
Abstract
The rich chemical information from tissue metabolomics provides a powerful means to elaborate tissue physiology or tumor characteristics at cellular and tumor microenvironment levels. However, the process of obtaining such information requires invasive biopsies, is costly, and can delay clinical patient management. Conversely, computed tomography (CT) is a clinical standard of care but does not intuitively harbor histological or prognostic information. Furthermore, the ability to embed metabolome information into CT to subsequently use the learned representation for classification or prognosis has yet to be described. This study develops a deep learning-based framework -- tissue-metabolomic-radiomic-CT (TMR-CT) by combining 48 paired CT images and tumor/normal tissue metabolite intensities to generate ten image embeddings to infer metabolite-derived representation from CT alone. In clinical NSCLC settings, we ascertain whether TMR-CT results in an enhanced feature generation model solving histology classification/prognosis tasks in an unseen international CT dataset of 742 patients. TMR-CT non-invasively determines histological classes - adenocarcinoma/squamous cell carcinoma with an F1-score = 0.78 and further asserts patients’ prognosis with a c-index = 0.72, surpassing the performance of radiomics models and deep learning on single modality CT feature extraction. Additionally, our work shows the potential to generate informative biology-inspired CT-led features to explore connections between hard-to-obtain tissue metabolic profiles and routine lesion-derived image data.
Similar content being viewed by others
Introduction
Numerous studies have developed imaging analysis pipelines to analyze and diagnose lung cancers. Tools such as radiomics have been helpful in extracting features from CT scans of lung cancer patients, followed by machine learning models to perform classification or prognosis tasks1,2,3. These features quantify the tumor’s spatial complexity, such as shape, size and intensity features and are becoming part of a routine investigation in the literature3. More recently, some studies have attempted to replace radiomic features with deep learning (DL) features extracted from convolutional neural networks (CNN) directly on lesions4,5,6,7. Of various CNN architectures, autoencoders are some of the most widely adopted and aim to find features that allow a model to reconstruct the original image in a different context4,7,8. In the context of diagnosing from CT scans, DL methods tend to outperform traditional radiomics feature extraction and selection methods9,10.
However, in practice, these features have limited clinical performance, e.g. classification C-index = 0.65, and the used framework precludes intuitive biological or clinical interpretability11. Current studies generate radiomic features and subsequently check if they associate with genes, metabolites, proteins and other biological factors, using for example, gene-set enrichment analysis12,13,14. In contrast, our present study develops a framework for generating features from images that have already learned specific biological representations of tissue metabolites.
A new and evolving field of computational biology combines two or more diverse modalities to improve the performance of each15,16,17. Gundersen and colleagues demonstrated the feasibility of developing multimodal pairings of pathology and genomic profiles to extract deep features from pathology images connected to the genomic profiles of the patient to obtain more explainability18. Once developed, the corollary of this learned representation approach, implies that one of the two modalities is sufficient to represent the other in the absence of both modalities being present18. Inspired by this approach, we investigate whether it is possible to generate deep features from hard-to-obtain tumor and normal tissue metabolome data, on the one hand, and the more routine CT scan image data on the other.
In characterizing tumors, the choice of metabolomics profiles as a benchmark is predicated on our recent work that expounds the use of tumor and adjacent tissue metabolome information in asserting the classification of histology subtypes, achieving an F1-score of 0.96, significantly outperforming most published models from imaging data in the field of lung cancer subtype classification or prognosis19,20. While the chemical information from tissue metabolomics is rich, the approach is not routinely used in patient management due to its invasiveness and analytical complexity21. Thus, we have developed a pipeline using an autoencoder to investigate, for the first time, whether the deep features of CT image reconstructions linked to chemical information from the metabolomics of patients will provide sensitive clinical information. These deep features extracted from the deep probabilistic canonical correlation analysis (DPCCA) model were named tissue metabolomic radiomic computed tomography (TMR-CT). The model aims to establish a connection between both data modalities.
