
Abstract
Anal squamous cell carcinoma (ASCC) is associated with immunosuppression and infection with human papillomavirus (HPV). Response to standard chemoradiotherapy (CRT) varies considerably. A comprehensive molecular characterization of CRT resistance is lacking, and little is known about the interplay between tumor immune contexture, host immunity, and immunosuppressive and/or immune activating effects of CRT. Patients with localized ASCC, treated with CRT at three different sites of the German Cancer Consortium (DKTK) were included. Patient cohorts for molecular analysis included baseline formalin fixed paraffin embedded biopsies for immunohistochemistry (n = 130), baseline RNA sequencing (n = 98), peripheral blood immune profiling (n = 47), and serum cytokine measurement (n = 35). Gene set enrichment analysis showed that pathways for IFNγ, IFNα, inflammatory response, TNFα signaling via NF-κB, and EMT were significantly enriched in poor responders (all p < 0.001). Expression of interferon-induced transmembrane protein 1 (IFITM1), both on mRNA and protein levels, was associated with reduced Freedom from locoregional failure (FFLF, p = 0.037) and freedom from distant metastasis (FFDM, p = 0.014). An increase of PD-L1 expression on CD4+ T-cells (p < 0.001) and an increase in HLA-DR expression on T-cells (p < 0.001) was observed in the peripheral blood after CRT. Elevated levels of regulatory T-cells and CXCL2 were associated with reduced FFLF (p = 0.0044 and p = 0.004, respectively). Inflammatory pathways in tissue in line with elevated levels of regulatory T-cells and CXCL2 in peripheral blood are associated with resistance to CRT. To counteract this resistance mechanism, the RADIANCE randomized phase-2 trial currently tests the addition of the immune checkpoint inhibitor durvalumab to standard CRT in locally advanced ASCC.
Similar content being viewed by others

Introduction
Anal squamous cell carcinoma (ASCC) is a rare disease with an annual incidence of 2–3 per 100,000 inhabitants1. In recent years, an increase in the incidence of ASCC has been reported2. The main risk factors for ASCC are immunosuppression and infection with human papillomavirus (HPV), which can be found in up to 95% of patients3. Following HPV viral DNA integration into host cells, oncoproteins E6 and E7 induce inactivation of tumor suppressors such as retinoblastoma protein and p53. Moreover, activating mutations of PIK3CA and mutations of genes important for DNA damage repair (ATM, BRCA 1/2, HUWE1), chromatin remodeling (EP300, SMARCB1), and the Wnt/β-catenin signaling (FAM123B9) have been frequently reported in ASCC, whereas clinically relevant alterations for KRAS, NRAS, BRAF, EGFR are rare4,5,6,7.
Standard treatment for localized ASCC is primary chemoradiotherapy (CRT). Radical surgery is reserved as salvage for locoregional failure. Several large randomized trials have tested various forms of induction or consolidation chemotherapy in addition to concurrent CRT, but failed to show oncological benefits. Thus, the use of 5-fluorouracil (5-FU) plus mitomycin c (MMC) CRT remains the standard of care8,9.
Smaller tumors without lymph node involvement (cT1-2N0M0) generally have a favorable prognosis after standard CRT, but incomplete responses or recurrences occur in up to 40% of patients with locally-advanced ASCC (cT3-4 and/or cN+)10,11. Well established clinical prognostic factors include primary tumor size, lymph node involvement, and sex11,12,13. In addition, expression of p16INK4A (surrogate for HPV) and CD8+ tumor infiltrating lymphocytes (TILs) have recently been established as molecular prognostic markers14,15. Given the relatively rare incidence of ASCC, a comprehensive molecular characterization of ASCC is lacking, and genomic profiling as part of The Cancer Genome Atlas (TCGA) project has not been reported. Here, we present results from multidimensional analyses of ASCC to gain a better molecular understanding of response to CRT, to develop prognostic/predictive markers for individual treatment efficacy and to help guide future treatment strategies based on subgroups identified.
Results
Patient characteristics and oncological outcome
Baseline demographics and clinical characteristics of the full cohort of 130 patients are summarized in Table 1; 70 (54%) patients had cT1-2N0M0 disease, and 60 (46%) patients were diagnosed with locoregionally advanced disease (cT3N0M0 or cTanyN+M0). A total of 22 patients (17%) were HIV-positive; these were mainly male (19/22), and significantly younger than HIV-negative patients (median 52 vs. 59 years; p < 0.001).
With a median follow-up of 48.5 (IQR, 24.5–70.25) months, cCR was achieved in 115/130 (88%) patients, a total of 27 (21%) patients either had residual tumor at first re-staging or developed a locoregional recurrence after initial cCR, 12 patients (9%) developed distant metastasis during follow-up; 5-year FFLF was 76.5% (95% CI, 69–85%), and 5-year FFDM was 89.2% (95% CI, 83–96%).
Interferon-associated pathways are enhanced in poor responders
After rigorous quality control of the RNA sequencing data in 130 baseline tumor samples, 98/130 (75%) were considered as suitable and selected for further analysis (Fig. 1a, GSEA254331). Patient characteristics and treatment outcomes of this group are summarized in Supplementary Table 1. Gene set enrichment analysis showed that pathways for interferon gamma and alpha response (IFNγ and IFNα, p < 0.001, respectively), inflammatory response (p < 0.001), epithelial mesenchymal transition (EMT, p < 0.001), and tumor necrosis factor alpha (TNFα) signaling via NF-κB (p < 0.001), were significantly enriched in poor responders to CRT (Fig. 1c). Also, DEG analysis revealed that interferon induced transmembrane protein 1 (IFITM1) was significantly upregulated in poor responders (log2FC −1.42; padj 0.005, Fig. 1b), and also part of the enriched hallmark gene sets IFNα response and inflammatory response (Supplementary Table 3).

