
Abstract
This comprehensive study delves into the intricate interplay between protons and organic polymers, offering insights into proton therapy in cancer treatment. Focusing on the influence of the spatial electron density distribution on stopping power estimates, we employed real-time time-dependent density functional theory coupled with the Penn method. Surprisingly, the assumption of electron density homogeneity in polymers is fundamentally flawed, resulting in an overestimation of stopping power values at energies below 2 MeV. Moreover, the Bragg rule application in specific compounds exhibited significant deviations from experimental data around the stopping maximum, challenging established norms.
Similar content being viewed by others
Introduction
In the last two decades, clinical therapy using proton beams to treat cancerous tumors has experienced steady growth1,2,3,4,5. Although this form of radiotherapy already counts with highly developed technology, it still retains significant challenges in terms of physical and clinical aspects6,7,8. One of these challenges is the precise accounting of relative biological effectiveness (RBE), which is the ratio between the doses required by two types of radiation to cause the same biological effect. This factor, measurable through linear energy transfer (LET) or microdosimetry, depends on how the energy is deposited on a micrometric scale9.
In proton therapy, relative biological effectiveness (RBE) is traditionally defined by a constant value of 1.1 (relative to X-ray dose) for all points along the beam path and all stopping points10,11. However, a comprehensive review of the available experimental data in the literature12 reveals that, despite a lack of experimental standardization and large uncertainties, there is evidence that RBE values vary considerably and can exceed 1.1 at the end of the beam range. These differences have clinical implications13,14. Therefore, it is important to accurately reduce experimental uncertainties to describe the effects of proton beams on tissues.
When an ion with kinetic energy moves through matter, it interacts with the target electrons and nuclei, leading to deceleration. These interactions are known as electronic and nuclear stopping, respectively15,16. The stopping power expresses a medium’s force on the ion, leading to the mean energy loss per unit path length while traveling in that medium. In the proton-matter interaction, the electronic stopping power is dominant. There is a very small contribution from nuclear-stopping power for \(v \ll v_{\text {F}}\) (with \(v_{\text {F}}\) being the Fermi velocity). For the current proposal, we will focus solely on calculations of electronic stopping power.
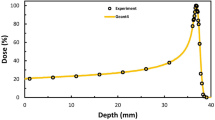
The energy of the proton transferred to the biological tissue is directly related to its velocity. As the proton slows down, the energy transferred to the tissue per unit path length, determined by the electronic stopping power, increases, resulting in maximum dose deposition at a specific depth. This region around the peak of maximum dose deposition is known as the Bragg peak17 and is closely related to the stopping maximum. It is the region of greatest interest in proton beam radiotherapy applications, and its precise positioning is crucial during the definition of the irradiation plan. This particular profile of proton beam energy deposition presents significant clinical advantages, especially for pediatric patients, by allowing optimal dose delivery to tumor tissue and by minimizing dose to organs at risk in surrounding areas, thus reducing the chances of future complications and induction of secondary tumors18,19,20. On the other hand, the high and relatively narrow dose peak makes quality control in dose monitoring and precise patient positioning even more crucial, with the risk of damaging health tissues with high radiation doses. Therefore, in-depth investigations of the uncertainties in the range and stopping power values are essential for a more accurate dose distribution in patients21,22.
Accurate knowledge of electronic stopping power is essential in proton therapy. It is also important in many fields of science and technological applications, such as outer space exploration (space weathering), nanotechnology (ion beam modeling), material modifications, and nuclear fusion research (plasma-wall interaction)23,24,25,26,27. However, as depicted above, its most critical application lies in dosimetry for cancer treatment using ions, given the increasing global use of protons and heavier ions in radiation therapy and the risks involved28,29. For convenience, stopping power can be normalized by the atomic density to eliminate the dependence on the material’s density. The resulting quantity is known as the stopping cross-section (SCS). Therefore, SCS is a fundamental quantity that requires a detailed understanding of energy-loss processes.
Perturbation theories employing the dielectric function were used to compute electronic stopping power30,31,32,33,34,35,36, presenting limitations in energy ranges relevant to studying biological damage caused by the radiation. Real-time time-dependent density functional theory (TDDFT) is a non-perturbative method based on modern quantum-mechanical simulations. It has been used as an alternative approach to investigate electronic stopping in complex systems like water and DNA37,38,39,40,41,42. However, using the precise atomic structure of complex chemical systems, such as DNA, within real-time TDDFT requires the computation of multiple trajectories to obtain an average (random) electronic stopping power and is highly computationally demanding.
