
Abstract
Study design:
Case report.
Setting:
Temple University Children's Medical Center in USA.
Objectives:
To report a patient whose recurrent and transient episodes of quadriplegia mimicked cervical cord neurapraxia. Cervical spine neuroimaging revealed congenital intersegmental fusion of C5 through C7 (consistent with Klippel–Feil anomaly), corresponding abnormal spinal cord signals on T2-weighted images and enhancing focal lesion opposite the C4 vertebral body. A posterior cervical decompression at C4–C5 and lateral mass fixation was performed, and the episodic quadriplegia has not recurred.
Conclusion:
Understanding of the biomechanics of Klippel–Feil anomaly may facilitate prompt referral for surgical management and avoidance of permanent disability.
Similar content being viewed by others

Introduction
Klippel–Feil (K–F) anomaly is characterized by the congenital fusion of any two of the seven cervical vertebrae. It is caused by a failure in the normal division or segmentation of the cervical vertebrae during the early weeks of fetal development.1 The clinical triad of short neck, low hairline, and restricted neck motion, in a patient with cervical vertebrae fusion, defines K–F syndrome, which may be associated with deformities of the skull, face, spine, and extremities, and abnormalities of the genitourinary, cardiac, respiratory, auditory, or central nervous system.2
First described in 1986, cervical cord neurapraxia (CCN) typically presents in young adult contact-sport participants, especially high school American-football players. These individuals experience acute neurological episodes of cervical cord origin immediately after hyperextension or flexion cervical injuries.3 Contrary to commonsense, and often contrary to medical advice, many athletes with CCN return to competition and experience second and third episodes of transient myelopathy; yet, serious spinal cord injury with persisting disability seems to be uncommon.4
We present the case of a non-athletic young male with non-traumatic, non-progressive, recurrent neurological events in the presence of the K–F anomaly.
Case report
A 16-year-old male presented to neurology clinic after six episodes of sudden onset of transient quadriplegia without altered sensorium. Each episode was preceded by sudden hyperextension of the neck followed by a full neurological recovery within 10 min. The patient had no other complaints or trauma. He was born at 37 weeks of gestation without perinatal problem. His motor development was normal, but he had a history of speech delay.
General examination revealed normal hairline and position of the scapula (no elevation or Sprengel anomaly). His neck motion was mildly restricted on hyperextension. Neurological examination was normal.
Routine laboratory blood tests, erythrocyte sedimentation rate, thyroid functions, serum folate and B-12, rheumatoid factor, antinuclear antibody, very long chain fatty acids, and magnetic resonance imaging (MRI) of the brain were normal. Cervical spine computerized tomography (CT) and MRI are shown in Figures 1 and 2, respectively. Median nerve somato-sensory-evoked potential findings suggested white matter pathway lesions.

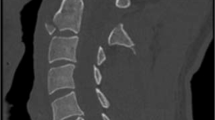
Cervical spine CT sagittal reconstruction shows loss of the normal curvature of the neck, fusion of vertebral bodies, posterior elements and decrease in the size of C5, C6, and C7 vertebrae (note the angulation at C4–C5, just cephalad to the C5, C6, and C7 block suggesting possible instability at this level)

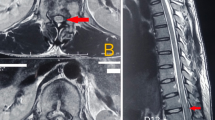
Cervical spine sagittal MRI. (a) T2-weighted image shows spinal canal narrowing at C3–C4 and C4–C5 levels owing to desiccated discs bulge, increase in signal intensity, and bulging of cervical spinal cord between central canal stenoses. (b) T1-weighted post-gadolinium image shows a focal enhancement of cervical spinal cord at the level of C4 and corresponding ventral K–F anomaly
Neurosurgical intervention
A posterior cervical decompression at C4–C5 and lateral mass fixation (shown in Figure 3) were performed by the consulting neurosurgeon.

