
Abstract
Macrophages are key inflammatory mediators in many pathological conditions, including cardiovascular disease (CVD) and cancer, the leading causes of morbidity and mortality worldwide. This makes macrophage burden a valuable diagnostic marker and several strategies to monitor these cells have been reported. However, such strategies are often high-priced, non-specific, invasive, and/or not quantitative. Here, we developed a positron emission tomography (PET) radiotracer based on apolipoprotein A1 (ApoA1), the main protein component of high-density lipoprotein (HDL), which has an inherent affinity for macrophages. We radiolabeled an ApoA1-mimetic peptide (mA1) with zirconium-89 (89Zr) to generate a lipoprotein-avid PET probe (89Zr-mA1). We first characterized 89Zr-mA1’s affinity for lipoproteins in vitro by size exclusion chromatography. To study 89Zr-mA1’s in vivo behavior and interaction with endogenous lipoproteins, we performed extensive studies in wildtype C57BL/6 and Apoe-/- hypercholesterolemic mice. Subsequently, we used in vivo PET imaging to study macrophages in melanoma and myocardial infarction using mouse models. The tracer’s cell specificity was assessed by histology and mass cytometry (CyTOF). Our data show that 89Zr-mA1 associates with lipoproteins in vitro. This is in line with our in vivo experiments, in which we observed longer 89Zr-mA1 circulation times in hypercholesterolemic mice compared to C57BL/6 controls. 89Zr-mA1 displayed a tissue distribution profile similar to ApoA1 and HDL, with high kidney and liver uptake as well as substantial signal in the bone marrow and spleen. The tracer also accumulated in tumors of melanoma-bearing mice and in the ischemic myocardium of infarcted animals. In these sites, CyTOF analyses revealed that natZr-mA1 was predominantly taken up by macrophages. Our results demonstrate that 89Zr-mA1 associates with lipoproteins and hence accumulates in macrophages in vivo. 89Zr-mA1’s high uptake in these cells makes it a promising radiotracer for non-invasively and quantitatively studying conditions characterized by marked changes in macrophage burden.
Similar content being viewed by others


Introduction
Macrophages are involved in diverse physiological and pathophysiological processes1,2. As key mediators of inflammation, they play a central role in deleterious conditions, including cardiovascular disease and cancer3,4. For example, after ischemic injury due to myocardial infarction (MI) or stroke, macrophages are recruited to repair and remodel the affected tissue. However, the release of pro-inflammatory mediators by these cells often aggravates the damage, culminating in cell death, necrosis, and in the case of atherosclerosis, dislodging of plaques5,6,7. Similarly, tumors produce growth factors and cytokines that recruit monocytes and induce their differentiation into tumor-associated macrophages (TAMs). These cells then become key regulators of the tumor microenvironment, frequently promoting tumor growth8, immunosuppression9, metastasis10, angiogenesis, and chemoresistance11. Consequently, a high TAM burden is typically associated with a poor prognosis4,12. Insights into macrophage burden and dynamics, therefore, facilitate diagnosing and stratifying cancer patients. TAM’s ability to modulate the immune system’s response to tumor growth has led to several TAM-targeting therapies4,13, some of which have already been trialed in humans14,15.
Several approaches to image macrophages have been reported in a number of pathological contexts and using various techniques, including magnetic resonance imaging (MRI), positron emission tomography (PET), and optical methods16,17,18,19. PET imaging often makes use of tracers targeting metabolic processes, such as 18F-FDG. These probes are taken up by activated macrophages due to their high metabolic and proliferative activity20. However, metabolic PET imaging lacks cell specificity and requires dietary preparation to control background signals from physiological glucose metabolism21. Nanoparticle-based imaging strategies have been introduced to enhance cellular specificity. These formulations typically rely on macrophages’ phagocytic properties and have been exploited in the context of PET, computed tomography (CT), and MRI16,22,23. Macrophage-specific PET imaging has also been achieved by targeting membrane proteins (over)expressed on activated macrophages using nanobody-based probes24,25,26, and peptide-based radiotracers, such as DOTATATE and pentixafor27,28. Small molecule tracers targeting translocator protein (TSPO) have also been successfully used for macrophage PET imaging29. Although some of these approaches led to promising results, there remains a pressing need for inexpensive, quantitative, and non-invasive macrophage-avid imaging agents for diagnosing/prognosing inflammatory diseases and TAM-driven tumors, such as gliomas, ovarian and breast cancer.
High-density lipoprotein (HDL) is a natural nanoparticle involved in reverse cholesterol transport30. It predominantly consists of phospholipids, cholesterol, and apolipoprotein A1 (ApoA1), through which it binds to several membrane receptors abundantly expressed on macrophages31. Capitalizing on this, we have previously used radiolabeled HDL to study macrophage burden in the context of atherosclerosis, cancer, and other diseases32. However, the high dispersity and manufacturing costs of these formulations complicate their clinical translation as PET tracers33. Here, we report an ApoA1-inspired PET radiotracer and its use as a tool for studying macrophage burden in vivo. The radiotracer consists of an ApoA1-mimetic peptide (mA1) that is labeled with zirconium-89 (89Zr) through desferrioxamine (DFO) to generate a macrophage-avid PET probe (89Zr-mA1). We anticipated that, upon intravenous administration, 89Zr-mA1 would associate with endogenous lipoproteins and subsequently accumulate in macrophages and other phagocytic cells. To test this hypothesis, we extensively studied tracer uptake and behavior in mouse models of normo- and hypercholesterolemia, cancer, and myocardial infarction. To achieve this, we made use of in vivo PET imaging and ex vivo assays, including CyTOF.