On the other side, in investigations into ion-matter interactions, it is customary to employ simplified models, exemplified by the homogeneous, free electron gas (FEG) model, to represent valence electrons within materials. This pragmatic approach facilitates straightforward predictions of stopping power and yields results that closely align with experimental data43,44,45,46,47,48,49. Although the FEG model is reliable for materials with simple electronic structures, its effectiveness diminishes when dealing with materials characterized by complex electronic excitations. Here, we demonstrate that these materials can still be treated as a collection of FEGs with high accuracy, ensuring simplicity and avoiding time-consuming full atomistic ab initio calculations. For this purpose, we utilized electronic stopping power for a FEG with different densities or plasmon frequencies from the real-time TDDFT calculations43,48,50,51. The results were averaged according to the Penn method52.
Knowledge of materials’ energy-loss function (ELF) is essential in this framework. The Penn approach53 introduced an algorithm to determine the electron inelastic mean free paths (IMFP) by utilizing a model dielectric function derived from the experimental ELF specific to the material under investigation. The same model has been applied to estimate the electron stopping power in various materials54 and has been extended to calculate the non-linear stopping power of ions52. This extension involves using the ELF to appropriately weight contributions from different electron gas components within a statistical ensemble that characterizes the material of interest. ELF functions at the optical limit can be found for different materials elsewhere55.
The Bragg rule has been used to calculate the stopping values for compounds such as hydrocarbons. According to this rule, the SCS per atom in a compound is the weighted average of the SCS of each of its constituent elements17, similar to a gas system of non-interacting atoms. The Bragg rule is considered relatively accurate for solids, with measurements of stopping powers for ions in compounds deviating less than 5% from its predictions56. However, the rule applicability has limitations, as the energy lost by the ions to the electrons in a material depends on its detailed orbital and excitation structure, which are affected by neighboring atoms. Additionally, these interactions can alter the charge state of the traveling ion, affecting the intensity of interactions with the medium.
According to Lodhi and Powers57, the Bragg rule may fail close to stopping power maximum for hydrocarbons compared to experimental data. The core and bounds (CAB) approach proposes that the stopping power of compounds can be predicted by combining the stopping caused by the atomic “core” electrons with the corresponding stopping of the bound electrons58,59. The core’s contribution to stopping power is determined by applying the Bragg rule to the atoms in the compound. In contrast, the bounding electrons in the compound would then include the necessary stopping correction. The CAB approach generates corrections to the Bragg rule for polymers containing light elements, such as H, C, N, and O. These light atoms have the most significant bonding effect on stopping powers. The application of these corrections is explained in reference 59.
By applying the proposed formalism, we aim to verify the validity of the Bragg rule and the FEG model, assuming homogeneous electron density in the context of organic polymers. Specifically, we examined the cases of polyethylene (PE), polystyrene (PS), poly(2-vinylpyridine) (P2VP), polyacetylene (PA), poly(methyl methacrylate) (PMMA) and polyimide (PI). The study of these polymers is important because virtually all phantoms used for dose verification and quality assurance in proton therapy treatments are manufactured with polymers such as PMMA. Furthermore, some components that make up the proton accelerators are constructed with PE or PS32,60,61,62,63,64,65.
Results and discussion
Real-time TDDFT-Penn calculations were performed according to Eqs. (3) to (5), and the electronic SCS results for PE, PS, P2VP, PA, PMMA, and PI to energetic protons are presented in Fig. 2, 3, 4, 5, 6 and 7, respectively. The data used to calculate SCS with the real-time TDDFT-Penn method are listed in Table 1, and the optical-ELF data for each polymer are shown in Fig. 1.
We compare the results of our approach with ICRU4968, ICRU3769, SRIM-201370, and real-time TDDFT with the homogeneous assumption. This comparison shows the need to completely break down the assumption of spatial homogeneity of the valence electron density in complex materials, such as polymers. For example, the homogeneous assumption leads to overestimating the SCS values for proton energies below approximately 2 MeV compared to the SRIM-2013 data. Our approach produces more realistic values and, at the same time, offers a physically sound approach to dealing with inhomogeneities.

We also included in the comparison the experimental data available at the IAEA database71,72 (uppercase letters), with which the real-time TDDFT-Penn results show an excellent agreement, as well as with semi-empirical SRIM-201370 results (red dashed line) using the Bragg rule17, as can be seen in Figs. 2 and 3. SCS results from the dielectric formalism, particularly the Mermin-Energy-Loss-Function Generalized Oscillator Strength model (MELF-GOS)31, are also included in the comparisons. This approach also considers inhomogeneities in the material’s electron density utilizing a similar optical ELF. Thus, both methods will give similar mean excitation energies (I) (occurring in the Bethe formula for fast projectiles). Several studies have employed this approach to calculate SCS in biological media31,32,38,73. However, even though such a linear model typically performs better at projectile energies above the maximum for protons, it underestimates the SCS by a significant amount at the maximum for the present polymers. Because this theoretical model is linear, it loses accuracy for ion energies around the stopping maximum and below. In this energy range, the non-linear effects become significant. Even though we have not presented MELF-GOS results for PE, we expect similar behavior to the others. This issue is expected to be significantly more severe for heavier projectiles than protons.