Cervical spine X-rays. Antero-posterior (a) and lateral views (b) show interval placement of rods and interpendicular screws spanning from C4 to C5 after neurosurgical posterior cervical decompression
Discussion
Quadriplegia without mental status change may be a presentation of central or peripheral nervous system disease, as seen in the Locked-In or Guillain–Barre syndromes, respectively. In the absence of cranial nerve dysfunction, the recurrent quadriplegia in the present case suggested a mid-cervical spinal cord origin. Because of the sudden onset of symptom, followed by complete neurological recovery, CCN was suspected. However, the neuroimaging findings of congenital cervical vertebral fusions, disc herniation, and a focal gadolinium-enhanced spinal cord lesion imparted to our patient a different diagnosis with a more ‘malignant’ prognosis. The isolated finding of fused cervical vertebrae and the absence of any other congenital abnormality made the diagnosis of K–F anomaly as opposed to K–F syndrome.
Treatable conditions, like thyrotoxic-hypokalemic periodic paralysis are worth considering in appropriate clinical situations.5
Pathophysiology
The pathophysiology of the recurrent and transient quadriplegia precipitated by hyperextension of the neck may be a result of the sudden approximation of the posterior-inferior aspect of a vertebral body with the superior aspect of the lamina of the next vertebral body below. This results in a sudden, brief decrease in the antero-posterior diameter of the canal, compressing the cord intermittently. This causes transient disruption of cell axonal membrane permeability, resulting in reversible depolarization.6
Patients with K–F anomaly or syndrome may be particularly vulnerable to canal compromise and spinal cord compression with sudden and unexpected neck movement because of inter-segmental fusions, so-called ‘block vertebrae’, and intervertebral disc degeneration, which occurs at an accelerated rate at the unfused levels, leading to instability and acquired spinal canal stenoses.7
What is the pathogenesis of the focal lesion of the spinal cord? It is likely to be an ‘old scar’ caused by repetitive trauma sustained during hyperextension of the neck owing to altered mechanical dynamics, primarily due to congenitally fused vertebrae.8
Neurosurgical consideration
The vulnerability of patients with K–F syndrome to catastrophic spinal cord injury after minor trauma has long been recognized.9, 10 Anomalies of the craniocervical junction, multisegmental block and adjacent block vertebrae, separated by a single motion segment have been judged to be particularly threatening.
Our patient's anomaly was limited to congenital fusion of two adjacent motion segments, a relatively mild lesion in the spectrum of K–F, but in view of the MRI signs of spinal cord injury, surgical intervention was required to prevent permanent disability.11 Whether prophylactic stabilization is indicated for the genuinely asymptomatic individual with radiographic hypermobility is a more debatable question, because surgical arthrodesis deliberately eliminates motion segments and accelerates the processes of disc degeneration and ligamentous instability at the motion segments that remain.12
Conclusions
The awareness of K–F anomaly, along with other clinical entities that cause recurrent and reversible episodes of quadriparesis or quadriplegia, will allow both the patient and physician to make an informed decision regarding the possibility of permanent neurological damage and the potential options of surgical intervention.
Conflict of Interest:
The author(s) have no financial considerations to disclose or competing interests in relation to this article.
References
Goetz CG . Textbook of Clinical Neurology, 2nd edn, Copyright © 2003 Saunders, an Imprint of Elsevier: Philadelphia, pp 578–579.
Behrman RE . Nelson Textbook of Pediatrics, 17th edn, Copyright © 2004 Saunders, an Imprint of Elsevier: Philadelphia, p 2289.
Torg JS et al. Cervical cord neurapraxia: classification, pathomechanics, morbidity, and management guidelines. J Neurosurg 1997; 87: 843–850.
Nagib MG, Maxwell RE, Chou SN . Identification and management of high-risk patients with Klippel–Feil syndrome. J Neurosurg 1984; 61: 523–530.
Silva MR, Chiamolera MI, Kasamatsu TS, Cerutti JM, Maciel RM . Thyrotoxic hypokalemic periodic paralysis, an endocrine emergency: clinical and genetic features in 25 patients. Arq Bras Endocrinol Metab 2004; 48: 196–215.
Torg JS et al. The pathomechanics and pathophysiology of cervical spinal cord injury. Clin Orthop 1995; 321: 259–269.
Pizzutillo PD, Woods M, Nicholson L, MacEwen GD . Risk factors in Klippel–Feil syndrome. Spine 1994; 19: 2110–2116.
Dickerman RD, Colle KO, Mittler MA . Intramedullary inflammatory mass dorsal to the Klippel–Feil deformity: error in development or response to an abnormal motion segment? Spinal Cord 2004; 42: 720–722.
Hensinger RN, Lang JE, MacEwen GD . Klippel–Feil syndrome; a constellation of associated anomalies. J Bone Joint Surg Am 1974; 56: 1246–1253.
Rouvreau P, Glorion C, Langlais J, Noury H, Pouliquen JC . Assessment and neurologic involvement of patients with cervical spine congenital synostosis as in Klippel–Feil syndrome: study of 19 cases. J Pediatr Orthop B 1998; 7: 179–185.
Vaidyanathan S, Hughes PL, Soni BM, Singh G, Sett P . Klippel–Feil syndrome – the risk of cervical spinal cord injury. BMC Fam Pract 2002; 3: 6.
Nagashima H, Morio Y, Teshima R . No neurological involvement for more than 40 years in Klippel–Feil syndrome with severe hypermobility of the upper cervical spine. Arch Orthop Trauma Surg 2001; 121: 99–101.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gupta, S., Piatt, J. & Belay, B. Cervical spinal cord neurapraxia in the setting of Klippel–Feil anomaly: a diagnostic and therapeutic challenge. Spinal Cord 45, 637–640 (2007). https://doi.org/10.1038/sj.sc.3101999
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.sc.3101999