a Top 25 differentially expressed genes between poor responders and good responders. b Volcano plot of the RNA Seq data dichotomized according to poor response (red) and good response (green). c Enrichment plots from the GSEA tool showing that in poor responders the pathways TNFα Signaling via NF-κB, Interferon gamma and alpha response, inflammatory response, and epithelial mesenchymal transition (EMT) are significantly enriched.
IFITM1 expression is associated with poor prognosis
Using the R survminer package to create an optimal cut-off, a high expression of IFITM1 was associated with significantly reduced FFLF (p < 0.0001, Fig. 2a) and FFDM (p = 0.025, Fig. 2b) in the RNAseq cohort. To validate this finding at the protein level, we stained pretreatment tumor samples of the whole cohort (n = 130) for IFITM1 (Fig. 2e). Median IFITM1 weighted score was 6. The cohort was divided accordingly into a high score (score >6) and a low score (≤6); 37 (28%) patients had a high expression, and 93 (72%) a low expression. IFITM1 was not associated with T-stage, N-stage, sex, and HIV-status (Table 2). A high immunohistochemical expression of IFITM1 was associated with reduced FFLF and FFDM (p = 0.037, p = 0.014, respectively, Fig. 2c, d).

After identification of an ideal cut-off using maximized rank statistics, a high expression of interferon induced transmembrane protein 1 (IFITM1) in RNA seq was associated with significantly reduced freedom from local failure (FFLF) (a), and freedom from distant metastasis (FFDM) (b). Using immunohistochemistry for IFITM1, a high semiquantitative weighted score for IFITM1 was associated with reduced FFLF (c) and FFDM (d). e Example stainings for IFITM1 high and low tumors. f Recurrence free survival in the The Cancer Genome Atlas (TCGA) cervical cancer cohort according to IFITM1 expression. The plot was generated using https://www.kmplot.com.
Next, we assessed IFITM1 using the TCGA cervical cancer dataset as an example for another HPV-driven squamous cell carcinoma. In line to the findings in our ASCC RNAseq cohort, we found a significant association between high expression of IFITM1 and reduced recurrence-free survival in the TCGA cervical cancer dataset (Fig. 2f).
Considering the potential relevance of IFITM1 for immunoregulation, we assessed whether it correlates with key markers of the ASCC immune contexture, such as p16INK4A, CD8 + TIL, and PD-L1 status. The median p16INK4A score was 6, and patients were divided in the same manner as for IFITM1; 75 (58%) patients had high expression, and 55 (42%) low expression of p16INK4A. Also, 29 (22%) of patients had high, and 101 (78%) had low CD8 + TIL infiltration. A high expression of IFITM1 was associated with a PD-L1 CPS ≥ 10 (p = 0.03), and a TPS ≥ 1 (p = 0.04) (Table 2), whereas no significant association between IFITM1 and expression of p16INK4A or CD8 + TIL was noted. In accordance to our previous reports, we confirmed that high p16INK4A and high CD8 + TIL expression were associated with improved FFLF (p = 0.006 and p = 0.042, respectively, Supplementary Fig. 3), while FFDM was not significantly impacted. After creating a combined variable with p16INK4A and CD8 we found that patients with a high expression in both markers had an excellent prognosis with no events of locoregional failure, while patients with a low expression of both parameters had very poor local control (p = 0.019, Supplementary Fig. 4A). We also analyzed combinations of IFITM1 with p16INK4A or CD8. The few patients with a low p16INK4A score and a high IFITM1 expression had a significantly decreased FFLF (p < 0.001, Supplementary Fig. 4B), whereas the combination of high IFITM1 expression and low CD8 TIL was associated with a significantly decreased FFLF (p = 0.0018, Supplementary Fig. 4C). Additionally, there was no significant association between p16INK4A and expression levels of IFITM1 in the RNA seq cohort (p = 0.68). Due to possible influences on the immune contexture, we also investigated if HIV-status affects any of the analyzed markers. We found no significant association between HIV-status and either p16INK4A (p = 0.5), CD8 (p = 0.35), IFITM1 (p = 0.41), TPS (p = 0.57) or CPS (p = 0.7). Additionally, there was no significant association between HIV-status und IFITM1 expression in the RNAseq cohort (p = 0.59). We also conducted multivariate cox regression analysis for FFLF and FFDM including established clinical parameters like T-stage and N-stage and included p16INK4A and IFITM1. A high IFITM1 score was associated with a reduced FFLF (HR 2.79; p = 0.009) and FFDM (HR 4.39; p = 0.012) independent of T-stage, N-stage and p16INK4A (Table 3).
To assess the role of EMT, we stained pretreatment samples for E-Cadherin and Vimentin (Supplementary Fig. 6). Scores were dichotomized in high (weighted score >6) and low (WS ≤ 6). We found a significantly worse OS in patients with a low expression of E-Cadherin (p = 0.017), while there was only a non-significant trend towards a worse FFLF (p = 0.09) and FFDM (p = 0.052). For Vimentin, a significantly worse FFLF (p = 0.014), FFDM (p = 0.028) and OS (p = 0.035) was noted for patients with a high intratumoral expression of vimentin (Supplementary Fig. 6). A low expression of E-Cadherin and a high expression of Vimentin was furthermore associated with advanced (cT3-4) tumors (p = 0.004, p = 0.013), while only a high expression of E-Cadherin was associated with cN+ tumors (p = 0.008). Additionally, there was no association of E-Cadherin or Vimentin with IFITM1, p16INK4A, TPS or CD8.