Results
Study outline
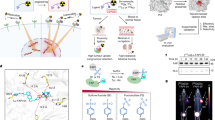
We have developed a PET tracer inspired by ApoA1 (89Zr-mA1), the predominant protein in HDL that has a natural ability to interact with phospholipids and cholesterol esters driven by its highly hydrophobic C-terminal domain. The ApoA1-mimetic 89Zr-mA1 also displays this ability, which enables it to bind lipoproteins in vivo and subsequently accumulate in phagocytes, as a result of lipoproteins’ propensity to interact with these cells. Consequently, 89Zr-mA1 enables non-invasive quantitative PET imaging of macrophage burden (Fig. 1A).

A 89Zr-mA1 binds lipoproteins in vivo. These nanoparticles are subsequently taken up by phagocytes, enabling PET imaging of macrophage burden. B Optimized 3D conformation of 89Zr-mA1 calculated using AlphaFold software. C Schematic overview of 89Zr-mA1 synthesis. mA1 has a C-terminus cysteine which facilitates site-specific conjugation with maleimide-functionalized deferoxamine (Mal-DFO) and subsequent radiolabeling with 89Zr to form 89Zr-mA1. D In vitro binding of 89Zr-mA1 to plasma constituents. Bare 89Zr and 89Zr-mA1 were incubated with either LDL, HDL or albumin before being analyzed by size-exclusion chromatography. E Blood pharmacokinetics for intravenously infused 89Zr-mA1 in C57BL/6 (n = 6) and Apoe−/− mice (n = 4) as well as the associated weighted half-lives and area under the curve values. F Representative size exclusion chromatograms of a plasma sample taken from an Apoe−/− mouse 30 min after administration of 89Zr-mA1, showing UV (black) and radioactivity (red) traces. AU arbitrary units, AUC area under the curve.
89Zr-mA1 synthesis
We used an ApoA1 mimetic polypeptide (37pA) that consists of two identical class A amphipathic helices linked by proline (Fig. 1B). We selected this sequence based on previously published literature data34,35. A cysteine residue was incorporated on the C terminus of our peptide to enable its site-specific conjugation with deferoxamine (DFO-Mal) via the maleimide-thiol reaction (Fig. 1C, left). HPLC analysis confirmed the purity of our mA1-DFO conjugate (Supplementary Fig. 1A). LC-MS corroborated the identity and purity of the mA1-DFO product (Supplemental Fig. 1B). mA1-DFO was subsequently radiolabeled with 89Zr (Fig. 1C, right). Chelation of 89Zr afforded the radiolabeled peptide with 75 ± 5% radiochemical yield (amount of activity bound to mA1 as a percentage of starting activity corrected for radioactive decay) and >98% radiochemical purity (n = 6), as assessed by radio-TLC (Supplementary Fig. 1C). The tracer’s molar activity was ~170 TBq/mol (n = 6).
89Zr-mA1 binds to plasma lipoproteins in vitro
We assessed 89Zr-mA1’s propensity to bind different plasma components by incubating 89Zr-mA1 with HDL, low-density lipoprotein (LDL), or albumin and using bare 89Zr as control. After incubation, 89Zr-mA1 efficiently bound to HDL (70% of activity bound) and, to a lesser degree, LDL (with 45% of activity bound) (Fig. 1D). Binding to albumin was minimal, as only 13% of activity was bound to this protein under the same assay conditions. These results imply that the peptide has a high propensity for binding to lipoproteins. In an additional experiment, tracer stability was evaluated by incubating bare 89Zr or 89Zr-mA1 with fetal bovine serum (FBS) at 37 °C and monitoring the free 89Zr fraction by radio-TLC (Supplementary Fig. 1D, left). Under these conditions, 89Zr did not dissociate from the tracer. We also confirmed that bare 89Zr does not bind to serum proteins (Supplementary Fig. 1D, right).
Pharmacokinetics and in vivo lipoprotein binding of 89Zr-mA1
To determine 89Zr-mA1’s blood circulation time in mice and how this depends on plasma lipoprotein concentrations, we performed pharmacokinetic studies in C57BL/6 and Apoe−/− (hypercholesterolemic) mice. The weighted tracer half-lives were 0.9 and 6.1 h in C57BL/6 and Apoe−/− mice, respectively (Fig. 1E). At 30 min post-injection (p.i.), Apoe−/− blood samples were taken and separated into cellular and plasma fractions. Plasma, which contained 97 ± 2% (n = 4) of blood radioactivity, was subjected to size exclusion chromatography (SEC) analysis. The radioactivity trace showed two peaks. The first peak had a retention time of 10 min, indicating compounds with a mass of >500 kDa, and was accompanied by a large peak in the UV spectrum. Based on these characteristics, we believe this signal stems from radiolabeled lipoproteins. The second peak eluted at ~20 min and matches well with the “free” radiolabeled peptide. The larger peak represented 89 ± 5% of the measured total radioactivity area (n = 4, Fig. 1F). These results, combined with our in vitro observations, strongly suggest that 89Zr-mA1 rapidly binds endogenous lipoproteins in vivo.
89Zr-mA1 PET imaging and biodistribution studies in C57BL/6 mice
We performed dynamic PET imaging in C57BL/6 mice during the first hour after i.v. 89Zr-mA1 administration (Fig. 2A). Images were initially dominated by a high blood pool signal, followed by increasing kidney uptake. The liver and spleen showed a significant uptake within the first ~5 min p.i. which remained nearly constant at a standardized uptake value (SUV) of ~2 g/mL (Fig. 2B). In a different subset of animals, a static scan was performed at 24 h p.i., showing strong signals in the liver and kidneys (Fig. 2C, Supplemental Table 1). Ex vivo gamma counting of selected tissues corroborated our PET imaging results (Fig. 2D).

A Representative 3D-rendered PET/CT images of C57BL/6 mice were scanned dynamically for 1 h following 89Zr-mA1 injection. B 89Zr-mA1 pharmacokinetics in specific tissues (n = 4). C Representative 3D-rendered PET/CT image of a C57BL/6 mouse 24 h after i.v. tracer injection. D Ex vivo biodistribution as assessed by gamma counting of tissues of interest 24 h p.i. (n = 5). E Ex vivo biodistribution as assessed by gamma counting of tissues of interest 24 h p.i. (n = 5) in mice treated with clodronate liposomes. Matched controls that did not receive clodronate injections are represented in gray.
