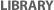
 Figure 1
Figure 1















« Prev Next »
You may find it hard to imagine that something as simple as a single base change—specifically, the appearance of a cytosine instead of a thymine somewhere along the 3-trillion-base-long human genome—could lead to a condition as profound as blindness. Yet, in certain cases of congenital blindness known as Leber congenital amaurosis, this is exactly what happens. On first glance, it seems tantalizingly simple to fix disorders that are caused by mutations in a single gene: Simply insert the correct form of the mutant gene into the cells that need the functioning gene, then stand back and watch the disease disappear. However, the field of gene therapy, in which scientists work to treat disorders at the level of the sequence of mutated genes, is much more complicated than it first appears.
How to Get Therapeutic DNA Inside Cells
One major obstacle facing effective gene therapy involves finding the best way to deliver therapeutic genes into target cells within the body. Scientists have tested a number of methods, some of which utilize liposomes or cell surface receptors. But the most common method of DNA delivery employs something that is perfectly evolved to enter cells: the virus.
Viruses are obligate intracellular parasites, designed through the course of evolution to infect cells, often with great specificity to particular cell types. Viruses efficiently transfect their own DNA into a host cell, then use the host's cellular machinery to replicate their DNA and synthesize certain viral proteins, thereby producing more viral particles. When it comes to gene therapy, scientists can capitalize on this process by removing the disease-causing portions of the viral genome and adding a foreign therapeutic gene. The engineered genome is then repackaged into the viral protein coat and allowed to infect the proper cell target. Now, instead of producing viral toxins and causing disease, the cell infected by this virus is able to produce the protein or enzyme it lacks.
One of the more common ways of introducing the wild-type version of a gene into cells with a defective copy is by way of an adenovirus. It is possible to use recombinant DNA technology to introduce a wild-type gene into an adenoviral vector. Once the adenovirus has infected the target cells, homologous recombination can occur, resulting in replacement of the native, mutant copy of the gene with the wild-type version from the vector (Figure 1).
LCA: A Disease Candidate for Gene Therapy
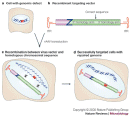
© 2001 Nature Publishing Group Acland, G. et al. Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics 28, 92 (2001). All rights reserved. 
As previously mentioned, the form of blindness known as Leber congenital amaurosis (LCA) arises from a single-base mutation. This mutation acts to disrupt the visual cycle, or the biological conversion of a photon of light to a chemical signal in the retina that is then sent to the visual cortex of the brain. Within this cycle, 11-cis-retinal protein, when struck by a photon of light, undergoes a conformational change that induces signal cascades that stimulate the visual cortex. The RPE65 gene codes for a protein that is critical for the regeneration of 11-cis-retinal protein into its starting form. Without RPE65, there is no 11-cis-retinal protein present to change light signals into chemical signals, and the result is blindness.
About 10% of cases of LCA bear a mutation in the RPE65 gene. Because this form of LCA is the result of a single-gene defect and there are no other treatments available, gene therapy was considered as a possibility for this disorder. Researchers began their work in this area using an animal model of LCA—specifically, a canine model. In dogs, a four-base-pair deletion in the RPE65 gene (denoted RPE65 -/-) alters that gene such that functional protein cannot be not made. Thus, in their initial studies aimed at determining whether adenoviral-based gene therapy might be a reasonable approach to treatment of LCA, investigators infected cells extracted from RPE65 -/- dogs with a recombinant adeno-associated virus (rAAV) that contained wild-type RPE65. The investigators were able to see a rescue of expression in the cells, suggesting that it was indeed possible to rescue expression of the RPE65 protein (Figure 2). It remained to be seen, however, whether this was sufficient to reverse blindness if used in the RPE65 -/- animals.
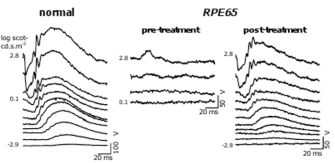
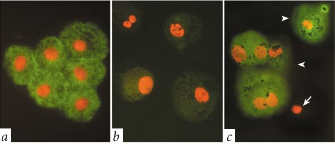
© 2001 Nature Publishing Group Acland, G. et al. Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics 28, 94 (2001). All rights reserved.
Next, in the second stage of research, the investigators applied the gene therapy treatment to RPE65 mutant dogs that were four years of age or younger, and they used a technique called functional magnetic resonance imaging (fMRI) to measure activity in the dogs' brains in response to light stimulation both before and after gene therapy. Following gene therapy, the researchers noted an almost-normal response in the dogs' visual cortices to light stimulation; before treatment, there had been no activity in this region of the dogs' brains. They also noted normal pupil function and normal electrical activity in the retinas of the treated dogs, both of which were abnormal before gene therapy. These findings were important, because they indicated that the visual pathway indeed retains the ability to respond to light and that the visual cortex of the brain can be activated even after years of visual deprivation.