Proton SCS in PE polymer. Real-time TDDFT results with a unique FEG (\(r_s=1.75\) au) are shown in the blue dash-dot line, and the real-time TDDFT-Penn is in the cyan short dash line. Experimental data (uppercase letters) around the stopping maximum71,72. Semi-empirical models ICRU4968 and SRIM-201370 presented.
In particular, the real-time TDDFT-Penn results for PS (refer to Fig. 3) agree better with the experimental data than SRIM-2013. SRIM-2013 employs the Bragg rule, resulting in an excitation energy (I) for PS of 65.5 eV74. However, electron energy loss spectroscopy (EELS) data from experiments67 for the compound PS suggest a lower I value (59.3 eV). This observation explains the higher values obtained with our approach around the stopping maximum.

Proton SCS in PS polymer. Real-time TDDFT results using a unique FEG (\(r_s=1.66\) au) and real-time TDDFT-Penn. Experimental data (uppercase letters) concentrated around the stopping maximum. Dielectric formalism results in purple dash-dot line32. Semi-empirical models ICRU4968 and SRIM-201370 showcased.
SCS results for P2VP and PA (see Figs. 4 and 5) agree well with SRIM-2013. However, it is important to note that we did not find any experimental data for comparison in this context. Furthermore, the dielectric approach, employing a similar ELF function as input31, deviates significantly below the position of the stopping maximum.

Interestingly, for PMMA and PI (see Figs. 6 and 7), the Bragg rule indicates 74 eV68 and 79.6 eV69, while the experimental ELF31 points to significantly lower values of 66 eV and 68 eV, respectively. It is worth noting that the core and bond (CAB) correction on SRIM-201370 is small but makes the deviation from our approach even higher, pointing to a correction in the opposite direction.

As shown in Figs. 2, 3, 4, 5, 6 and 7, the differences between real-time TDDFT-Penn and SRIM-2013 are small but more pronounced for PMMA and PI at the position of the stopping maximum. They can be attributed to a stronger breakdown of the Bragg rule due to the complex molecular structures of PMMA and PI, which feature bonds between C, O, and N.
Table 2 shows the different types of chemical bonds in the respective polymeric monomers of PE, PS, P2VP, PA, PMMA, and PI. PMMA and PI exhibit higher chemical structure complexity than other polymers. While PMMA has one \(\hbox {C}=\hbox {O}\) and two \(\hbox {C}-\hbox {O}\) bonds, PI has four \(\hbox {C}=\hbox {O}\), two \(\hbox {C}-\hbox {O}\) bonds, and four \(\hbox {N}-\hbox {C}\) bonds. The electronegativity of the atoms in these bonds varies, with oxygen being more electronegative than nitrogen, which is more electronegative than carbon.

Analogously, in TiN compounds, there is a transfer of 1.51 electrons from titanium to nitrogen49, and the transferred charges. The transfer of charges is expected to be more noticeable in the double bonds between carbon and oxygen in polymers like PMMA and PI. Therefore, our results suggest the possibility of charge transfer occurring from carbon to oxygen or nitrogen on these polymers. If this transfer occurs, employing the Bragg rule will likely lead to a decreased accuracy in predicting the SCS. Indeed, the SRIM-2013 results (red dashed line) for the PMMA and PI compounds are numerically lower in the position of the stopping maximum compared to real-time TDDFT-Penn predictions.
Finally, it should be pointed out that the consideration of neutral hydrogen (H\(^0\)) charge states may affect the calculated stopping power around the stopping power maximum. The presence of neutral hydrogen will reduce the energy loss for target ionization and excitation but will add other energy-loss mechanisms, such as capture and electron loss. Simulations using the CasP program75 show that these mechanisms compensate for the reduction in ionization and excitation.

Conclusion
Theorists have been working for decades to develop methods to model the physical processes responsible for electronic SCS. Although these methods have been successful, they have yet to be able to cover a wide range of energy using a single approach. Some models explain a limited energy range, while others achieve good agreement with experimental data by treating inner and valence electrons differently. This work presents a theoretical framework based on ab initio calculations of different FEGs and the Penn method. This framework achieves excellent agreement with experimental data and reference data tables across a wide range of energy using a single approach for all electrons.
To showcase our approach’s efficiency, we investigated the electronic stopping power of organic polymers for protons. Generally, a single FEG model cannot accurately describe the electronic stopping power around the stopping maximum. Our new method provides a theoretical framework to tackle electron density variations. We have demonstrated the effectiveness of our method by obtaining excellent agreement with the experimental data and the reference data tables for polymers such as PE, PS, P2VP, PA, PMMA, and PI (as shown in Figs. 2, 3, 4, 5, 6 and 7). Our findings highlight the importance of considering the intricate electronic structures of polymers in the theoretical modeling of stopping power.