Regulatory T cells in the peripheral blood are associated with poor prognosis
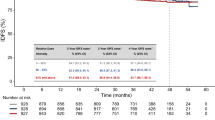
To assess the peripheral blood immune profile, we conducted FACS analysis in 47 consecutive ASCC patients with available peripheral blood samples (Supplementary Tables 4–7). The proportion of CD3+HLA-DR+ T-cells was significantly higher in male than in female patients at baseline (median 25.3% vs. 13.8%, p = 0.0021). Additionally, in HIV positive patients, CD3+HLA-DR+ T-cells were also significantly elevated at baseline (median 40.4% vs. 13.9%, p < 0.001), whereas no differences in the expression of PD1 on CD4+ or CD8+ T-cells was noted (p = 0.08 and p = 0.8, respectively). There were no other significant associations with T-stage, N-stage, or sex. The longitudinal measurements showed a significant reduction of CD3+ T-cells during CRT (p < 0.001, Fig. 3a), while the proportion of CD3+ T-cells expressing HLA-DR increased significantly from start of treatment until the first follow-up visit (p < 0.001, Fig. 3b). Additionally, the expression of PD1 on CD4+ T-cells increased significantly during CRT, while the PD1 expression on CD8+ T-cells showed no significant changes (Fig. 3c, d). These changes during treatment were significant regardless of the HIV- status (Supplementary Fig. 5). In this subset of 47 patients, 6 cases of locoregional failure, and 6 cases of distant metastasis were diagnosed during follow-up. Using the proportion of CD4+ T-cells that express CD25 and FoxP3 (regulatory T cells; Tregs) as a continuous variable, a cox regression showed a significantly reduced FFLF and FFDM for patients with a higher proportion at baseline (LF: HR 1.44; 95% CI: 1.05–1.97; p = 0.024; DM: HR 1.63; 95% CI: 1.15–2.31; p = 0.007). Using maximally selected rank statistics the ideal cut-off for Tregs was calculated at 5.2% (of CD3+ T-cells). After dichotomization, 7 (15%) patients were deemed to have high Tregs at baseline, and this was associated with a reduced FFLF (p = 0.0044, Fig. 3e) and FFDM (p = 0.0047, Fig. 3f). There were no significant differences between the levels of Tregs and HIV-status (p = 0.49).

From start to the end of CRT, and first follow-up, the amount of CD3+ T cells in the peripheral blood was significantly reduced (a), while the expression of human leukocyte antigen—DR isotype (HLADR) on CD3+ T-cells increased significantly (b). PD1 expression on CD4+ T-cells was significantly higher at the end of CRT (c), while PD1 expression on CD8+ T-cells remained stable (d). A high proportion of CD4+ T cells that express CD25 and FoxP3 (Tregs—regulatory T cells) is associated with significantly decreased freedom from local failure (FFLF) and freedom from distant metastasis (FFDM) (e, f).
CXCL2 levels in the peripheral blood are associated with neutrophilia and poor prognosis
We conducted baseline multiplex cytokine/chemokine measurements in 35 consecutive patients for whom peripheral blood serum samples were available (Fig. 4a). A high level of CXCL2 was significantly associated with higher T-stage (p = 0.0095) and neutrophil levels in the peripheral blood at baseline (R = 0.53, p = 0.0019, Fig. 4b). There was no significant correlation between CXCL2 levels and neutrophil to lymphocyte ratio (NLR) at baseline (R = 0.32, p = 0.075). Due to the small sample size, no significant correlation of CXCL2 levels was noted for FFLF or FFDM, however, combined locoregional and distant failure was significantly associated with CXCL2 levels (HR for an increase of 10 pg/ml 1.02; 95% CI 1.01–1.03, p = 0.004). Moreover, high expression of CXCL2 in tumor tissue (RNAseq dataset) was associated with a reduced FFLF (p = 0.0031, Fig. 4c).

a Heatmap showing the concentration of several cytokines for 35 patients before initiation of treatment. On the top, N-stage and T-stage are annotated. b Serum CXCL2 levels at baseline are significantly correlated with neutrophil levels. c Kaplan–Meier plots that show that an increased expression of CXCL2 in the RNAseq dataset (n = 98) is associated with impaired freedom from local failure (FFLF).
Discussion
To the best of our knowledge, we present the largest RNA sequencing cohort in patients treated with standard CRT for localized ASCC for whom clinically annotated data and long-term oncological outcome is available. Gene set enrichment analysis revealed that hallmark pathways associated with chronic inflammation (IFNγ; IFNα, inflammatory response, TNFα signaling via NF-κB) and EMT are significantly enriched in patients with poor response and prognosis. IFN signaling is at the core of immune regulation and generally works in an immune-stimulatory, proapoptotic and antiproliferative fashion to confer antiviral and antitumor properties to the host. However, it is well known that chronic IFN signaling can also lead to treatment resistance in tumors, e.g. via regulation of multigenic resistance programs to immune checkpoint blockade, including upregulation of PD-L1, on cancer cells16,17.
We here showed that the interferon-induced transmembrane protein 1 (IFITM1) is an adverse prognostic factor for FFLF and FFDM in ASCC, both on mRNA and protein expression levels, and acts independently of established prognostic factors like T-stage and N-stage. IFITM1 is part of the IFITM family of transmembrane proteins that exert antiviral properties by inhibiting the entry of viruses into cells, but may also play a pivotal role regulating innate antiviral and inflammatory as well as adaptive T-cell and B-cell responses18,19. Recent evidence showed that high expression of IFITM1 was associated with therapeutically resistant and aggressive disease courses for lung, esophageal and colon cancer, while a protective role was reported in cervical cancer and gastric cancer20,21,22,23.