89Zr-mA1 in vivo distribution in a macrophage depletion model
To evaluate tracer affinity for macrophages, we employed the clodronate liposome mouse model of macrophage depletion36,37. Ex vivo 89Zr-mA1biodistribution experiments showed lower radiometric signal in the spleen 1.6 (IQR, 1.4–1.7), vs. 2.1 (IQR, 2.0-2.4) in untreated controls (p = 0.008), an organ targeted by these liposomes (Fig. 2E). The spleen functions as a natural reservoir of immune cells, harboring macrophage populations responsible for cytokine production (innate immunity activators), apoptotic cell clearance, antigen presentation and pathogen scavenging38, further evidencing 89Zr-mA1’s ability to target these cells.

A Schematic overview of the experimental design. B Representative PET/CT images of infarcted animals scanned 24 h after tracer injection. Arrow indicates infarcted region. C PET-based quantification of 89Zr-mA1 uptake in the myocardium 24 h post tracer injection (n = 4). D 89Zr-mA1 uptake in the myocardium measured by ex vivo gamma counting (n = 4–5). Control: tissue from C57BL/6 mice. E Autoradiographs showing tracer distribution in the myocardium of infarcted mice 24 h p.i. Images show whole heart followed by a representative sectioned slice of ~1 mm thick. Corresponding TTC-stained tissues are also shown. F Autoradiography (left) and qualitative histology of the infarcted myocardium showing H&E staining (top right) and regions of macrophage accumulation as determined by Mac-3 staining (bottom). Scale bars represent 1 mm in the main figure and 20 μm in the magnification. G Mice were injected with natZr-mA1 two days after myocardial infarction. Twenty-four hours after injection, mice were euthanized and CyTOF performed on the infarct zone (n = 3). t-SNE plot of viable leukocytes in the infarct, color-coded for natZr-mA1 uptake and the expression of various myeloid cell markers. H Quantification of cell subsets in the infarct as a percentage of the total number of viable cells. I Percentage of natZr-positive cells within each subset. J Relative abundance of natZr-positive cell subsets in the infarct (right). Control: healthy myocardium of non-infarcted animals, DC dendritic cells, remote remote myocardium. *P < 0.05.
Employing 89Zr-mA1 in a mouse model of myocardial infarction
Myocardial infarction promotes inflammation due to tissue damage and this is strongly mediated by resident and recruited macrophages39. We used a mouse model of MI to evaluate 89Zr-mA1’s applicability toward imaging this increased macrophage burden in the infarcted area. Two days after left anterior descending (LAD) coronary artery ligation, mice were injected with the radiotracer, and PET-scanned statically at 24 h p.i. (Fig. 3A). Images revealed an overall 89Zr-mA1 biodistribution similar to that observed in naive C57BL/6 mice, including a high kidney and liver uptake (Supplementary Fig. 2A, B). Ex vivo gamma counting of these organs at 1- and 24-h post injection corroborated our PET observations (Supplementary Fig. 2C). Tracer accumulation in the infarcted area was significantly higher at 24 h p.i. (Fig. 3B) as indicated by a twofold stronger signal compared to the remote myocardium (p = 0.03) (Fig. 3C, Supplementary Table 1). MI-to-blood and MI-to-muscle (skeletal) ratios were 0.2 (IQR, 0.2–0.3) and 17 (IQR, 16.1–19.6) at 1 h p.i., respectively, and 2 (IQR, 1.7–2.2) and 15.1 (IQR, 10.9–15.2) at 24 h p.i., respectively (Supplementary Fig. 2D). Ex vivo gamma counting data confirmed the imaging results, although the contrast between inflamed and remote tissue was more pronounced due to the intrinsically higher sensitivity of gamma counting assays. Tracer uptake in the infarcted area was ~3-fold higher than in remote myocardium at both 1 and 24 h (p = 0.03 and 0.008, respectively) and 4-fold higher than in healthy myocardium of C57BL/6 mice (p = 0.03 and 0.008, respectively, Fig. 3D). These data were also corroborated by autoradiography of whole hearts and 1 mm thick slices stained with TTC, showing a similarly increased radioactivity accumulation in the infarcted area compared to remote myocardium (Fig. 3E). 89Zr-mA1 clearly accumulated in the infarcted area and tracer uptake correlates with regions of high macrophage accumulation, as demonstrated by H&E and Mac-3 histology staining (Fig. 3F). Ex vivo CyTOF analyses using mA1 labeled with non-radioactive natZr (natZr-mA1, Fig. 3G, Supplementary Fig. 3A) showed a pronounced presence of viable macrophages in the infarcted myocardium (Fig. 3H), as reported in previous studies40,41. Both macrophages and dendritic cells showed marked uptake of natZr-mA1, in comparison to other immune cell subsets (Fig. 3I). More importantly, the most abundant natZr-positive immune cell population—when expressed as a percentage of total viable cells– were macrophages, which indicates that the imaging signal is mostly due to tracer uptake by this population even in the presence of more abundant non-leukocyte cells (Fig. 3J). Further analysis showed that MHCII-CD206+ macrophages were preferentially targeted over their MHCII+CD206− counterparts (Supplementary Fig. 3B, C).