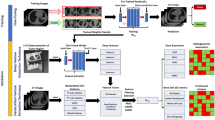
The model comprises a two-stage neural network and PCCA. The neural network part first finds separate embeddings for each data modality (CT scan, metabolites). These embeddings are subsequently combined to maximize correlation and minimize reconstruction loss for both modalities18. The benefit of this structure is two-fold. First, the deep features captured for each data view maximize the shared variation. Second, the generative structure of the model allows cross-data modality imputation. This is particularly important given the difficulty in obtaining the paired datasets from both modalities19. We explore the aforementioned benefit in this manuscript by utilizing the embeddings derived from CT scans for histology subtype classification and prognosis of non-small cell lung cancer (NSCLC) patient data. Our approach achieves an enhanced feature generation model in both tasks while also providing valuable biological insights. We accomplish this by employing TMR-CT, which encompasses reconstructed representations of tissue metabolite types and intensities. This work demonstrates the potential for the method to enhance the practitioner’s – radiologist’s, respiratory physician’s, or oncologist’s – ability to determine histology subtype classification, as well as prognosis, using algorithms derived from the current work, as illustrated in Fig. 1.

a Dataset collection for generating deep features, evaluating feature stability, histology subtype classification and prognosis. b The DPCCA model is used to find a shared latent space between the CT scans and metabolomics. An enlarged version of this model is shown, with the purple box highlighting the section responsible for generating TMR-CT features. In this model, Xa, Xb is the original paired image and metabolomics; from these, we create ya,yb image and metabolomics embeddings, respectively. The PCCA model then combines them into za,zab,zb latent variables. The latent variables za,zb capture view-specific variation while zab captures the covariance. From the latent variables, we use a generative process of the model-sampling from the low dimension PCCA to reconstruct image and metabolomics embeddings \({\hat{y}}^{a},{\hat{y}}^{b}\). Each embedding \({\hat{y}}^{a},{\hat{y}}^{b}\) is then decoded to produce the \({\hat{X}}^{a},{\hat{X}}^{b}\) using view-specific decoders. During implementation, when we only have the CT image, we extract the learned representation ya, which we have defined as the TMR-CT features. c Convolutional neural networks within the purple box were used to generate TMR-CT features on external datasets and to test the stability of the features using the RIDER dataset. d TMR-CT features are utilized for histology subtype classification, with random forest (RF)-based approaches displayed, as they exhibited superior performance in both tasks.
Results
Datasets
The datasets in our study can be split into three parts, as seen in Fig. 1 (data source): developing deep features, testing feature stability, histology classification and prognosis.
The dataset from 48 patients with both tissue and CT scans used to develop TMR-CT was obtained from the University Hospital Reina Sofia (UHRS), Spain. The research study was conducted in accordance with the Helsinki Declaration and was approved by the Cordoba Clinical Research Ethics Committee, all patients provided a signed written informed consent for participation in the study. Paired CT and tissue, obtained from both the tumor and non-tumor adjacent tissues, were collected from patients with NSCLC. Tissue samples were stored by the Andalusian Health Services Biobank, and the metabolomic profiling was performed under contract by Metabolon19. The patients did not receive any radiation or chemotherapy treatments before surgical resection, and the clinicopathological information was obtained prospectively and shown in Table 1. All tissue data were processed as previously reported19. CT scans were segmented by a board-certified clinical radiologist (MC).
Metabolite analyses were performed as previously described19. Samples were extracted by an aqueous methanol extraction process and analyzed with ultra‐performance liquid chromatography/tandem mass spectrometry (UPLC/MS/MS; positive mode), UPLC/MS/MS (negative mode), and GC/MS by Metabolon. Tissue metabolites were identified by comparison with library entries of purified standards or recurrent unknown entities. Based on the literature and KEGG/HMDB databases, metabolites were annotated to one of eight ‘super pathways’ corresponding to their general metabolic processes (amino acid, lipid, carbohydrate, nucleotide, peptide, energy, cofactors and vitamins, and xenobiotics), and to one of 73 ‘sub pathways’ representing more specific metabolic pathways or biochemical subclasses; in the aggregate, 851 metabolites were identified through this approach for both lung adenocarcinoma (AC) and squamous cell carcinoma (SCC) subtypes, and normal lung tissues19.