The molecular mechanism of IFITM 1 in cancer generally remains poorly examined, but may include, among others, regulation of matrix metalloproteinases, caveolin-1, and other proteins associated with cell migration and adhesion24,25,26. Interestingly, a recent study in oral squamous cell carcinoma (also HPV-related) found that siRNA knockdown of IFITM1 sensitized tumor cells to ionizing radiation in vitro, indicating that IFITM1 may also be involved in DNA damage repair, although the mechanism remains unclear27. Here we demonstrate that high expression of IFITM1 in ASCC was associated with high PD-L1 expression on tumor cells (TPS), and on immune and tumor cells combined (CPS), which is in line with the notion of an immunoregulatory role of IFITM1 in ASCC18,28. This may, directly or indirectly, limit tumor immune activation. The lack of association between IFITM1 and p16 was surprising as it has been reported that IFITM1 is downregulated by HPV29. It is possible that the occurrence of infection is distinctively different to the state of tumorigenesis. It is also noteworthy that we found no differences in the composition of the tumor immune microenvironment according to HIV status, which is in line with the literature30.
Additionally, we further examined the role of EMT via immunostaining for classical EMT markers E-Cadherin and Vimentin. To the best of our knowledge this has not been analyzed in ASCC yet. A high expression of Vimentin was associated with impaired FFLF and FFDM in line with similar data in head and neck squamous cell carcinoma31,32.
In general, baseline leukocytosis and neutrophilia in the peripheral blood is a well-established negative prognostic factor in many tumor types. Recently, we could show that peripheral leukocytosis was inversely correlated with intratumoral CD8+ T-cells infiltration and associated with worse outcome after CRT in ASCC33,34. No detailed prospective immune monitoring studies have been reported for ASCC patients to date. Therefore, we assessed serial peripheral blood samples at baseline, during, at completion, and at first response assessment 6–8 weeks after CRT by use of multiparametric flow cytometry of PBMC. In line with reports in other tumor types35,36,37, a reduction of CD3+ T-cells and lymphopenia were observed after CRT. Interestingly, the remaining T-cells strongly expressed the activation marker HLA-DR which mediates antigen presentation and was suggested as possible biomarker to predict response to chemotherapy in breast cancer38,39. Conversely, CD4+ T-cells showed a marked increase in PD-L1 expression at CRT completion, in line with the observation that fractionated radiotherapy can lead to an upregulation of PD-L140,41. Thus, the observed HLA-DR activation and PD-L1 upregulation on CD4+ T-cells following CRT could be reflections of both immunosuppressive and immune activating effects of fractionated radiotherapy42. Whereas there were no differences in the tumor microenvironment between HIV-positive and -negative patients, we found a marked increase of CD3+HLA−DR+ cells at baseline in HIV+ patients. Nevertheless, the changes in the peripheral blood immune profile were again similar regardless of HIV status, supporting the notion that HIV+ patients under antiretroviral therapy can be treated like HIV- patients with CRT. Moreover, a higher proportion of CD25+/FoxP3+CD4+ T-cells (Tregs) at baseline was associated with a significantly reduced FFLF and FFDM in our series, indicating that a systemic immunosuppressive state is associated with poor prognosis in ASCC. Of note, in a previous work we found that a high intratumoral expression of FoxP3+ was associated with improved disease-free survival14—suggesting that there are differences in the role of systemic and intratumoral Tregs. Regarding other types of immune cells, the role of tumor associated macrophages in the microenvironment or myeloid derived suppressor cells in the peripheral blood and tumor microenvironment in anal cancer is still unclear. The differences in baseline CD3+ T-cells between male and female patients were most likely due to the fact that the vast majority of HIV+ patients in this cohort were male. Nevertheless, we found no differences in the changes of CD3+ T-cells, CD3+HLA-DR+ T-cells and CD4+PD1+ T-cells according to HIV-status. To gain more insights into the peripheral blood immune profile, several other markers, e.g. immune checkpoint molecules like LAG3, CTLA-4 or TIM-3, should be assessed in further flow cytometry studies of ASCC patients.
Finally, multiplex cytokine/chemokine measurements were conducted in a subset of 35 consecutive patients. CXCL2, a neutrophil chemo-attractant that is involved in chronic inflammation and tumor progression, was identified as negative prognostic factor after CRT in peripheral blood. Moreover, high expression of CXCL2 in tumor tissue in the RNAseq dataset was associated with significantly reduced FFLF. This again sheds light on the link between chronic inflammation in the tumor tissue and changes in the peripheral blood leading to worse treatment outcome in ASCC.
This study has several limitations. First, the retrospective nature of data collection could lead to selection bias. Second, immune phenotyping and cytokine measurements of blood samples were only available in a subset of patients, and cytokine measurement was restricted to baseline analysis to identify predictive markers. Third, the markers Foxp3 and CD25 alone are limited to demonstrate that these cells are Tregs, as a transient upregulation of FoxP3 on T-cells is possible43. Fourth, due to the rarity of the disease all patients were treated within a large timeframe, and it is known that PD-L1 staining decreases with sample age44. Fifth, p16INK4A is only a surrogate for HPV infection but could also be upregulated due to other causes. Nevertheless, it is already known that infection with HPV16 either as a mono infection or dual/triple infection is the main driver of p16INK4A expression in ASCC accounting for more than 90% of cases45.