Finally, the research team sought to test these findings in adult human patients with LCA. After using MRI to verify that these patients' optic nerves and visual cortices were anatomically normal, the investigators again turned to fMRI to measure what response, if any, the patients' visual cortices would have to light stimulation. Surprisingly, the researchers noted a normal response to bright light (but not dim light) in these patients (Aguirre et al., 2007). These findings suggested that gene therapy might restore some functional vision to people with LCA, and they therefore served as encouragement to scientists and doctors planning human gene therapy trials for LCA.
Moving from Animals to Humans: Important Considerations
Despite the promise associated with the use of viral vectors in gene therapy, there are also significant risks. One such risk is the possibility that a virus may recover its ability to cause disease once inside the body. Another is the threat of insertional mutagenesis when using a retrovirus. This type of virus inserts its genome within a host cell's chromosomes, thereby potentially disrupting important genes. In the worst-case scenario, the viral genome is inserted into a tumor suppressor gene, thereby causing the development of cancer. There is also the possibility of immune and inflammatory responses whenever any foreign particle, such as a virus, is introduced into the body.
In addition to these physical considerations, scientists in the early days of gene therapy had to address ethical issues beyond those dealing with the safety of their research subjects. Early ethical debate centered on "playing God" and other philosophical and theological issues. By the time gene therapy in humans became a reality in 1990, most people saw gene therapy as simply a different kind of treatment for disease. However, debate continues about the ethical implications of changing germ-line cells (eggs or sperm), which would alter the genetic makeup of future generations.
Beyond safety and efficacy issues, many of the ethical concerns associated with modern-day somatic gene therapy research are also present in many other clinical settings. These include matters related to weighing potential harms and benefits, establishment of procedural fairness in selection of patients for research, assurance that consent to experimental treatments is informed and voluntary, and protection of privacy and confidentiality of medical information.
A Major Setback to the Field of Gene Therapy
Even when all pertinent ethical considerations are met, adverse events can still occur with gene therapy. For instance, in 1999, just nine years after the first human trial for gene therapy was conducted, an 18-year-old gene therapy trial volunteer died. Jesse Gelsinger suffered from ornithine transcarbamylase (OTC) deficiency, an enzyme deficiency that causes an inability to break down ammonia (Somia & Verma, 2000). Four days after receiving an injection of adenovirus carrying a corrected version of the OTC gene, Gelsinger died due to multiple organ failure. His death is believed to have been triggered by a severe immune response to the adenovirus carrier. Questions quickly surfaced about severe side effects and other risks that were undisclosed to Gelsinger on the research consent form, along with suggestions that investigators ignored high ammonia levels in Gelsinger's blood that would have excluded him from the trial. In response, the U.S. Food and Drug Administration temporarily halted all gene therapy trials while the National Institutes of Health conducted thorough reviews of all adverse reactions and deaths associated with this type of research.
Moving Forward: Human Clinical Trials with RPE65 and Additional Target Genes
Gene Therapy Restores Vision to Blind Mice
Learn about another example of how investigators are using gene therapy as a treatment for blindness. Listen to Dr. Masland of Harvard Medical School discuss recent advances from his laboratoy looking at gene therapy to restore vision in mice. Specifically by using an AAV vector expressing melanopsin in murine retinas, his laboratory was able to see restored retinal responses in blind mice.
© 2009 Genetic Engineering and Biotechnology News All rights reserved.
It is important to consider that RPE65 is not the only potential therapeutic gene that could be a target for gene therapy approaches. Other investigators, like those in the Masland laboratory, have been able to restore some retinal function in blind mice using gene therapy for the melanopsin gene (Lin et al., 2008). Thus despite some setbacks, this field is growing and has promise to yield positive therapeutic results for patients in the future.
References and Recommended Reading
Acland, G. M., et al. Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics 28, 92-95 (2001) doi:10.1038/ng0501-92 (link to article)
Aguirre, G. K., et al. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Medicine 4, e230 (2007)
Bainbridge, J. W., et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. New England Journal of Medicine 358, 2231-2239 (2008)
Bemelmans, A. P., et al. Lentiviral gene transfer of Rpe65 rescues survival and function of cones in a mouse model of Leber congenital amaurosis. PLoS Medicine 3, e347 (2006) doi:10.1371/journal.pmed.0030347
Lin, B. et al., Restoriation of visual function in retinal degeneration mice by ectopic expression of melanopsin. PNAS 105, 16009-16014 (2008)
Somia, N., & Verma, I. M. Gene therapy: Trials and tribulations. Nature Reviews Genetics 1, 91-99 (2000) doi:10.1038/35038533 (link to article)
Vasileva, A., & Jessberger, R. Precise hit: Adeno-associated virus in gene targeting. Nature Reviews Microbiology 3, 837-847 (2005) doi:10.1038/nrmicro1266 (link to article)