To test the stability of the TMR-CT features, we used the open-sourced RIDER dataset consisting of 32 patients with NSCLC who underwent two sequential chest CT scans within 15 mins, employing the same imaging protocols22. In this study, three radiologists measured the two greatest diameters of each lesion on both scans obtaining highly reproducible measurements, all with concordance correlation coefficients (CCC) greater than 0.96. Thus, this dataset has been shown to be useful in determining the reproducibility of deep learning features for NSCLC23.
To test how useful TMR-CT is for histology classification and prognosis prediction, we used four different datasets summarized in Table 2. To train our models, we used the open-source TCIA (The Cancer Imaging Archive), from which we selected 203 patients diagnosed with either AC or SCC24. The TCIA was split into 120 for training and validation and 83 for external validation. Then, to evaluate how well the developed model generalize to new NSCLC datasets, we used three geographically distinct datasets from the OCTAPUS-AI study (ClinicalTrials.gov identifier: NCT04721444) as external test sets (GSTT, Imperial and RMH); OCTAPUS-AI represents a study from multiple UK cancer centers (Guy’s and St Thomas’ NHS Foundation Trust, Imperial College Healthcare NHS Trust and the Royal Marsden NHS Foundation Trust respectively) collected for the explicit purpose of developing robust predictive lung cancer algorithms25. As the data were deidentified, patient consent was not required as per the respective Health Research Authority and Research Ethics Committee approvals.
Overview of metabolic profiles for NSCLC
With many more metabolomic features than patients, we first filtered the metabolomics by only including those profiled in all 48 patients for both tumor and non-tumor adjacent tissue from the UHRS hospital; this reduced the number of metabolites to 174. The super pathway of these features is summarized in Fig. 2a, and we observed a high degree of positive and negative correlation between several of the features from the tumor tissue samples, as shown in Fig. 2b.

a Distribution of metabolites super pathway, which were present in all patient’s data used in the current study. b Pearson correlation heat map between tumor metabolomics for all patients.
Despite the large number of metabolomic features compared to our sample size, we did not need to perform feature reduction, as principal component analysis (PCA) is known to be robust to correlated features26. Furthermore, we reasoned that when incorporating the metabolomics feature into the DPCCA model, we would perform data augmentation as specified in “Pre-processing data for training and testing DPCCA” to mitigate overfitting.
We were unable to use permutation importance to identify the most important metabolomic features due to feature collinearity phenomenon; permuting any single feature would have little effect on the random forest (RF) performance. As an alternative, we performed hierarchical clustering on the Pearson rank-order correlation and chose a single feature from every cluster, as suggested by Rosato and co-workers in a systems biology-enhanced analytical framework for metabolomics data27. This approach allowed us to reduce the number of features to six metabolomics (1,5-anhydroglutocitol (1,5-AG), 1-arachidonoylglycerophosphoethanol-amine*, 1-stearoylcerol (1-monostearin), 3-hydroxybutyrate(BHBA), 3-phosphogylcerate and alanine) while still maintaining the same performance (F1-score of 1) in discriminating AC from SCC tissue.
Ordinarily metabolites discriminate histology subtypes but are unconnected to radiomic features
We investigated the data structure of the metabolomic profiles obtained from 48 tumor and non-tumor tissue samples in relation to the radiomic features from CT scans. For each CT scan, we extracted 438 radiomic features using the TextLab 2.0 software related to shape, size, intensity, and wavelet decomposition1. After pairing metabolomic and radiomic features for the 48 samples, we compared the predictive power and connection between both modalities. To determine the predictive power of both modalities, we examined 2-dimensional PCA with all variables. We found that the metabolomics provided more informative predictions of tissue subtypes compared to CT radiomics Fig. 3a–c. Determining the most important metabolomic features, proved challenging as described in “Overview of Metabolic Profiles for NSCLC” and we used hierarchical clustering based on the Pearson rank-order correlation.