These findings emphasize the importance of considering the complex electronic structures of polymers in predicting stopping power accurately. The agreement between the real-time TDDFT-Penn approach and the experimental data, indicated by uppercase letters in Figs. 2 and 3, demonstrates the theoretical framework’s precision. However, further validation is necessary, particularly for polymers with limited data.
Differences in predicted mean excitation energies and SCS magnitudes between the real-time TDDFT-Penn approach and semi-empirical models such as SRIM-2013, particularly around the stopping maximum, highlight the potential influence of molecular structure on these predictions. The distinct chemical compositions of PMMA and PI, which have varying electronegativities in their bonds, likely contribute to charge transfer effects not adequately accounted for by the Bragg rule. The primary consequence of the breakdown of the Bragg rule is a reduction in the mean excitation energy by approximately 10% and a decrease in the projectile range by about 3 mm for 200 MeV protons.
In conclusion, the results of this research challenge established assumptions and emphasize the need for precise modeling of materials with complex electronic structures. This study enhances our understanding of ion-polymer interactions. It provides a solid foundation for future applications in proton therapy and other fields where accurate predictions of stopping power are essential.
Simulation methods
Real-time TDDFT approach
Real-time TDDFT is a highly effective ab initio tool for describing electronic stopping power in spherical jelliums. The jellium model assumes a positive background (representing the ion cores) that provides a charge balancing for the electron gas. Compared to fully atomistic models, the advantage of such representation is computational efficiency. The real-time TDDFT in a FEG has been shown to provide accurate results for near-free-electron systems. On the other hand, using an atomistic representation requires careful consideration of trajectories to calculate the random stopping power76,77.
The polymeric media are modeled as jellium spheres in all the DFT and real-time TDDFT calculations. The positive background density of the jellium with radius \(R_\text{cl}\) is defined by \(n_{0}^{+}(\textbf{r})=n_{0}^{+}(r_s) \Theta (R_\text{cl}-r)\), where \(\Theta (x)\) denotes the Heaviside step-function and \(n_{0}^{+}(r_s)\) is the constant bulk density, which depends only on the Wigner-Seitz radius \(r_{s}\): (\(4\pi r_{s}^{3}/3)=1/n_{0}\). The total number of electrons in the neutral clusters, \(N_{e}\), is then given by \(N_{e}=(R_{\text {cl}}/r_{s})^{3}\). Thus, the size of each closed-shell cluster is determined by the density parameters \(r_s\) and the total number of electrons, \(N_{e}=588\). According to the polymer’s ELFs (see Fig. 1), and using the relation \(\omega _p^2=4\pi n_o\), the most important Wigner-Seitz radii vary from \(r_s = 1.00\) to 5.00 au. The jellium spheres corresponding to this range have sizes varying from \(R_{\text {cl}} = 8.38\) to 1.68 au.
Although there have been minor refinements in terms of accuracy, the approach adopted in this work reflects the methodology used in43,48,50,78, and as such, it will be briefly explained in this section. In this approach, the time evolution of electronic density incorporates, in a non-perturbative manner, the complete dynamic interaction between an external field and the medium. This computational framework has been used to analyze various issues in condensed matter systems, such as dynamic charge screening in metallic media79, energy loss of atomic particles in matter43,48,80, as well as many-body effects associated with hole screening in photoemission78.
A static density functional theory (DFT) calculation is performed to obtain the system’s ground state. The time evolution of the complete electronic density, \(n(\textbf{r},t)\), in response to an external field (in this case, a proton), is conducted within the framework of real-time TDDFT in the Kohn–Sham regime (atomic units are used throughout unless specified otherwise):
where \(\psi _j(\textbf{r},t)\) are the Kohn-Sham orbitals and T is the kinetic energy operator. The Kohn-Sham effective potential, \(V_{\text {eff}}([n],\textbf{r},t)\), is a function of the electronic density of the system: \(n(\textbf{r},t)= \sum _{j\in occ.}{\left| \psi _{j}(\textbf{r},t)\right| ^{2}}\). The effective potential \(V_{\text {eff}}=V_\mathrm{{ext}}^+(\textbf{r})+V_\mathrm{{H}}([n],\textbf{r},t)+V_\mathrm{{xc}}([n],\textbf{r},t)+V_\mathrm{{p}}(\textbf{r},t)\) is obtained as the sum of the external potential created by the positive background of the jellium sphere \(V_{\text {ext}}^+(\textbf{r})\), the Hartree potential \(V_{\text {H}}([n],\textbf{r},t)\), the exchange-correlation potential \(V_{\text {xc}}([n],\textbf{r},t)\), and the potential representing the projectile \(V_\mathrm{{p}}(\textbf{r},t)\), which is modeled as a bare Coulomb charge. \(V_{\text {xc}}([n],\textbf{r},t)\) is treated within a standard adiabatic local density approximation (ALDA) approach. The numerical procedure is that employed in Refs.43,79,80,81, where additional details can be found.
