Our approach should be of utility in real scenarios, where the identity of inbred individuals is unknown.
Although the AFLP-based estimator of inbreeding coefficients works well on simulated data, when applied to real data from a mouse pedigree, the gradient of the relationship between known f and fAFLP was significantly steeper than the slope of unity expected. There are a number of possible reasons for this difference. First, the simulations were based on unlinked biallellic dominant markers. While it is expected that most AFLP markers fit these three criteria (Krauss, 1999; Questiau et al., 1999; Parsons and Shaw, 2001) it is likely that a few of the 94 AFLP markers used may not. Second, there will be some scoring error although this should be low in our study as only easily scoreable loci were used. Third, as the calculation of fAFLP involved estimating Pexp and pi from the genotypes of only 35 individuals, fAFLP values are likely to be imprecise, though the resulting error is unlikely to be either large or systematic. Fourth, there will be some bias in the loci used in the analysis. A proportion of AFLP markers with very low present or absent allele frequencies would be excluded from the analyses as they would appear to be absent or monomorphic in the genotyped samples. While the individual influence of each factor on fAFLP is expected to be small, the cumulative effect is probably responsible for the significant departure of the empirical data from theoretical expectations. Finally, allele frequencies may have been influenced by natural selection, particularly given that wild-caught mice may be adapting to captive conditions.
A small number of studies have compared pedigree-based inbreeding coefficients with heterozygosity estimates based on microsatellite genotypes, reviewed in Slate et al. (2004). Three of the seven systems examined show significant correlations between the two parameters. However, the population sizes for some of these systems are small (wolves, Canis lupus, n=30; Hedrick et al., 2001), while in some of the larger systems there is some evidence of inaccuracies in the pedigrees (Coopworth sheep; Slate et al., 2004). Our sample size of 179 individuals is greater than those in most previously reported studies. In addition, our experimental animals were kept under laboratory conditions and the sexes kept apart unless matings were required for the purposes of the study (Lacy et al., 1996). Under these conditions, inaccuracies in the pedigree that lead to errors in the calculation of inbreeding coefficients are expected to be very low or non-existent. Thus, results obtained from our dataset are unlikely to be affected by methodological inaccuracies, other than those caused by the assumption that the wild-caught founders were unrelated and non-inbred.
Comparing AFLPs and microsatellites we find that while both perform well at estimating both f and relatedness, per locus scored, microsatellites in general outperformed AFLP markers. Molecular markers carry information about f and relatedness through allelic state. For f this is the identity of alleles within an individual, while for relatedness it is through the identity of alleles between individuals. However, in both cases what is important is the identity of alleles by descent. Therefore, loci with higher allelic diversity carry more information due to the greater probability that when alleles are identical by state they are also identical by descent. The information carrying content of each locus will also be affected by the allele frequency distribution, loci with highly skewed distributions in general being less informative. In our study there were on average 10.3 alleles per microsatellite locus compared to only two alleles per AFLP locus. The dominant nature of AFLP markers combined with low allelic diversity account for the lower per locus performance of AFLP compared to microsatellite markers. However, the low per locus information content of AFLP markers is compensated for by the fact that large numbers of polymorphic AFLP loci are usually readily amplified.
One possible way to improve fAFLP might be to weight each locus according to its information content. This can be conveniently achieved by multiplying the contribution of each locus by the inverse of the variance of the single locus estimate, p/2−p (where p is the null allele frequency; K Ritland, Personal Communication). In practice, in our simulations we find that this does reduce the variance of the fAFLP values when mean f is low or zero by ∼10%. However, as f increases the benefit reduces and even reverses, particularly when applied to our method for estimating f without an extrinsic set of unrelated individuals. Presumably this is related to the fact that the weighting factor assumes f=0. In addition, the weighting appears to introduce a small but consistent negative bias (∼1%). For these reasons, we do not present results that incorporate locus weight, but this is clearly an interesting avenue for future research.