Employing 89Zr-mA1 in a melanoma mouse model
We also studied 89Zr-mA1’s potential in monitoring TAM burden using a B16F10 melanoma mouse model. PET scans were performed statically at 24 h p.i. (Fig. 4A). Imaging signatures were similar to those obtained in C57BL/6 mice, with strong kidney and liver signals (Supplementary Fig. 4A, B). At 24 h p.i, a clear tracer signal was observed in the tumor (Fig. 4B). Tumor uptake reached an SUV of 0.5 (IQR, 0.4–0.6, n = 10) at 24 h p.i. (Fig. 4C, Supplementary Table 1). These data were corroborated by ex vivo gamma counting of animal tissues at 1- and 24-h post tracer injection, which showed increased uptake in the tumor over time, with 2.1%ID/g (IQR, 1.9–2.4, n = 4) at 1 h p.i. and 4.5%ID/g (IQR, 4.2–4.9, n = 11) at 24 h p.i. (Fig. 4D). Ex vivo gamma counting also indicated high radioactivity concentrations in the kidney and liver, followed by the spleen and bone marrow (Supplementary Fig. 4C). Tumor-to-blood and tumor-to-muscle ratios were 0.1 (IQR, 0.1-0.1) and 5 (IQR, 4.7–5.2) at 1 h p.i., respectively, and increased to 2.4 (IQR, 2.1–2.7) and 14.2 (IQR, 10.6–15.7) at 24 h p.i., respectively (Supplementary Fig. 4D). Our CyTOF results with natZr-mA1 (Fig. 4E, Supplementary Fig. 5A) show that macrophages are the main immune cell population in this tumor model (Fig. 4F), and corroborate our tracer’s affinity for myeloid cells, especially macrophages and dendritic cells (Fig. 4G). Similar to our results in the MI model, the vast majority of viable natZr-positive cells were macrophages (Fig. 4H) implying that macrophages underly the majority of the signal. Macrophage subtype analysis showed that CD206+ macrophages were preferentially targeted, similar to the MI model, albeit to a lesser degree (Supplementary Fig. 5B, C).

A Schematic overview of the experimental design. B Representative PET/CT images of tumor-bearing animals scanned 24 h after tracer injection. Arrow indicates the tumor, delineated in red. C PET-based quantification of 89Zr-mA1 tumor uptake 24 h post tracer injection (n = 10). D 89Zr-mA1 uptake in the tumor measured by ex vivo gamma counting, (n = 4–10). E Mice were injected with natZr-mA1 14 days after tumor inoculation. 24 h after injection, mice were euthanized and CyTOF performed on the tumor (n = 3). t-SNE plot of viable leukocytes in the tumor, color-coded for natZr-mA1 uptake and the expression of various myeloid cell markers. F Quantification of cellular subsets in the tumor. G Percentage of natZr-positive cells within each subset. H Relative abundance of natZr-positive cell subsets in the tumor (right). **P < 0.01, ***P < 0.005.
Discussion
We have developed an apolipoprotein A1 mimetic-based radiotracer for monitoring macrophage burden. In vitro experiments show that 89Zr-mA1 efficiently binds lipoproteins. In a serum incubation experiment, tracer labeling displayed great stability with no evidence of 89Zr dissociation. The tracer exhibited longer circulation time in hypercholesterolemic mice (Apoe−/−) than in C57BL/6 mice, further demonstrating 89Zr-mA1’s ability to bind endogenous lipoproteins in vivo as Apoe−/− mice have higher HDL and LDL plasma levels than wild type mice42,43. Moreover, our SEC analysis of plasma samples from Apoe−/− mice collected at 30 min post-tracer administration suggests rapid in vivo lipoprotein binding, although these studies did not allow proper distinction between lipoprotein fractions. Macrophage targeting was also evaluated using clodronate liposome injected animals. We hypothesized that upon macrophage depletion, 89Zr-mA1 would show diminished uptake in macrophage-harboring organs. As expected, decreased tracer uptake was observed in the spleen, an organ with a large population of macrophages38 and a typical site for clodronate liposome accumulation. We did not observe different 89Zr-mA1 uptake in other organs, indicating that clodronate liposomes did not affect the tracer’s general behavior.
We used CyTOF and natZr-labeled mA1 as an innovative way to investigate our tracer’s cellular fate in the cancer and MI models included in this study. In contrast to fluorescence-based biodistribution analyses, this approach allowed us to use the same molecular species at a radiotracer-relevant dose. These studies showed a similar percentage of natZr+ macrophages and dendritic cells. However, the much higher abundance of macrophages means that the observed signal mainly stems from these cells. Our analyses also show that, although non-leukocytes outnumbered macrophages in both models, tracer uptake in these cells was low, resulting in limited background signal. Additionally, we explored macrophage subtype specificity by studying tracer uptake in CD206+ (MRC1, mannose receptor) versus CD206− cells. In both models, we observed that CD206+ macrophages are more likely to be natZr-mA1-positive than CD206− macrophages, which suggests that our tracer is more prone to accumulate in resident-like/alternatively activated macrophages. However, this analysis is based on a limited number of markers and, nevertheless, both natZr+CD206+ and natZr+CD206− macrophage subpopulations were sizeable suggesting that 89Zr-mA1 may not have a marked subtype selectivity.
We also demonstrated 89Zr-mA1’s applicability toward imaging macrophage accumulation in the infarcted myocardium. In this setting, tissue damage in the MI lesion results in monocyte recruitment and their subsequent differentiation into macrophages39. In our study design, we imaged these mice at 3 days post-LAD ligation, which is when infiltration of inflammatory macrophages peaks in this animal model44. Increased tracer uptake was observed in the infarcted area both in vivo and ex vivo. A similar assessment has been performed previously using 18F-FDG imaging45,46,47,48. However, 18F-FDG lacks cell specificity and displays high uptake in the remote myocardium49 and other metabolically active immune and non-immune cells, creating a substantial background signal50,51. For these reasons, the use of 18F-FDG for macrophage and inflammation quantification in the myocardium is often challenged by the need for complex dietary restrictions lasting several days prior to PET scanning52,53. Due to its ability to interact with endogenous lipoproteins, it is likely that 89Zr-mA1 imaging would also require certain dietary adaptations, such as overnight fasting or avoiding fat-rich meals prior to the imaging session. Gratifyingly, 89Zr-mA1’s infarct-to-remote myocardium uptake ratios were similar or higher compared to other reported tracers, such as 18F-Macroflor, 64Cu-Macrin, and 68Ga-Pentixafor54,55,56.