The energy loss is calculated by integrating the time-dependent induced force over the proton:
where v is the (constant) velocity at which the proton traverses the jellium. Once the induced force on the proton is calculated, the average or effective stopping power is computed as the energy loss per unit path length, i.e.,
Recently, an alternative non-linear method has been introduced to characterize the stopping power of light and heavy ions in materials52. This method incorporates the influence of non-free electron distributions within a theoretical model for stopping power calculations, such as real-time TDDFT. For a low energy proton (\(v < v_F\)), the Penn approach has been used recently in the transport cross section (TCS)49. This approach considers the combination of electron-gas responses characterized by inhomogeneous densities, similar to the approach outlined in the Penn method53.
Real-time TDDFT-Penn approach
To achieve this goal, each free electron density is analyzed based on the material’s ELF at the optical limit, as follows52:
The stopping power depends on the plasmon frequency \(\omega _p\), a value determined by the individual electron gas contributions obtained from \(r_s\); \(\omega _p=\sqrt{3}r_s^{-3/2}\). Therefore, the stopping power is now calculated as follows52:
In the above equation, the term \(\left[ dE/dz(v,\omega _p)\right] _{\text {TDDFT}}\) is calculated in the real-time TDDFT framework using Eq. (3). Because of that, we named the Eq. (5) as the real-time TDDFT-Penn approach.
Data availibility
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Delaney, G., Jacob, S., Featherstone, C. & Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 104(6), 1129–1137 (2005).
Bernier, J., Hall, E. J. & Giaccia, A. Radiation oncology: A century of achievements. Nat. Rev. Cancer 4(9), 737–747 (2004).
Baskar, R., Lee, K., Yeo, R. & Yeoh, K. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 9(3), 193–199 (2012).
Nogueira, L. M., Jemal, A., Yabroff, K. R. & Efstathiou, J. A. Assessment of proton beam therapy use among patients with newly diagnosed cancer in the US, 2004–2018. JAMA Netw. Open 5(4), e229025 (2022).
Kotecha, R., La Rosa, A. & Mehta, M. P. How proton therapy fits into the management of adult intracranial tumors. Neuro-Oncology 26, S26–S45. https://doi.org/10.1093/neuonc/noad183 (2024).
Loeffler, J. S. & Durante, M. Charged particle therapy-optimization, challenges and future directions. Nat. Rev. Clin. Oncol. 10(7), 411–24 (2013).
Durante, M., Orecchia, R. & Loeffler, J. S. Charged-particle therapy in cancer: Clinical uses and future perspectives. Nat. Rev. Clin. Oncol. 14(8), 483–495 (2017).
Durante, M., Debus, J. & Loeffler, J. S. Physics and biomedical challenges of cancer therapy with accelerated heavy ions. Nat. Rev. Phys. 3(12), 777–790 (2021).
Grassberger, C., Trofimov, A., Lomax, A. & Paganetti, H. Variations in linear energy transfer within clinical proton therapy fields and the potential for biological treatment planning. Int. J. Radiat. Oncol. Biol. Phys. 80(5), 1559–66. https://doi.org/10.1016/j.ijrobp.2010.10.027 (2011).
Paganetti, H. et al. Relative biological effectiveness (RBE) values for proton beam therapy. Int. J. Radiat. Oncol. Biol. Phys. 53(2), 407–421. https://doi.org/10.1016/S0360-3016(02)02754-2 (2002).
Paganetti, H. et al. Report of the AAPM TG-256 on the relative biological effectiveness of proton beams in radiation therapy. Med. Phys. 46(3), e53–e78. https://doi.org/10.1002/mp.13390 (2019).
Paganetti, H. Relative biological effectiveness (RBE) values for proton beam therapy variations as a function of biological endpoint, dose, and linear energy transfer. Phys. Med. Biol. 59(22), R419–R472 (2014).
Peeler, C. R. et al. Clinical evidence of variable proton biological effectiveness in pediatric patients treated for ependymoma. Radiother. Oncol. 121(3), 395–401 (2016).
Underwood, T. et al. OC-0245: Clinical evidence that end-of-range proton RBE exceeds 1.1: Lung density changes following chest RT. Radiother. Oncol. 123, S123–S124 (2017).
Sigmund, P. Particle Penetration and Radiation Effects Vol. 2 (Springer, 2014).
Sigmund, P. Stopping power in perspective. Nucl. Instrum. Methods Phys. Res. Sect. B 135, 1–15. https://doi.org/10.1016/S0168-583X(97)00638-1 (1998).
Bragg, W. & Kleeman, R. On the \(\alpha \) particles of radium, and their loss of range in passing through various atoms and molecules. Philos. Mag. 10, 318 (1905).