a Two-dimensional PCA of metabolomic features from UHRS dataset. b Two-dimensional PCA of radiomic features from UHRS dataset. c Two-dimensional PCA of radiomic features from a larger TCIA dataset. d Pearson correlation heatmap between CT radiomic features and six metabolomic features important for classification of histology subtypes from UHRS dataset.
Due to the large number of radiomic features, it is common practice to perform a feature reduction step prior to model building. While there is no established set way, various task-dependent strategies have been proposed28. For comparison, we only retained features with an intra-class-correlation coefficient (ICC) greater than 0.75, resulting in 438 features29.
To investigate the connection between the two sets of features within the tumor metabolomics and imaging datasets, we conducted a Pearson correlation analysis between the radiomics and the top ten metabolomics features. A heatmap of the data is shown in Fig. 3d. With a maximum absolute correlation coefficient of 0.45 and a mean correlation of 0.02, the results suggested a weak correlation between the two data modalities and techniques. Consequently, traditional canonical correlation analysis might be inappropriate.
The DPCCA model can be trained on CT scans and metabolomics to define TMR-CT
DPCCA was trained to expound the shared latent space between metabolomics and CT scan data. To evaluate the performance of DPCCA, we assessed the following. Firstly, we investigated if our model could reconstruct both modalities from the shared latent space. Thus, we examined the reconstructed CT slices and the metabolomic covariance matrix on the held-out test dataset. As seen in Fig. 4, DPCCA successfully reconstructed both views. As enshrined in a similar inference framework by Gundersen and co-workers for gene expression and latent pathology space18, we wanted to verify that our end-to-end model, composed of a neural network and DPCCA, makes use of both components. To test this, we computed the expected complete negative log-likelihood on the held-out dataset. A limitation of our work relates to the restricted training sample size, which could lead to overfitting when using DPCCA for training. As shown in Supplementary Fig. 1, the loss function of the validation set decreased during training for two modalities similar to those observed in a previously published study that reported DPCCA. As a baseline, we compared its performance to how well the image component of the DPCCA image autoencoder can reconstruct the image modality. As expected, and noted in an earlier study, the single modality is faster to train and has smaller reconstruction loss than the DPCCA which aims to reconstruct both views18.

To test the quality of our latent space model developed by DPCCA, we examined CT and metabolomics reconstruction. The images above were obtained from the test data for different patients (Top row). The original metabolomics expression covariance matrix and random CT slices from test data and (Bottom row) the reconstruction of the CT image of unseen test dataset when both the original image and metabolites are provided as inputs to the model.
Tumor size, CT scan thickness, and manufacturer can impact the outcomes. Principal component analysis (explained variance) outputs of TMR-CT, to assess congruence of data from TNM8 stage and CT scan thickness are illustrated in Supplementary Figs. 2 and 3, and show that these variables have no impact on the TMR-CT. We also examined the correlation between tumor size and the TMR-CT features and found that the maximum absolute correlation was weak (0.32, p = 0.03).
To gain a good understanding of metabolomic features that are the focus of the study, we plotted the maximum absolute correlation between the metabolomic features and the ten TMR-CTs in Fig. 5a. The two metabolites with the lowest correlation are 2-hydroxyglutarate and urea, with a maximum correlation of 0.14 and 0.16, respectively. The two metabolites with the highest correlation are sedoheptulose-7-phosphate and uridine, with a correlation of 0.72 and 0.64, respectively.