Macrophages also play an important role in tumor development, making TAM burden monitoring highly relevant. Furthermore, numerous immunotherapeutic interventions are being developed or already in use in the clinic. For instance, immune checkpoint blockade has afforded promising results, leading to persistent remission in 10–20% of patients with certain cancers57. This limited success is at least partly due to an immunosuppressive tumor environment in which macrophages are key players9. Indeed, a high TAM burden is associated with poor prognosis in several cancers. For these reasons, a non-invasive macrophage quantification tool like the one presented in this study could be tremendously valuable for monitoring TAM-targeting therapies. We recently showed that response to a colony-stimulating factor 1 receptor inhibitor TAM-burden reducing therapy could be non-invasively assessed by PET imaging with a 89Zr-HDL nanotracer58. However, peptide-based radiotracers are generally easier to clinically translate compared to nanomaterial-based variants. In addition, molecular radiotracers usually require microdoses in Phase 0 clinical trials (≤100 μg)59, simplifying initial regulatory requirements and potentially reducing costs.
Noteworthy, in addition to 18F-FDG and HDL-based nanotracers, other classes of PET tracers have been developed for the monitoring macrophage burden. However, low tracer specificity, patient variability and lack of translatability remain a challenge in the field, underscoring the need for the engineering of new strategies. For instance, Gaemperli et al. 29 investigated the use of 11C-PK1195 for the imaging of carotid plaques, showing good histological colocalization of the tracer with plaque macrophages. However, the presence of genetic polymorphisms affecting TSPO expression reportedly affected tracer’s aptness. Despite their complex production and high costs, radiolabeled nanobodies targeting specific immune cell epitopes have also been employed for the imaging of leukocytes60,61 and macrophages26,62 in different disease contexts. In comparison, 89Zr-mA1 is produced through chemical synthesis, using a biocompatible mimetic peptide, offering reduced manufacturing costs.
In this study, we chose to radiolabel our tracer using 89Zr as this isotope has a relatively long physical half-life (t1/2 = 78.4 h), which simplifies extensive in vivo and ex vivo studies. While 89Zr is increasingly used in the clinic, a shorter-lived isotope, such as 64Cu (t1/2 = 12.7 h), might be desirable for human use, as a short half-life helps reduce patients’ radiation exposure, including due to high kidney radioactivity retention63. Our future efforts will therefore focus on developing mA1-based radiotracers labeled with 64Cu.
In conclusion, 89Zr-mA1’s high specificity for phagocytes makes it an effective probe for quantitatively and non-invasively assessing macrophage-driven inflammation. 89Zr-mA1 holds potential as an imaging tool for studying diseases characterized by abundant macrophage burden and infiltration.
Methods
Chemistry
The apolipoprotein A1 mimetic peptide 37pA34,35 modified with a cysteine at the C terminus [mA1] was purchased from Peptide 2.0 (Chantilly, VA) and had the following amino acid sequence: DWLKAFYDKVAEKLKEAFPDWLKAFYDKVAEKLKEAFC. Deferoxamine-maleimide (DFO-Mal) was purchased from Macrocyclics (Plano, TX). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Radiochemistry
89Zr in 1 M oxalic acid was purchased from 3D imaging (Little Rock, AR). Activity measurements were performed using a Capintec CRC-15R Dose Calibrator (Capintec, Ramsey, NJ).
Functionalizing mA1 with DFO
The terminal cysteine on mA1 allows its site-specific conjugation with the chelator DFO via maleimide chemistry. mA1 was dissolved in degassed DMSO (1.9 mg in 200 μL, 1.0 eq), and a DFO-Mal solution in DMSO (0.45 mg in 200 μL, 1.5 eq.) was added. The mixture was degassed using N2 and allowed to react at room temperature for 48 h in the dark under a nitrogen atmosphere. Water (2 mL) was added, and the mixture was transferred to a 2000 molecular weight cut-off (MWCO) 3 mL dialysis bag (Slide-A-Lyzer, Thermo Scientific). The solution was dialyzed against water (~1 L) in the dark and overnight to remove DMSO and any unreacted DFO-Mal. The DFO functionalized peptide (mA1-DFO) was purified using a Shimadzu HPLC system equipped with an SPD-M10AVP photodiode array detector. Runs were carried out on a C18 Phenomenex Gemini column (6 × 250 mm, 5 μm) using water and acetonitrile as eluents (each containing 0.1%TFA) with a gradient of 70% to 40%, 10 min, and 40% to 70% 1 min and flow rate of 1 mL/min. The obtained product was dialyzed against water (~1 L) in the dark and overnight to remove acetonitrile (2000 MWCO, 3 mL dialysis bag from Slide-A-Lyzer, Thermo Scientific). Electrospray ionization mass spectrometry spectra were recorded with a Waters Acquity UPLC (Milford, CA) with an electrospray ionization SQ detector. The compound was lyophilized and stored at −20 °C until further use, in which case it was redissolved in PBS.
Radiolabeling of mA1-DFO
A solution of 89Zr oxalate (1 M aqueous oxalic acid) was neutralized with sodium carbonate 1 M to reach a pH between 6.8 and 7.4. The DFO-bearing peptide was reacted with the 89Zr solution using a thermomixer (600 rpm) for 1 h at 37 °C. The solution was allowed to cool to room temperature and radiolabeled mA1 purified by gel filtration using a PD-10 column and PBS as eluent. Radiochemical yield and purity were assessed by radio-TLC using a Lablogic Scan-RAM Radio-TLC/HPLC detector. The same labeling procedure was performed with natural zirconium to obtain natZr-mA1 for mass cytometry experiments. Optimized 3D conformation of 89Zr-mA1 was determined using AlphaFold software (Alphabet Inc., Mountain View, CA)64 and further modified using PyMol molecular visualization system (Schrödinger Inc, New York, NY) and structural information on Zr-DFO by Allot et al. 65.