Hall, E. J. Intensity-modulated radiation therapy, protons, and the risk of second cancers. Int. J. Radiat. Oncol. Biol. Phys. 65(1), 1–7. https://doi.org/10.1016/j.ijrobp.2006.01.027 (2006).
Merchant, T. E. Proton beam therapy in pediatric oncology. Cancer J. 15(4), 298–305 (2009).
Zhang, R. et al. A comparative study on the risks of radiogenic second cancers and cardiac mortality in a set of pediatric medulloblastoma patients treated with photon or proton craniospinal irradiation. Radiother. Oncol. 113(1), 84–8 (2014).
Charlie Ma, C.-M. & Tony, L. Proton and Carbon Ion Therapy (CRC Press, 2012).
Paganetti, H. Range uncertainties in proton therapy and the role of Monte Carlo simulations. Phys. Med. Biol. 57(11), R99-117. https://doi.org/10.1088/0031-9155/57/11/R99 (2012).
Brunetto, R. & Strazzulla, G. Elastic collisions in ion irradiation experiments: A mechanism for space weathering of silicates. Icarus 179, 265 (2005).
Kim, S., Lee, S. & Hong, J. An array of ferromagnetic nanoislands nondestructively patterned via a local phase transformation by low-energy proton irradiation. ACS Nano 8(5), 4698–704 (2014).
Papaléo, R. M. et al. Confinement effects of ion tracks in ultrathin polymer films. Phys. Rev. Lett. 114, 118302 (2015).
Meisl, G. et al. Implantation and erosion of nitrogen in tungsten. New J. Phys. 16, 093018 (2014).
Mayer, M. et al. Ion beam analysis of fusion plasma-facing materials and components: Facilities and research challenges. Nucl. Fusion 60, 025001. https://doi.org/10.1088/1741-4326/ab5817 (2020).
Newhauser, W. D. & Zhang, R. The physics of proton therapy. Phys. Med. Biol. 60, R155 (2015).
Durante, M. & Loeffler, J. S. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 7(1), 37 (2010).
Emfietzoglou, D., Garcia-Molina, R., Kyriakou, I., Abril, I. & Nikjoo, H. A dielectric response study of the electronic stopping power of liquid water for energetic protons and a new I-value for water. Phys. Med. Biol. 54, 3451. https://doi.org/10.1088/0031-9155/54/11/012 (2009).
de Vera, P., Abril, I. & Garcia-Molina, R. Inelastic scattering of electron and light ion beams in organic polymers. J. Appl. Phys. 109, 094901 (2011).
de Vera, P. et al. Water equivalent properties of materials commonly used in proton dosimetry. Appl. Radiat. Isotop. 83, 122–127 (2013).
Montanari, C. C. & Miraglia, J. E. Neonization method for stopping, mean excitation energy, straggling, and for total and differential ionization cross sections of CH\(_4\), NH\(_3\), H\(_2\)O and FH by impact of heavy projectiles. J. Phys. B 47, 015201. https://doi.org/10.1088/0953-4075/47/1/015201 (2013).
Incerti, S. et al. The geant4-dna project. Int. J. Model. Simul. Sci. Comput. 01, 157–178. https://doi.org/10.1142/S1793962310000122 (2010).
Incerti, S. et al. Geant4-DNA example applications for track structure simulations in liquid water: A report from the Geant4-DNA Project. Med. Phys. 45, e722–e739. https://doi.org/10.1002/mp.13048 (2018).
Tran, H. N., Chappuis, F., Incerti, S., Bochud, F. & Desorgher, L. Geant4-DNA modeling of water radiolysis beyond the microsecond: An on-lattice stochastic approach. Int. J. Mol. Sci.https://doi.org/10.3390/ijms22116023 (2021).
Yao, Y., Yost, D. C. & Kanai, Y. \(K\)-shell core-electron excitations in electronic stopping of protons in water from first principles. Phys. Rev. Lett. 123, 066401. https://doi.org/10.1103/PhysRevLett.123.066401 (2019).
Alcocer- Ávila, M. E., Quinto, M. A., Monti, J. M., Rivarola, R. D. & Champion, C. Proton transport modeling in a realistic biological environment by using TILDA-V. Sci. Rep. 9, 14030. https://doi.org/10.1038/s41598-019-50270-5 (2019).
Gu, B. et al. Efficient ab initio calculation of electronic stopping in disordered systems via geometry pre-sampling: Application to liquid water. J. Chem. Phys. 153, 034113. https://doi.org/10.1063/5.0014276 (2020).
Gu, B. et al. Bragg’s additivity rule and core and bond model studied by real-time TDDFT electronic stopping simulations: The case of water vapor. Radiati. Phys. Chem. 193, 109961. https://doi.org/10.1016/j.radphyschem.2022.109961 (2022).
Shepard, C., Yost, D. C. & Kanai, Y. Electronic excitation response of DNA to high-energy proton radiation in water. Phys. Rev. Lett. 130, 118401. https://doi.org/10.1103/PhysRevLett.130.118401 (2023).