Despite the novelty of the findings, more detailed mechanistic insight is lacking, mainly due to the lack of preclinical in vitro and in vivo models of ASCC. Of note, we have an established patient-derived organoid (PDO) research program for anorectal cancers in our center46,47 and have, hence, attempted to establish a PDO platform for ASCC to facilitate preclinical mechanistic studies to better understand the role of IFITM1 in ASCC (Goethe University of Frankfurt, Ethics committee protocol number 206/18). However, unlike PDO in rectal cancer patients, culture and growth of PDO from patients with ASCC was unsuccessful to date.
In conclusion, we found that chronic inflammatory pathways in tumor tissue, as measured by RNA sequencing (IFNγ; IFNα, TNFα signaling via NF-κB, IFITM1) and immunohistochemistry (IFITM1, PD-L1, CD8 + TIL), in line with elevated levels of regulatory T-cells and CXCL2 in peripheral blood, are associated with CRT resistance and poor prognosis in ASCC. One possible way to counteract the herein proposed resistance mechanism in ASCC could be the addition of immune checkpoint inhibitors to CRT. This hypothesis is currently tested in our ongoing, randomized phase-2 trial (RADIANCE) of standard CRT with or without addition of the immune checkpoint inhibitor durvalumab in locally advanced ASCC (NCT04230759)48.
Methods
Patients and treatment
Patients with histologically confirmed, localized ASCC, treated between 1998 and 2019 at 3 different sites (Frankfurt, Berlin, Tübingen) of the German Cancer Consortium (DKTK) were included. All patients received standard 5-FU/MMC-based CRT. Patient cohorts for molecular analysis were defined based on availability and quality of samples, including baseline formalin fixed paraffin embedded tissue for immunohistochemistry (FFPE, n = 130), baseline RNA sequencing of FFPE (n = 98), peripheral blood immune profiling (n = 47), and baseline serum cytokine measurement (n = 35). Patients and tumor characteristics of these cohorts are summarized in Supplementary Table 1. Due to the unavailability of sufficient tissue samples from the pretreatment biopsies, only a subset of 24 patients of the peripheral blood immune profiling cohort were available for tissue analysis. A flow chart depicting the composition and overlaps between these subsets is provided in Supplementary Fig. 1.
This study was performed in line with the principles of the Declaration of Helsinki. Approval for the collection of patient tissue and data within the DKTK-ROG was granted by the Ethics Committee of University of Frankfurt (Protocol Number 458/17). Taking of sequential blood samples during CRT was approved by the Ethics Committee of University of Frankfurt (Protocol Number 75/17). Informed consent was obtained from all individual participants included in the study.
Immunohistochemistry
Immunohistochemical staining of pretherapeutic biopsies was performed by a horseradish-peroxidase technique using a fully automated DAKO Omnis staining system (Agilent, Santa Clara, CA, US) with standardized DAKO EnVision™ FLEX TRS Blocking reagent for antigen retrieval (DAKO, pH high, 97 °C) and polyconal IFITM1 (Sigma-Aldrich, Munich, Germany, dilution 1:250), monoclonal E-Cadherin (clone NCH-38, GA05961-2, Agilent DAKO, ready to use), Vimentin (clone V9, GA63061-2, Agilent DAKO, ready to use), CD8 (clone C8/144B, GA62361-2, Agilent DAKO; ready to use), PD-L1 (clone ZR-3, GE00621-2, Agilent DAKO, ready to use) and p16INK4a antibodies (clone MX007, MAD000690QD12, Vitro-Master Diagnóstica, Sevilla, Spain, ready to use). Next, dextran polymer conjugated horseradish-peroxidase (EnVision FLEX HRP, DAKO) and 3,3’-diaminobenzidine (DAB) chromogen was used for visualization and DAKO hematoxylin solution (Agilent, Santa Clara, CA, US) for counterstaining. Negative control slides in the absence of primary antibodies were included for each staining.
For scoring of IFITM1, E-Cadherin and Vimentin, a weighted score was created by multiplication of the proportion of positive tumor cells (4: 75%–100%, 3: 50–75%, 2: 25–50%, 1: 0–25%) and the staining intensity (3: high, 2: medium, 1: low). The scores generated this way are between 1 and 12. The scoring of expression of CD8 + TIL was done semi-quantitatively as described before14. Categories for scoring the intratumoral component were: (1) no, or sporadic cells; (2) moderate number of cells; (3) abundant occurrence of cells; (4) highly abundant occurrence of cells. Patients were then divided into either having a high infiltration (score 3 or 4) or a low amount of infiltration (score 1 or 2). Analysis of p16INK4a detection was done as described previously45. For PD-L1 tumor proportion score (TPS), immune cell score (IC) and combined positive score (CPS) were assessed49. Samples were independently evaluated by three investigators (DM, PKZ, FR) who were blinded to patient-specific clinical information. Images were acquired with the AxioScan Z.1 slide scanner and ZEN software (Carl Zeiss Microscopy, Jena, Germany).
RNA sequencing
Baseline FFPE tumor samples were processed for total RNA isolation, library preparation and raw counts generation by GENEWIZ, LLC (South Plainfield, NJ, USA). Total RNA was isolated from FFPE samples using Qiagen RNeasy FFPE Kit (Qiagen) and depleted from ribosomal RNA (rRNA) to prepare sequencing libraries After quality control (TapeStation, Agilent Technologies, Palo Alto, CA, USA) samples were sequenced using a 2 × 150 paired end configuration (HiSeq 2 × 150 PE HO) and quality guarantee ≥80% of bases ≥Q3.