While both AFLP-based inbreeding coefficient and microsatellite heterozygosity correlate strongly with f, there is a large amount of unexplained variance with only ∼30 and ∼40% of the variance in f being explained by each marker type, respectively. Much of the unexplained variance is likely due to ‘noise’ caused by the same problem of alleles in an individual being identical by state but not identical by descent. This problem should be greater for AFLP markers as there are only two alleles per locus. The ability of AFLP markers to estimate f will also be adversely affected by the fact that being dominant markers, at each locus the presence of a band could indicate either a homozygote or a heterozygote. The fact that a model including both microsatellite heterozygosity and AFLP-based inbreeding coefficient explained significantly more variance in f than either predictor separately suggests that using more markers, whatever they are, will tend to improve estimates of f, and that data from different markers can be combined. When data from AFLP and microsatellites are combined, the variance explained exceeds 50%, suggesting that very large numbers of markers would yield excellent estimates of f.
In systems with random mating, simulation studies indicate that incest occurs too rarely to be detectable using genetic markers (Balloux et al., 2004; Slate et al., 2004). Thus, in a homogenous population of 1000 individuals, use of around 10 microsatellites cannot detect inbreeding, a situation that does not change even when the number of markers is raised to 200 (Balloux et al., 2004; Slate et al., 2004). This leads to the question of whether or not AFLP or microsatellite loci can detect inbreeding in wild populations. For the most part, the answer is probably no. However, there are a number of exceptions. Large predators and organisms living on islands or in fragmented habitats often exist in small populations. In addition, most populations are not homogenous and instead have some level of population structuring making incestuous matings more probable. In particular, overlapping generations, high reproductive skew and philopatry may create circumstances in which close inbreeding may occur naturally (Hoffman et al., 2004). Moreover, in many plant species, the absence of opportunities for cross-fertilisation can lead to self-fertilisation and other forms of close inbreeding (Stebbins, 1950; Lande, 1985). Thus, the combination of small population sizes with particular population structures and mating systems may give rise to circumstances under which levels of inbreeding in wild populations are high enough to be detected using molecular markers.
Given uncertainties about the frequency of close inbreeding in natural populations, the question arises as to whether AFLP or microsatellite loci can detect inbreeding in wild P. polionotus. We did not find any significant correlation between our AFLP and microsatellite-based estimators of inbreeding for the 35 founding wild mice, suggesting that levels of inbreeding in these mice are too low to be detected. However, the sample size for this comparison is small and a significant correlation might not be detectable without many more samples. Furthermore, the pairwise relatedness estimates between the wild mice based on AFLP and microsatellites do correlate significantly with one another. This is encouraging because it implies that there are detectable levels of relatedness between some of the wild mice. Since molecular estimates of f are directly analogous to estimates of parental relatedness, but based on half the information (one allele from each parent per locus instead of four alleles across the two individuals) it seems reasonable to conclude that the conditions we deployed are at least close to being able to detect inbreeding in this system.
A number of recent studies have employed microsatellite markers to investigate correlations between heterozygosity and fitness (Coltman et al., 1998; Slate et al., 2000; Amos et al., 2001), several of which report marker specific effects (Heath et al., 2002; Merila et al., 2003; Acevedo-Whitehouse et al., 2006) with a few loci contributing excessively to the overall heterozygosity-fitness correlation (HFC). These loci are thought to be linked to genes experiencing balancing selection. The chance of finding single locus heterosis with microsatellite markers is limited by the number of loci genotyped. Although the current move in HFC studies is towards greater genome coverage using larger numbers of loci, such studies are still constrained by the number of microsatellite loci available, typically only 10–15 loci (Hoffman et al., 2004; Acevedo-Whitehouse et al., 2006). Unless the organism being studied is a model system, the need for large numbers of microsatellites requires extensive microsatellite cloning effort, or the screening of loci isolated in other species which often suffer from higher null allele frequencies.
We propose that AFLP loci offer a useful alternative class of markers to microsatellites for estimating f and for the investigation of HFCs. Large numbers of polymorphic AFLP loci can be readily amplified in the majority of systems without the need for time consuming cloning. AFLP markers will allow a larger proportion of an organism's genome to be covered, potentially allowing investigation of linkage to greater numbers of genes than is usual using microsatellites. However, as AFLPs are dominant markers with only two alleles per locus, the problem of lower information content per locus compared to microsatellites will reoccur. This may mean that HFCs are not detectable unless large sample sizes are used or the correlation is strong. Although the utility of AFLP markers in HFC detection has yet to be tested, they may prove to be a valuable additional molecular tool in such studies, especially as they are easily amplifiable in any species without requiring optimisation or primer development.