Koval, N. E. et al. Nonlinear electronic stopping of negatively charged particles in liquid water. Phys. Rev. Res. 5, 033063. https://doi.org/10.1103/PhysRevResearch.5.033063 (2023).
Quijada, M., Borisov, A. G., Nagy, I., Muiño, R. D. & Echenique, P. M. Time-dependent density-functional calculation of the stopping power for protons and antiprotons in metals. Phys. Rev. A 75, 042902. https://doi.org/10.1103/PhysRevA.75.042902 (2007).
Goebl, D., Roessler, W., Roth, D. & Bauer, P. Influence of the excitation threshold of d electrons on electronic stopping of slow light ions. Phys. Rev. A 90, 042706 (2014).
Roth, D. et al. Electronic stopping of slow protons in transition and rare earth metals: Breakdown of the free electron gas concept. Phys. Rev. Lett. 118, 103401 (2017).
Roth, D. et al. Electronic stopping of slow protons in oxides: Scaling properties. Phys. Rev. Lett. 119, 163401 (2017).
Sortica, M. A. et al. Electronic energy-loss mechanisms for H, He, and Ne in TiN. Phys. Rev. A 96, 032703 (2017).
Matias, F. et al. Ground- and excited-state scattering potentials for the stopping of protons in an electron gas. J. Phys. B 50, 185201. https://doi.org/10.1088/1361-6455/aa843d (2017).
Matias, F. et al. Nonlinear stopping effects of slow ions in a no-free-electron system: Titanium nitride. Phys. Rev. A 100, 030701(R). https://doi.org/10.1103/PhysRevA.100.030701 (2019).
Koval, N. E., Sánchez-Portal, D., Borisov, A. G. & Muiño, R. D. Dynamic screening and energy loss of antiprotons colliding with excited ai clurters. Nucl. Instrum. Methods Phys. Res. B 317, 56–60 (2013).
Koval, N. E. et al. Vicinage effect in the energy loss of h2 dimers: Experiment and calculations based on time-dependent density-functional theory. Phys. Rev. A 95, 062707 (2017).
Vos, M. & Grande, P. L. Extension schemes of the dielectric function, and their implications for ion stopping calculations. J. Phys. Chem. Solids 133, 187–196. https://doi.org/10.1016/j.jpcs.2019.03.010 (2019).
Penn, D. R. Electron mean-free-path calculations using a model dielectric function. Phys. Rev. B 35(2), 482–486 (1987).
Shinotsuka, H., Tanuma, S., Powell, C. & Penn, D. Calculations of electron stopping powers for 41 elemental solids over the 50 eV to 30 keV range with the full Penn algorithm. Nucl. Instrum. Methods Phys. Res. B 270, 75–92. https://doi.org/10.1016/j.nimb.2011.09.016 (2012).
Sun, Y., Xu, H., Da, B., Mao, S. F. & Ding, Z. J. Database of Fitted Energy-Loss Function for 26 Materials. http://micro.ustc.edu.cn/ELF/ELF.html (2016).
Paul, H. & Schinner, A. Statistical analysis of stopping data for protons and alphas in compounds. Nucl. Instrum. Methods Phys. Res. B 249, 1–5. https://doi.org/10.1016/j.nimb.2006.03.010 (2006).
Lodhi, A. S. & Powers, D. Energy loss of \(\alpha \) particles in gaseous C-H and C-H-F compounds. Phys. Rev. A 10, 2131 (1974).
Both, G., Krotz, R., Lohman, K. & Neuwirth, W. Density dependence of stopping cross sections measured in liquid ethane. Phys. Rev. A 28, 3212 (1983).
Ziegler, J. & Manoyan, J. The stopping of ions in compounds. Nucl. Instrum. Methods Phys. Res. B 35, 215–228. https://doi.org/10.1016/0168-583X(88)90273-X (1988).
Kirby, D. et al. LET dependence of GafChromic films and an ion chamber in low-energy proton dosimetry. Phys. Med. Biol. 55(2), 417–33 (2010).
Söderman, C. et al. Image quality dependency on system configuration and tube voltage in chest tomosynthesis: A visual grading study using an anthropomorphic chest phantom. Med. Phys. 42(3), 1200–12 (2015).
Battaglia, M. C. et al. Dosimetry for low energy protons with ionization chambers and EBT3 films in the Bragg peak region. Phys. Med. 32, 204 (2016).
Casolaro, P. et al. Real-time dosimetry with radiochromic films. Sci. Rep. 9(1), 5307 (2019).
Rezaeian, P., Kashian, S. & Mehrara, R. Investigation of the effective atomic number dependency on kinetic energy using collision stopping powers for electrons, protons, alpha, and carbon particles. Sci. Rep. 13(1), 3573 (2023).