Overall analysis was done by a custom RNAseq analysis workflow, making use of public software packages. In this context, quality of raw reads was assessed prior and after trimming by FastQC (https://github.com/s-andrews/FastQC), while read quality trimming was performed by Trimmomatic version 0.3950. Trimmed and filtered reads were aligned on the Human Genome (Ensemble version hg38; homo_sapiens.104.mainChr.gtf) using the STAR mapper software (2.7.9a), using default parameters plus: --outFilterMismatchNoverLmax 0.1 --outFilterScoreMinOverLread 0.66 --outFilterMatchNminOverLread 0.66 --outFilterMatchNmin 20 --alignEndsProtrude 10 ConcordantPair --alignMatesGapMax 2000 --limitOutSAMoneReadBytes 10000000 --outMultimapperOrder Random --sjdbOverhang 100 --alignIntronMax 20000051. Counting of reads aligned to genes by STAR was performed by the featureCounts tool (included in the Subread package version 2.0.2)52. All reads that map at least partially within a given exon are aggregated to gene counts (homo_sapiens.104.mainChr.gtf). Multimapping reads were excluded. Following raw counting, gene expression values were normalized by the DESeq2 tool (1.30.0)53, and log transformed for further analysis. Differentially expressed genes were identified by DESeq2 as well (corrected p value of ≤0.05; Log2FC ≥ 0.58 and ≤−0.58 were considered to be differentially regulated).
Heatmaps were generated, using the normalized gene values and the complexheatmap R package, making use of z-score transformation per row. All groups of differential gene expression (DEG) lists were analyzed for enrichment of gene sets by the KOBAS tool (version 3.0) and the GSEA tool (version 4.3.2)54. For further exploration of the RNA sequencing data and DESeq2 analysis, patients were divided into two groups: poor responders who did either not have a clinical complete response (cCR) after treatment or developed locoregional or distant recurrence after initial cCR, and good responders who had a cCR and no recurrence during follow-up.
Multiparametric flow cytometry of peripheral blood mononuclear cells
The prospective analysis of immune cells in the peripheral blood was done on a subset of 47 consecutive patients with available blood samples that were treated with standard CRT at Frankfurt. This prospective study was approved by the local ethics committee (Goethe University of Frankfurt, protocol Nr. 75/17) and written consent was obtained. Blood was taken before the initiation of treatment, on day 8 of CRT, on day 38 (last day of CRT), and on the first follow-up visit 6–8 weeks after completion of CRT.
Immune phenotyping was performed from peripheral blood mononuclear cells (PBMCs) following density gradient centrifugation (Biocoll, Biochrom, Berlin, Germany) by multi-color flow cytometry (CytoFLEX S, Beckman Coulter, Krefeld, Germany) using 3 panels of fluorescence-labeled antibodies (Supplementary Table 2). Briefly, 1 × 106 cells were blocked with human FcR binding inhibitor (eBioscience, Frankfurt am Main, Germany), incubated with antibodies specific for lymphatic lineages (panel 1: CD45 [clone 2D1, 655873, dilution 1:10], CD3 [clone SK7, 332771, dilution 1:10], HLA-DR [clone L243, 655874, dilution 1:80]), T lymphocytes and activation markers (panel 2: CD3 [clone SK7, 332771, dilution 1:10], CD4 [clone SK3, 651849, dilution 1:80], CD8 [clone SK1, 641400, dilution 1:40), PD-1 [clone eBioJ105, 25-2799-42, dilution 1:40], Perforin [clone dG9, 556577, dilution 1:10]), or regulatory T cells (panel 3: CD3 [clone SK7, 332771, dilution 1:10], CD4 [clone SK3, 651849, dilution 1:80], CD8 [clone SK1, 641400, dilution 1:40], CD25 [clone 2A3, 740198, dilution 1:40], PD-1 [clone eBioJ105, 25-2799-42, dilution 1:40], FoxP3 [clone 236A/E7, 17-4777-42, dilution 1:40]) after fixation and permeabilization (Fix & Perm, Nordic MUbio, Susteren, Netherlands, and FoxP3 staining set, Invitrogen, Frankfurt am Main, Germany) for panels 2 and 3. Next, a total of 100,000 CD45+ cells were gated in case of panel 1, and of CD3+ cells in case of panels 2 and 3 (see Supplementary Fig. 2), and data were analyzed by using the CytExpert software (Beckman Coulter) with the ratio of positively stained cells defined as the percentage of cells within a defined leukocyte phenotype gate from total CD45+/CD3+ cells.
Chemokines, cytokines and growth factor measurements in serum
Quantification of baseline chemokines, cytokines and growth factors protein levels in baseline blood serum (1:4 dilution) was conducted in a subset of 35 consecutive patients with available pretreatment serum samples. The Bio-Plex Pro human chemokine panel 40-plex Assay kit (Bio-Rad) was used according to the manufacturer’s instructions manual (#171AK99MR2) and analyzed using a Bio-PlexTM 100/200 reader (Bio-Rad).
Statistical analysis
Differences between groups were assessed using Pearson’s Chi-squared test for categorical variables, and the nonparametric Wilcoxon rank sum test for continuous variables. Correlations between continuous variables were assessed using the Spearman’s rank correlation coefficient. Survival times were calculated from start of CRT to the date of respective events or last follow-up. Freedom from local failure (FFLF) was calculated using the date of diagnosis of incomplete response at restaging or locoregional recurrence after initial cCR as event. Freedom from distant metastasis (FFDM) was calculated using the date of diagnosis of distant metastases as event. Differences were calculated using the log-rank test or Cox regression analysis as necessary. The assumption of proportional hazard was verified by assessing the scaled Schoenfeld residuals. Cut-offs for IFITM1 and CXCL2 reads were calculated using the maximally selected rank statistics, which calculate the most optimal cut-off for a continuous variable55. All statistical analysis was performed with R (Version 4.2.2)56. A p value ≤ 0.05 was considered significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA Sequencing data are deposited under GEO accession GSE254331. Other data are available upon reasonable request.