References
Acevedo-Whitehouse K, Spraker TR, Lyons E, Melin SR, Gulland F, Delong RL et al. (2006). Contrasting effects of heterozygosity on survival and hookworm resistance in California sea lion pups. Mol Ecol 15: 1973–1982.
Amos W, Wilmer JW, Fullard K, Burg TM, Croxall JP, Bloch D et al. (2001). The influence of parental relatedness on reproductive success. Proc R Soc Lond B 268: 2021–2027.
Ballou JD (1997). Ancestral inbreeding only minimally affects inbreeding depression in mammalian populations. J Hered 88: 169–178.
Balloux F, Amos W, Coulson T (2004). Does heterozygosity estimate inbreeding in real populations? Mol Ecol 13: 3021–3031.
Blouin MS, Parsons M, Lacaille V, Lotz S (1996). Use of microsatellite loci to classify individuals by relatedness. Mol Ecol 5: 393–401.
Bonnet E, Van de Peer Y (2002). Zt: a software for simple and partial Mantel tests 7: 1–12.
Chirhart SE, Honeycutt RL, Greenbaum IF (2000). Microsatellite markers for the deer mouse Peromyscus maniculatus. Mol Ecol 9: 1669–1671.
Coltman DW, Bowen WD, Wright JM (1998). Birth weight and neonatal survival of harbour seal pups are positively correlated with genetic variation measured by microsatellites. Proc R Soc Lond B 265: 803–809.
Coltman DW, Pilkington JG, Smith JA, Pemberton JM (1999). Parasite-mediated selection against inbred Soay sheep in a free-living island population. Evolution 53: 1259–1267.
Coulson T, Albon S, Slate J, Pemberton JM (1999). Microsatellite loci reveal sex-dependent responses to inbreeding and outbreeding in red deer calves. Evolution 53: 1951–1960.
Crnokrak P, Roff DA (1999). Inbreeding depression in the wild. Heredity 83: 260–270.
Crow JF, Kimura M (1970). An Introduction to Population Genetic Theory. Harper & Row: New York.
Ellegren H (1999). Inbreeding and relatedness in Scandinavian grey wolves Canis lupus. Hereditas 130: 239–244.
Frankham R, Ballou JD, Briscoe DA (2002). Introduction to Conservation Genetics. Cambridge University Press: Cambridge.
Greenwood P, Harvey P, Perrins C (1978). Inbreeding and dispersal in the great tit. Nature 271: 52–54.
Hardy OJ (2003). Estimation of pairwise relatedness between individuals and characterization of isolation-by-distance processes using dominant genetic markers. Mol Ecol 12: 1577–1588.
Hauser TP, Loeschke V (1996). Drought stress and inbreeding depression in Lychnis flos-cucli (Caryophyllaceae). Evolution 50: 1119–1126.
Heath DD, Bryden CA, Shrimpton JM, Iwama GK, Kelly J, Heath JW (2002). Relationships between heterozygosity, allelic distance (d2), and reproductive traits in chinook salmon, Oncorhynchus tshawytscha. Can J Fish Aquat Sci 59: 77–84.
Hedrick P, Fredrickson R, Ellegren H (2001). Evaluation of d2, a microsatellite measure of inbreeding and outbreeding, in wolves with a known pedigree. Evolution 55: 1256–1260.
Hoffman JI, Boyd IL, Amos W (2004). Exploring the relationship between parental relatedness and male reproductive success in the Antarctic fur seal Arctocephalus gazella. Evolution 58: 2087–2099.
Holsinger KE, Lewis PO, Dey DK (2002). A Bayesian approach to inferring population structure from dominant markers. Mol Ecol 11: 1157–1164.
Houle D (1989). Allozyme-associated heterosis in Drosophila melanogaster. Genetics 123: 789–801.
Jeffreys AJ, Wilson V, Thein SL (1985). Hypervariable ‘minisatellite’ regions in human DNA. Nature 314: 67–73.