In vitro 89Zr-mA1 binding to lipoproteins and albumin
We used SEC to assess 89Zr-mA1’s ability to bind common serum components, namely HDL (human), LDL (human), and albumin (bovine).89Zr-mA1 or free 89Zr (2.22 MBq) were incubated with a solution of the corresponding component at 0.2 mg/mL (HDL and LDL) or 0.5 mg/mL (albumin) for 1 h at 37 °C. Then, radio-SEC was performed using a Shimadzu HPLC system with an SPD-20A detector coupled to a Superdex 75-5/150 GL column (GE Healthcare Life Sciences). 100% PBS was used as eluent at a flow rate of 0.5 mL/min. The retention profile of each aliquot was compared to bare 89Zr and the UV trace of the bare serum constituent.
Animals
All animal experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committees at the Icahn School of Medicine at Mount Sinai and Memorial Sloan Kettering, following National Institutes of Health guidelines for animal welfare. Female C57BL/6 mice (7 weeks old, n = 50) were purchased from the Jackson Laboratory (Bar Harbor, ME) and randomly assigned to the different experimental groups. Female Apoe−/− mice (8 weeks old, n = 8) were also purchased from Jackson Laboratory. Details on the mouse models of hypercholesterolemia, macrophage depletion, myocardial infarction, and melanoma can be found in the Supplementary Material.
Pharmacokinetics of 89Zr-mA1
C57BL/6 (n = 6) and Apoe−/− mice (n = 4) were injected with 89Zr-mA1 (~1.1 and 3.5 MBq, respectively) via the lateral tail vein. Blood radioactivity half-life was determined by serial blood draws from the tail vein at 1, 5, 10, 15, 30, 60 min, and 2, 6, and 24 h after injection. Blood was weighed, and gamma counted using a Wizard2 2480 automatic gamma counter (Perkin Elmer, Waltham, MA). Results were corrected for decay, and radioactivity concentration was reported as a percentage of injected dose per gram (%ID/g). A time-activity curve was generated, and the resulting values fitted to a biphasic decay to obtain blood radioactivity half-lives for 89Zr-mA1. Weighted half-life is defined as: (% fast × t1/2 fast + % slow × t1/2 slow)/100.
Biodistribution of 89Zr-mA1
Tissue radioactivity distribution was determined at different time points after 89Zr-mA1 administration to healthy (n = 5), macrophage depleted (n = 5), LAD ligated (n = 9) and melanoma (n = 14) C57BL/6 mice. Mice were injected with ~3.5 MBq (or 7.4 MBq for macrophage depleted mice) of 89Zr-mA1 via the lateral tail vein, and the tracer was allowed to circulate for 1 (n = 4) and/or 24 h (n = 5–10). After euthanasia and perfusion (20 mL saline), tissues were harvested (brain, heart, lungs, bone marrow, spleen, liver, kidneys, skeletal muscle, and bone) and weighed before radioactivity counting. The values were corrected for decay and radioactivity concentration converted to %ID/g.
In vivo 89Zr-mA1 binding to blood plasma components
Female Apoe−/− mice were intravenously injected with 89Zr-mA1 (~3.7 MBq, n = 4). 30 min after administration, animals were euthanized, and blood (0.5–0.7 mL) was collected by cardiac puncture in a heparinized tube. The sample was rapidly centrifuged at 15,000 rpm for 2 min, and the two phases obtained were carefully separated. The plasma fraction was analyzed by SEC using a Superdex 10/300 column (GE Healthcare Life Sciences) and PBS as eluent at a flow rate 1 mL/min.
In vivo PET/CT imaging
Dynamic PET imaging was performed on a cohort of C57BL/6 animals (n = 4 per group) for 60 min after 89Zr-mA1 administration. These mice were anesthetized with isoflurane (2% in medical air for induction, 1% for maintenance) and placed on the bed of a nanoScan PET/CT scanner (Mediso, Budapest, Hungary). Following a scout scan, a full-body CT was performed, and a 60-min dynamic PET scan with a field of view aligned with the full positioning of the mouse was initiated right after injection of 89Zr-mA1 (3.4 MBq) via the lateral tail vein. Upon completion of the scan, a total of 5 dynamic frames were individually reconstructed for the following timepoints: 1, 5, 15, 30, and 60 min. ROIs were individually analyzed for each timepoint. In a different cohort of C57BL/6, LAD-ligated, and B16F10-inoculated animals, 89Zr-mA1 (3.5 MBq, n = 5–10 per group) was allowed to circulate for 24 h after intravenous injection. Animals were then anesthetized with isoflurane (2% in medical air for induction, 1% for maintenance) and subsequently imaged. PET acquisition time for the static scans was 20 min. For both dynamic and static scans, a high-resolution CT scan was acquired at 50 kVp and 300 ms exposure per projection. CT-contrast agent (isovue-370, Bracco Diagnostics) was continuously infused through a tail vein catheter. Reconstruction was performed using the TeraTomo 3D reconstruction engine for eight iterations and six subsets per iteration. The voxel size was isotropic at 0.3 mm. Immediately after the PET/CT scan, animals were euthanized for ex vivo analysis.
Statistical analysis
All results are presented as median and interquartile range (IQR) unless otherwise stated. Unpaired data were analyzed with nonparametric Mann-Whitney tests. For all tests, α < 0.05 represents statistical significance. Levels of significance are indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Epelman, S., Lavine, K. J. & Randolph, G. J. Origin and functions of tissue macrophages. Immunity 41, 21–35 (2014).
Wynn, T. A. & Vannella, K. M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721 (2013).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Frangogiannis, N. G., Smith, C. W. & Entman, M. L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 53, 31–47 (2002).