Penner, C. et al. A multi-point optical fibre sensor for proton therapy. Electronicshttps://doi.org/10.3390/electronics13061118 (2024).
Tanuma, S., Powell, C. J. & Penn, D. R. Calculations of electron inelastic mean free paths. V. Data for 14 organic compounds over the 50–2000 eV range. Surf. Interface Anal. 21, 165–176. https://doi.org/10.1002/sia.740210302 (1994).
Inagaki, T., Arakawa, E. T., Hamm, R. N. & Williams, M. W. Optical properties of polystyrene from the near-infrared to the x-ray region and convergence of optical sum rules. Phys. Rev. B 15, 3243 (1977).
ICRU49. ICRU Report 49: Stopping powers and ranges for protons and alpha particles. J. ICRU os-25 (1993).
ICRU37. ICRU Report 37: Stopping powers for electroncs and positrons. J. ICRU os-19 (1984).
Ziegler, J. F., Ziegler, M. & Biersack, J. Srim: The stopping and range of ions in matter. Nucl. Instrum. Methods Phys. Res. B 268, 1818–1823 (2010).
IAEA Stopping Power Database. Electronic Stopping Power of Matter for Ions. https://nds.iaea.org/stopping (version 2024-03).
Montanari, C. & Dimitriou, P. The IAEA stopping power database, following the trends in stopping power of ions in matter. Nucl. Instrum. Methods Phys. Res. B 408, 50–55. https://doi.org/10.1016/j.nimb.2017.03.138 (2016).
Garcia-Molina, R., Abril, I., de Vera, P., Kyriakou, I. & Emfietzoglou, D. A study of the energy deposition pro?le of proton beams in materials of hadron therapeutic interest. Appl. Radiat. Isotop. 83, 109–114 (2014).
Dalton, P. & Turner, J. E. New evaluation of mean excitation energies for use in radiation dosimetry. Health Phys. 15(3), 257–262 (1968).
Grande, P. & Schiwietz, G. Convolution Approximation for Swift Particles (CasP) Program. http://www.casp-program.org/ (CasP version 6.0) (2021).
Gu, B. et al. Efficient ab initio calculation of electronic stopping in disordered systems via geometry pre-sampling: Application to liquid water. J. Chem. Phys. 153(3), 034113 (2020).
Kononov, A., Hentschel, T. W., Hansen, S. B. & Baczewski, A. D. Trajectory sampling and finite-size effects in first-principles stopping power calculations. npj Comput. Mater.https://doi.org/10.1038/s41524-023-01157-7 (2023).
Koval, N. E., Sánchez-Portal, D., Borisov, A. G. & Muiño, R. D. Dynamic screening of a localized hole during photoemission from a metal cluster. Nanoscale Res. Lett. 7, 447 (2012).
Borisov, A. G., Sánchez-Portal, D., Muiño, R. D. & Echenique, P. M. Building up the screening below the femtosecond scale. Chem. Phys. Lett. 387, 95–100 (2004).
Pruneda, J. M., Sánchez-Portal, D., Arnau, A., Juaristi, J. I. & Artacho, E. Electronic stopping power in LiF from first principles. Phys. Rev. Lett. 99, 235501 (2007).
Quijada, M., Muiño, R. D., Borisov, A. G., Alonso, J. A. & Echenique, P. M. Lifetime of electronic excitations in metal nanoparticles. New J. Phys. 12, 053023 (2010).
Acknowledgements
This work was partially supported by IPEN (project number 2020.06.IPEN.32) and CNPq (project number 406982/2021-0). The authors acknowledge FAPESP for supporting the computer cluster (process numbers 2012/04583-8 and 2020/04867-2). TFS and MHT acknowledge the financial support provided by CNPq-INCT-FNA (project number 464898/2014-5).
Author information
Authors and Affiliations
Contributions
F.Matias: Conceptualization, Methodology, Software, Validation, Writing—Original Draft. T. F. Silva: Funding acquisition, Methodology, Validation, Writing—Review & Editing. N. E. Koval: Data Curation, Writing—Review & Editing. J. J. N. Pereira: Data Curation, Writing - Review & Editing. P. C. G. Antunes: Data Curation, Writing - Review & Editing. P. T. D. Siqueira: Data Curation, Writing—Review & Editing. M. H. Tabacniks: Funding acquisition, Data Curation, Writing - Review & Editing. H. Yoriyaz: Funding acquisition, Data Curation, Writing—Review & Editing. J. M. B. Shorto: Funding acquisition, Data Curation, Writing - Review & Editing. P.L. Grande: Methodology, Validation, Writing - Review & Editing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matias, F., Silva, T.F., Koval, N.E. et al. Efficient computational modeling of electronic stopping power of organic polymers for proton therapy optimization. Sci Rep 14, 9868 (2024). https://doi.org/10.1038/s41598-024-60651-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-60651-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