Jones CJ, Edwards KJ, Castaglione S, Winfield MO, Sala F, vandeWiel C et al. (1997). Reproducibility testing of RAPD, AFLP and SSR markers in plants by a network of European laboratories. Mol Breed 3: 381–390.
Keller LF (1998). Inbreeding and its fitness effects in an insular population of song sparrows (Melospiza melodia). Evolution 52: 240–250.
Keller LF, Grant PR, Grant BR, Petren K (2007). Environmental conditions affect the magnitude of inbreeding depression in survival of Darwin's finches. Evolution 56: 1229–1239.
Kempenaers B, Adriaensen F, van Noordwijk AJ, Dhondt AA (1996). Genetic similarity, inbreeding and hatching failure in blue tits: are unhatched eggs infertile? Proc R Soc Lond B 263: 179–185.
Krauss SL (1999). Complete exclusion of nonsires in an analysis of paternity in a natural plant population using amplified fragment length polymorphism (AFLP). Mol Ecol 8: 217–226.
Kuhnlein U, Zadworny D, Dawe Y, Fairfull RW, Gavora JS (1990). Assessment of inbreeding by DNA fingerprinting: development of a calibration curve using defined strains of chickens. Genetics 125: 161–165.
Lacy RC, Alaks G, Walsh A (1996). Hierarchical analysis of inbreeding depression in Peromyscus polionotus. Evolution 50: 2187–2200.
Lacy RC, Ballou JD (1998). Effectiveness of selection in reducing the genetic load in populations of Peromyscus polionotus during generations of inbreeding. Evolution 50: 2187–2200.
Lande R (1985). The evolution of self-fertilization and inbreeding depression in plants. Evolution 39: 24–40.
Lynch M, Milligan BG (1994). Analysis of population genetic structure with RAPD markers. Mol Ecol 3: 91–99.
Lynch M, Ritland K (1999). Estimation of pairwise relatedness with molecular markers. Genetics 152: 1753–1766.
Madden JR, Lowe TJ, Fuller HV, Coe RL, Dasmahapatra KK, Amos W et al. (2004). Neighbouring male spotted bowerbirds are not related, but do maraud each other. Anim Behav 68: 751–758.
Marshall TC, Coltman DW, Pemberton JM, Slate J, Spalton JA, Guinness FE et al. (2002). Estimating the prevalence of inbreeding from incomplete pedigrees. Proc R Soc Lond B 269: 1533–1539.
Menotti-Raymond M, O'Brien SJ (1995). Evolutionary conservation of ten microsatellite loci in four species of Felidae. J Hered 86: 319–322.
Merila J, Sheldon B, Griffith S (2003). Heterotic effects on fitness in a wild bird population. Ann Zool Fenn 40: 269–280.
Mueller UG, Wolfenbarger LL (1999). AFLP genotyping and fingerprinting. Trends Ecol Evol 14: 389–394.
Parsons YM, Shaw KL (2001). Species boundaries and genetic diversity among Hawaiian crickets of the genus Laupala identified using amplified fragment polymorphism. Mol Ecol 10: 1765–1772.
Pemberton JM, Albon S, Guinness FE, Clutton-Brock TH (1991). Countervailing selection in different fitness components in female red deer. Evolution 45: 93–103.
Pray LA, Goodnight CJ (1995). Genetic variation in inbreeding depression in the red flour beetle Tribolium castaneum. Evolution 49: 176–188.
Primmer CR, Moller AP, Ellegren H (1996). A wide-ranging survey of cross-species microsatellite amplification in birds. Mol Ecol 5: 365–378.
Prince K, Glenn T, Dewey M (2002). Cross-species amplification among Peromyscines of new microsatellite DNA loci from the oldfield mouse (Peromyscus polionotus subgriseus). Mol Ecol Notes 2: 133–136.
Queller DC, Goodnight KF (1989). Estimating relatedness using genetic markers. Evolution 43: 258–275.
Questiau S, Eybert MC, Taberlet P (1999). Amplified fragment length polymorphism (AFLP) markers reveal extra-pair parentage in a bird species: the bluethroat (Luscinia svecica). Mol Ecol 8: 1331–1339.
Ralls K, Ballou JD, Templeton A (1988). Estimation of lethal equivalents and the cost of inbreeding in mammals. Conserv Biol 2: 185–193.