Code availability
Custom code that was used in the generation or analysis of the datasets is available upon reasonable request.
References
Jemal, A. et al. Annual Report to the Nation on the Status of Cancer, 1975-2009, featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J. Natl. Cancer Inst. 105, 175–201 (2013).
Shiels, M. S., Kreimer, A. R., Coghill, A. E., Darragh, T. M. & Devesa, S. S. Anal Cancer Incidence in the United States, 1977-2011: distinct patterns by histology and behavior. Cancer Epidemiol. Biomark. Prev. 24, 1548–1556 (2015).
De Vuyst, H., Clifford, G. M., Nascimento, M. C., Madeleine, M. M. & Franceschi, S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int. J. Cancer 124, 1626–1636 (2009).
Cacheux, W. et al. Mutational analysis of anal cancers demonstrates frequent PIK3CA mutations associated with poor outcome after salvage abdominoperineal resection. Br. J. Cancer 114, 1387–1394 (2016).
Casadei Gardini, A. et al. KRAS, BRAF and PIK3CA status in squamous cell anal carcinoma (SCAC). PLoS ONE 9, e92071 (2014).
Chung, J. H. et al. Comprehensive genomic profiling of anal squamous cell carcinoma reveals distinct genomically defined classes. Ann. Oncol. https://doi.org/10.1093/annonc/mdw152 (2016).
Iseas, S. et al. A clinical and molecular portrait of non-metastatic anal squamous cell carcinoma. Transl. Oncol. 14, 101084 (2021).
James, R. D. et al. Mitomycin or cisplatin chemoradiation with or without maintenance chemotherapy for treatment of squamous-cell carcinoma of the anus (ACT II): a randomised, phase 3, open-label, 2 × 2 factorial trial. Lancet Oncol. 14, 516–524 (2013).
Gunderson, L. L. et al. Long-term update of US GI intergroup RTOG 98-11 phase III trial for anal carcinoma: survival, relapse, and colostomy failure with concurrent chemoradiation involving fluorouracil/mitomycin versus fluorouracil/cisplatin. J. Clin. Oncol. 30, 4344–4351 (2012).
Martin, D., Rödel, C. & Fokas, E. Chemoradiotherapy for anal cancer: are we as good as we think? Strahlenther. Onkol. Organ Dtsch. Rontgengesellschaft Al 195, 369–373 (2019).
Gunderson, L. L. et al. Anal carcinoma: impact of TN category of disease on survival, disease relapse, and colostomy failure in US Gastrointestinal Intergroup RTOG 98-11 phase 3 trial. Int. J. Radiat. Oncol. Biol. Phys. 87, 638–645 (2013).
Ajani, J. A. et al. Prognostic factors derived from a prospective database dictate clinical biology of anal cancer: the intergroup trial (RTOG 98-11). Cancer 116, 4007–4013 (2010).
Glynne-Jones, R. et al. Prognostic factors for recurrence and survival in anal cancer: generating hypotheses from the mature outcomes of the first United Kingdom Coordinating Committee on Cancer Research Anal Cancer Trial (ACT I). Cancer 119, 748–755 (2013).
Balermpas, P. et al. Human papilloma virus load and PD-1/PD-L1, CD8+ and FOXP3 in anal cancer patients treated with chemoradiotherapy: rationale for immunotherapy. OncoImmunology 6, e1288331 (2017).
Serup-Hansen, E. et al. Human papillomavirus genotyping and p16 expression as prognostic factors for patients with American Joint Committee on Cancer stages I to III carcinoma of the anal canal. J. Clin. Oncol. 32, 1812–1817 (2014).
Benci, J. L. et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554.e12 (2016).
Minn, A. J. & Wherry, E. J. Combination cancer therapies with immune checkpoint blockade: convergence on interferon signaling. Cell 165, 272–275 (2016).
Diamond, M. S. & Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13, 46–57 (2013).
Gómez-Herranz, M., Taylor, J. & Sloan, R. D. IFITM proteins: understanding their diverse roles in viral infection, cancer, and immunity. J. Biol. Chem. 299, 102741 (2023).
Borg, D. et al. Expression of IFITM1 as a prognostic biomarker in resected gastric and esophageal adenocarcinoma. Biomark. Res. 4, 10 (2016).
Sakamoto, S. et al. Interferon-induced transmembrane protein 1 (IFITM1) promotes distant metastasis of small cell lung cancer. Int. J. Mol. Sci. 21, 4934 (2020).
Sari, I. N. et al. Interferon-induced transmembrane protein 1 (IFITM1) is required for the progression of colorectal cancer. Oncotarget 7, 86039–86050 (2016).
Zheng, W. et al. Down-regulation of IFITM1 and its growth inhibitory role in cervical squamous cell carcinoma. Cancer Cell Int. 17, 88 (2017).
Hatano, H. et al. IFN-induced transmembrane protein 1 promotes invasion at early stage of head and neck cancer progression. Clin. Cancer Res. 14, 6097–6105 (2008).
Yu, F. et al. IFITM1 promotes the metastasis of human colorectal cancer via CAV-1. Cancer Lett. 368, 135–143 (2015).
Yu, F. et al. Knockdown of interferon-induced transmembrane protein 1 (IFITM1) inhibits proliferation, migration, and invasion of glioma cells. J. Neurooncol. 103, 187–195 (2011).