Sager, H. B., Kessler, T. & Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 9, S30–S35 (2017).
Liu, J., Wang, H. & Li, J. Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: a double-edged sword. Clin. Med. Insights Cardiol. 10, 79–84 (2016).
Coffelt, S. B., Hughes, R. & Lewis, C. E. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim. Biophys. Acta 1796, 11–18 (2009).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 1, 369–382 (2019)
Condeelis, J. & Pollard, J. W. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124, 263–266 (2006).
De Palma, M. & Lewis, C. E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23, 277–286 (2013).
Tang, X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 332, 3–10 (2013).
Ries, C. H. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25, 846–859 (2014).
Cassier, P. A. et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: A dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 16, 949–956 (2015).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Weissleder, R., Nahrendorf, M. & Pittet, M. J. Imaging macrophages with nanoparticles. Nat. Mater. 13, 125–138 (2014).
Foss, C. A., Sanchez-Bautista, J. & Jain, S. K. Imaging Macrophage-associated Inflammation. Semin. Nuclear Med. 48, 242–245 (2018).
Li, Y., Liu, T. M. Discovering macrophage functions using in vivo optical imaging techniques. Front. Immunol. 9, 502 (2018).
Toner, Y. C. et al. Systematically evaluating DOTATATE and FDG as PET immuno-imaging tracers of cardiovascular inflammation. Sci. Rep. 12, 6185 (2022).
Li, X., Rosenkrans, Z. T., Wang, J. & Cai, W. PET imaging of macrophages in cardiovascular diseases. Am. J. Transl. Res. 12, 1491–1514 (2020).
Blomberg, B. A. & Høilund-Carlsen, P. F. [18F]-fluorodeoxyglucose PET imaging of atherosclerosis. PET Clin. 10, 1–7 (2015).
Serkova, N. J. Nanoparticle-based magnetic resonance imaging on tumor-associated macrophages and inflammation. Front. Immunol. 8, 590 (2017).
Dadfar, S. M. et al. Iron oxide nanoparticles: diagnostic, therapeutic and theranostic applications. Adv. Drug Deliv. Rev. 138, 302–325 (2019).
Senders, M. L. et al. Nanobody-facilitated multiparametric PET/MRI phenotyping of atherosclerosis. JACC Cardiovasc. Imaging 12, 2015–2026 (2019).
Bao, G., Tang, M., Zhao, J. & Zhu, X. Nanobody: a promising toolkit for molecular imaging and disease therapy. EJNMMI Res. 11, 6 (2021).
Movahedi, K. et al. Nanobody-based targeting of the macrophage mannose receptor for effective in vivo imaging of tumor-associated macrophages. Cancer Res. 72, 4165–4177 (2012).
Malmberg, C. et al. 64Cu-DOTATATE for noninvasive assessment of atherosclerosis in large arteries and its correlation with risk factors: Head-to-head comparison with 68Ga-DOTATOC in 60 patients. J. Nucl. Med. 56, 1895–1900 (2015).
Li, X. et al. [68Ga]Pentixafor PET/MR imaging of chemokine receptor 4 expression in the human carotid artery. Eur. J. Nucl. Med. Mol. Imaging 46, 1616–1625 (2019).
Gaemperli, O. et al. Imaging intraplaque inflammation in carotid atherosclerosis with 11C-PK11195 positron emission tomography/computed tomography. Eur. Heart J. 33, 1902–1910 (2012).
Favari, E. et al. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 224, 181–206 (2015).
Van Eck, M., Pennings, M., Hoekstra, M., Out, R. & Van Berkel, T. J. Scavenger receptor BI and ATP-binding cassette transporter A1 in reverse cholesterol transport and atherosclerosis. Curr. Opin. Lipidol. 16, 307–315 (2005).
Mulder, W. J. M. et al. High-density lipoprotein nanobiologics for precision medicine. Acc. Chem. Res. 51, 127–137 (2018).
Metselaar, J. M. & Lammers, T. Challenges in nanomedicine clinical translation. Drug Deliv. Transl. Res. 10, 721–725 (2020).
Owens, R. J. et al. Apolipoprotein A-I and its amphipathic helix peptide analogues inhibit human immunodeficiency virus-induced syncytium formation. J. Clin. Investig. 86, 1142–1150 (1990).
Remaley, A. T. et al. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J. Lipid Res. 44, 828–836 (2003).
Van Rooijen, N., Kors, N. & Kraal, G. Macrophage subset repopulation in the spleen: differential kinetics after liposome-mediated elimination. J. Leukoc. Biol. 45, 97–104 (1989).
Naito, M. et al. Liposome-encapsulated dichloromethylene diphosphonate induces macrophage apoptosis in vivo and in vitro. J. Leukoc. Biol. 60, 337–344 (1996).
Den Haan, J. M. M. & Kraal, G. Innate immune functions of macrophage subpopulations in the spleen. J. Innate Immun. 4, 437–445 (2012).
Swirski, F. K. & Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 339, 161–166 (2013).
Nahrendorf, M., Pittet, M. J. & Swirski, F. K. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121, 2437–2445 (2010).
Sager, H. B. et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 119, 853–864 (2016).
Jiang, F., Gibson, A. P. & Dusting, G. J. Endothelial dysfunction induced by oxidized low-density lipoproteins in isolated mouse aorta: a comparison with apolipoprotein-E deficient mice. Eur. J. Pharmacol. 424, 141–149 (2001).
Phenotypes of LDLR & APOE Knockout Mice | The Jackson Laboratory. https://www.jax.org/jax-mice-and-services/strain-data-sheet-pages/phenotype-information-for-002052-and-002207.
Thackeray, J. T. & Bengel, F. M. Molecular imaging of myocardial inflammation with positron emission tomography post-ischemia: a determinant of subsequent remodeling or recovery. JACC Cardiovasc. Imaging 11, 1340–1355 (2018).