Raymond M, Rousset F (1995). GENEPOP (version 1.2) population genetics software for exact test and ecumenicism. J Hered 86: 248–249.
Ritland K (1996). Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res 67: 175–185.
Ritland K (2005). Multilocus estimation of pairwise relatedness with dominant markers. Mol Ecol 14: 3157–3165.
Ritland K, Travis S (2004). Inferences involving individual coefficients of relatedness and inbreeding in natural populations of Abies. For Ecol Manage 197: 171–180.
Rosendahl S, Taylor JW (1997). Development of multiple genetic markers for studies of genetic variation in arbuscular mycorrhizal fungi using AFLP.™ Mol Ecol 6: 821–829.
Sambrook J, Fritsh EF, Maniatis T (1989). Molecular Cloning: A Laboratory Manual 2nd edn. Cold Spring Harbor Laboratory Press: New York.
Schmidt CA (1999). Variance and congruence of microsatellite markers for Peromyscus leucopus. J Mammal 80: 522–529.
Siemer BL, Harrington CS, Nielsen EM, Borck B, Nielsen NL, Engberg J et al. (2004). Genetic relatedness among Campylobacter jejuni serotyped isolates of diverse origin as determined by numerical analysis of amplified fragment length polymorphism (AFLP) profiles. J Appl Microbiol 96: 795–802.
Slate J, David P, Dodds KG, Veenvliet BA, Glass BC, Broad TE et al. (2004). Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expectations and empirical data. Heredity 93: 255–265.
Slate J, Kruuk LEB, Marshall TC, Pemberton JM, Clutton-Brock TH (2000). Inbreeding depression influences lifetime breeding success in a wild population of red deer (Cervus elephas). Proc R Soc Lond B 267: 1657–1662.
Stebbins GL (1950). Variation and Evolution in Plants. Columbia University Press: New York.
Stephens JC, Gilbert DA, Yuhki N, O'Brien SJ (1992). Estimation of heterozygosity for single-probe multilocus DNA fingerprints. Mol Biol Evol 9: 729–743.
Vos P, Hogers R, Bleeker M, Reijans M, Lee Tvd, Hornes M et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23: 4407–4414.
Walsh P, Metzger DA, Higuchi R (1991). Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10: 506–513.
Wang JL (2002). An estimator for pairwise relatedness using molecular markers. Genetics 160: 1203–1215.
Wang JL (2004). Estimating pairwise relatedness from dominant genetic markers. Mol Ecol 13: 3169–3178.
Williams J, Kubelik A, Livak K, Rafalski J, Tingey S (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18: 6531–6535.
Wright S (1922). Coefficients of inbreeding and relationship. Am Nat 56: 330–338.
Acknowledgements
Excellent animal care was provided by Glen Alaks and Allison Walsh. We thank Jinliang Wang, Joseph Hoffman, Kermit Ritland the handling editor and three anonymous referees for valuable advice and comments that have substantially improved the manuscript. Funding for the study was provided by the Biotechnology and Biological Sciences Research Council and the Chicago Zoological Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dasmahapatra, K., Lacy, R. & Amos, W. Estimating levels of inbreeding using AFLP markers. Heredity 100, 286–295 (2008). https://doi.org/10.1038/sj.hdy.6801075
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6801075
Keywords
This article is cited by
-
Origin, demographics, inbreeding, phylogenetics, and phenogenetics of Karamaniko breed, a major common ancestor of the autochthonous Greek sheep
Tropical Animal Health and Production (2022)
-
Spatial patterns of AFLP diversity in Bulbophyllum occultum (Orchidaceae) indicate long-term refugial isolation in Madagascar and long-distance colonization effects in La Réunion
Heredity (2016)
-
Sexual reproduction efficiency and genetic diversity of endangered Betula humilis Schrk. populations from edge and sub-central parts of its range
Folia Geobotanica (2016)
-
From glacial refugia to wide distribution range: demographic expansion of Loropetalum chinense (Hamamelidaceae) in Chinese subtropical evergreen broadleaved forest
Organisms Diversity & Evolution (2016)
-
Dispersal constraints for the conservation of the grassland herb Thymus pulegioides L. in a highly fragmented agricultural landscape
Conservation Genetics (2015)