Yang, J. et al. Combination of IFITM1 knockdown and radiotherapy inhibits the growth of oral cancer. Cancer Sci. 109, 3115–3128 (2018).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015).
Ma, W. et al. Human papillomavirus downregulates the expression of IFITM1 and RIPK3 to escape from IFNγ- and TNFα-mediated antiproliferative effects and necroptosis. Front. Immunol. 7, 496 (2016).
Yanik, E. L. et al. Association of HIV status with local immune response to anal squamous cell carcinoma: implications for immunotherapy. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2017.0115 (2017).
Nijkamp, M. M. et al. Expression of E-cadherin and vimentin correlates with metastasis formation in head and neck squamous cell carcinoma patients. Radiother. Oncol. 99, 344–348 (2011).
Zhou, J., Tao, D., Xu, Q., Gao, Z. & Tang, D. Expression of E-cadherin and vimentin in oral squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 8, 3150–3154 (2015).
Martin, D. et al. Peripheral leukocytosis is inversely correlated with intratumoral CD8+ T-cell infiltration and associated with worse outcome after chemoradiotherapy in anal cancer. Front. Immunol. 8, 1225 (2017).
Schernberg, A. et al. Leukocytosis and neutrophilia predicts outcome in anal cancer. Radiother. Oncol. 122, 137–145 (2017).
Cho, O., Chun, M., Chang, S.-J., Oh, Y.-T. & Noh, O. K. Prognostic value of severe lymphopenia during pelvic concurrent chemoradiotherapy in cervical cancer. Anticancer Res. 36, 3541–3547 (2016).
Kroese, T. E. et al. Severe lymphopenia acquired during chemoradiotherapy for esophageal cancer: incidence and external validation of a prediction model. Radiother. Oncol. 163, 192–198 (2021).
Liu, F. et al. Hypofractionated concurrent chemoradiotherapy related lymphopenia and its association with survival in locally advanced non-small cell lung cancer patients. Front. Oncol. 12, 979384 (2022).
Saraiva, D. P., Jacinto, A., Borralho, P., Braga, S. & Cabral, M. G. HLA-DR in cytotoxic T lymphocytes predicts breast cancer patients’ response to neoadjuvant chemotherapy. Front. Immunol. 9, 2605 (2018).
Viallard, J. F. et al. HLA-DR expression on lymphocyte subsets as a marker of disease activity in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 125, 485–491 (2001).
Deng, L. et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Invest. 124, 687–695 (2014).
Dovedi, S. J. et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 74, 5458–5468 (2014).
Sharabi, A. B., Lim, M., DeWeese, T. L. & Drake, C. G. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 16, e498–509 (2015).
Wang, J., Ioan-Facsinay, A., van der Voort, E. I. H., Huizinga, T. W. J. & Toes, R. E. M. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur. J. Immunol. 37, 129–138 (2007).
Fernandez, A. I., Gaule, P. & Rimm, D. L. Tissue age affects antigenicity and scoring for the 22C3 immunohistochemistry companion diagnostic test. Mod. Pathol. 36, 100159 (2023).
Rödel, F. et al. Human papillomavirus DNA load and p16 INK4a expression predict for local control in patients with anal squamous cell carcinoma treated with chemoradiotherapy: HPV16 DNA and p16 INK4a expression. Int. J. Cancer 136, 278–288 (2014).
Michels, B. E. et al. Pooled in vitro and in vivo CRISPR-Cas9 screening identifies tumor suppressors in human colon organoids. Cell Stem Cell 26, 782–792.e7 (2020).
Nicolas, A. M. et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell 40, 168–184.e13 (2022).
Martin, D. et al. RADIANCE—radiochemotherapy with or without Durvalumab in the treatment of anal squamous cell carcinoma: A RANDOMIZED Multicenter phase II trial. Clin. Transl. Radiat. Oncol. 23, 43–49 (2020).
Witte, H. M. et al. Prognostic IMPACT of PD-L1 expression in malignant salivary gland tumors as assessed by established Scoring criteria: tumor proportion score (TPS), combined positivity score (CPS), and immune cell (IC) infiltrate. Cancers 12, 873 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. 102, 15545–15550 (2005).
Lausen, B. & Schumacher, M. Maximally selected rank statistics. Biometrics 48, 73–85 (1992).
R Development Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2008).
Acknowledgements
D.M. and P.K.Z. were supported by the Else-Kröner-Fresenius-Foundation. M.D. and M.F. were supported by the LOEWE FCI.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
D.M.: conception, data collection, data analysis and interpretation, drafting the article; F.R.: conception, data collection, data analysis and interpretation; S.H.: data collection, conduction of experiments; M.L.: data analysis and interpretation; P.K.Z.: data collection; M.F.: data collection; M.D.: data collection, data analysis; L.F.: conduction of experiments; G.K.: data collection; I.T.: data collection; D.Z.: data collection; C.G.: data collection; C.R.: conception, data analysis and interpretation, critical revision of the article; E.F.: conception, data analysis and interpretation, critical revision of the article; all authors: final approval of the version to be published.
Corresponding author
Ethics declarations
Competing interests
E.F. received funding from AstraZeneca. D.M., F.R., S.H., M.L., P.K.Z., M.F., M.D., L.F., G.K., I.T., D.Z., C.G., and C.R. declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martin, D., Rödel, F., Hehlgans, S. et al. Inflammatory pathways confer resistance to chemoradiotherapy in anal squamous cell carcinoma. npj Precis. Onc. 8, 93 (2024). https://doi.org/10.1038/s41698-024-00585-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00585-y