Andrej, Ć. et al. Novel positron emission tomography tracers for imaging vascular inflammation. Curr. Cardiol. Rep. 22, 1–11 (2020).
Thackeray, J. T. et al. Targeting post-infarct inflammation by PET imaging: comparison of 68Ga-citrate and 68Ga-DOTATATE with 18F-FDG in a mouse model. Eur. J. Nucl. Med. Mol. Imaging 42, 317–327 (2015).
Lee, W. W. et al. PET/MRI of inflammation in myocardial infarction. J. Am. Coll. Cardiol. 59, 153–163 (2012).
Vasudevan, P. et al. 18F-FDG PET-based imaging of myocardial inflammation following acute myocardial infarction in a mouse model. Int. J. Mol. Sci. 21, 3340 (2020).
Tarkin, J. M. et al. 68Ga-DOTATATE PET identifies residual myocardial inflammation and bone marrow activation after myocardial infarction. J. Am. Coll. Cardiol. 73, 2489–2491 (2019).
Manabe, O. et al. Radiopharmaceutical tracers for cardiac imaging. J. Nucl. Cardiol. 25, 1204–1236 (2018).
Bengel, F. M. Imaging of Post-Infarct Inflammation: Moving Forward Toward Clinical Application. Circ. Cardiovasc. Imaging. 9, e0047131–3 (2016).
Kumar, P., Patel, C. D., Singla, S. & Malhotra, A. Effect of duration of fasting and diet on the myocardial uptake of F-18-2-fluoro-2-deoxyglucose (F-18 FDG). at rest.Indian J. Nucl. Med. 29, 140–145 (2014).
Surasi, D. S., Bhambhvani, P., Baldwin, J. A., Almodovar, S. E. & O’Malley, J. P. 18F-FDG PET and PET/CT patient preparation: a review of the literature. J. Nucl. Med. Technol. 42, 5–13 (2014).
Thackeray, J. T. et al. Molecular imaging of the chemokine receptor CXCR4 after acute myocardial infarction. JACC Cardiovasc. Imaging 8, 1417–1426 (2015).
Keliher, E. J. et al. Polyglucose nanoparticles with renal elimination and macrophage avidity facilitate PET imaging in ischaemic heart disease. Nat. Commun. 8, 14064 (2017).
Nahrendorf, M. et al. Imaging cardiovascular and lung macrophages with the positron emission tomography sensor 64Cu-macrin in mice, rabbits, and pigs. Circ. Cardiovasc. Imaging 13, e010586 (2020).
O’Donnell, J. S., Long, G. V., Scolyer, R. A., Teng, M. W. L. & Smyth, M. J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 52, 71–81 (2017).
Mason, C. A. et al. An 89Zr-HDL PET tracer monitors response to A CSF1R inhibitor. J. Nucl. Med. 61, 433–436 (2020).
Burt, T. et al. Phase 0/microdosing approaches: time for mainstream application in drug development? Nat. Rev. Drug Discov. 19, 801–818 (2020).
Rashidian, M. et al. Noninvasive imaging of immune responses. Proc. Natl. Acad. Sci. USA 112, 6146–6151 (2015).
Maier, A. et al. Multiparametric immunoimaging maps inflammatory signatures in murine myocardial infarction models. JACC Basic Transl. Sci. 8, 801–816 (2023).
Put, S. et al. SPECT imaging of joint inflammation with nanobodies targeting the macrophage mannose receptor in a mouse model for rheumatoid arthritis. J. Nucl. Med. 54, 807–814 (2013).
Pérez-Medina, C. et al. In Vivo PET imaging of HDL in multiple atherosclerosis models. JACC Cardiovasc. Imaging 9, 950–961 (2016).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Allott, L. et al. Evaluation of DFO-HOPO as an octadentate chelator for zirconium-89. Chem. Commun. 53, 8529–8532 (2017).
Acknowledgements
This work was supported by a grant from the Leducq Foundation, the National Institutes of Health grant R01 CA220234, the American Heart Association (16SDG31390007), as well as a Vici grant from the Dutch Research Council (NWO), a European Research Council (ERC) Advanced Grant (TOLERANCE) (W.J.M.M.) and an I + D + i project grant from the Spanish Ministry of Science and Innovation (PID2020-114902RB-I00|AEI/10.13039/501100011033) (CPM). The funders played no role in the study design, data collection, analysis and interpretation of data, or the writing of this manuscript. Parts of the figures were drawn by using pictures from Servier Medical Art by Servier, licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Author information
Authors and Affiliations
Contributions
Specific author contributions are as follows: Y.C.T, G.P., M.M.T.v.L., T.R., Z.A.F., W.J.M.M., A.J.P.T, C.P.M. engaged in the conception and design of the experiments. Y.C.T, G.P., M.M.T.v.L, J.M., R.O., A.V.V., Y.v.E., V.P., R.J.F.M, A.R., J.M-F., W.W., M.U., A.d.D., J.C.F., N.S., A.M., C.M. participated in the data acquisition process and Y.C.T, G.P., M.M.T.v.L, J.M., R.O., A.V.V., Y.v.E., V.P., R.J.F.M., A.R., J.M-F., A.d.D., J.C.F., N.S., A.M., C.M., A.J.P.T., C.P.M. performed the analysis and interpretation of the data. All authors contributed to the drafting or revision of the manuscript. The final content of the manuscript was seen and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
W.J.M.M. is a founder and CSO of Trained Therapeutix Discovery. Z.A.F. is a founder and member of the board of Trained Therapeutix Discovery. T.R. is now an employee of and shareholder in Novartis AG. No other potential financial or non-financial conflicts of interest relevant to this article exist.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toner, Y.C., Prévot, G., van Leent, M.M.T. et al. Macrophage PET imaging in mouse models of cardiovascular disease and cancer with an apolipoprotein-inspired radiotracer. npj Imaging 2, 12 (2024). https://doi.org/10.1038/s44303-024-00009-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44303-024-00009-3