a Correlation of performance of the TMR-CT model developed by DPCCA, the bar plot shows the highest absolute correlation between metabolites and the TMR-CT features; we expand the top ten least correlated metabolites and the top ten most correlated metabolites in the yellow boxes. b Unsupervised hierarchical clustering of the TMR-CT features with the metabolomic profile from nodule UHRS identified three distinct subgroups. The blue rectangle contains a summary of the performance of the distinct cluster for classification and prognosis. The pentose phosphate metabolite sedoheptulose-7-phosphate, an important source for ribonucleotides and reduced nicotinamide adenine dinucleotide phosphate (NADPH), adenosine, reported in our previous lung metabolomics publication to be high in tumor tissue are emphasized in both the highly correlated metabolite set (a) and cluster 2 (b). Other metabolites such as asparagine, an important regulator of cancer cell amino acid homeostasis, anabolic metabolism and proliferation are emphasized only by the clustering approach19,45,46.
It is important to note that the focus of the DPCCA model was to find a shared latent space and then reconstruct the learned representation; thus, the ‘metabolomic features’ that the model focuses on are not necessarily those with the greatest classification or prognosis power, but rather those that the model can use in representing the CT-images. To interpret the influence of metabolomics on the shared embeddings, we plotted an unsupervised hierarchical clustering of the TMR-CT correlations in Fig. 5b, which shows the presence of three clusters with relevant metabolite super pathway information on the test dataset of the UHRS.
We see how the three clusters correlate very differently to the metabolomic features, showing that they complement themselves and model different parts of the metabolomic features. This shows that the DPCCA has achieved the goal of generating different features that capture the entire metabolomic profile by through correlation, though the exact biological pathways used in the correlation is not intuitive. A small subset of metabolites at the bottom of the map has a low correlation with all three clusters, suggesting that the DPCCA model could not correctly identify them in the CT image. The performance of each cluster on classification and prognosis appears unique; clusters C1 and C2, while showing opposite correlation values to most metabolites demonstrated similar performance in both histology classification and prognosis, which C3 performed less well in both tasks.
Reliability and reproducibility of TMR-CT
To assess the stability of our encoder algorithm, we tested it in a test-retest context using the publicly available dataset, RIDER, consisting of 32 patients with lung cancer. Each patient underwent two chest CT thorax scans (within 15 min apart) using the same imaging protocol22. We evaluated the stability of the encoder by examining the TMR-CT features between the test and retest scans. Our results demonstrated a high level of stability with an ICC of 0.86 for TMR-CT showing that our model had been well-regularized.
To account for inter-reader stability, we adopted an approach of relocating the input seed points to the center of the tumor. This aimed to simulate various radiologists annotating the tumor, which would cause variability between them. In this case, we showed a high correlation with a Spearman’s rank-order correlation of 0.85 between the TMR-CT, showing strong inter-reader stability.
Exploiting TMR-CT features from DPCCA for classification and prognosis of CT scans without metabolomic profiles
We aimed to determine if our latent variables captured meaningful, held-out biological information such as histology subtype and overall survival (OS). To have an overview of the information captured in the shared and view specific embedding space from the DPCCA model we plotted the PCA of the TMR_CT features in Fig. 6. Similarly, we plotted the PCA for the CT_emb features in Supplementary Fig. 4 and observed how the image embeddings are less informative compared to Fig. 6. However, to get a better understanding of how these features can be useful in downstream tasks, we trained separate models and feature selection were tuned on TCIA data as seen in Fig. 7, to select the best model for each task. In the case of the radiomics features we also selected the best feature selection technique. With many radiomic features (438), the feature selection technique is particularly important to optimize the model and make it directly comparable to the TMR-CT and CT_emb features which both have 10 features respectively. After performing feature reduction using the techniques showed in Fig. 7 the number of remaining features ranged from four to thirteen depending on the feature reduction method. More specifically the best radiomic models for histology classification and prognosis used six and eight features, respectively, thus being the same order of magnitude as TMR-CT features. The features used in the best feature selection model combination are the following: six features for histology classification (FD_max_LLH_25HUgl, GLRLM_RP_LHL_25HUgl, GLCM_Entrop_HLH_25HUgl, GLCM_AutoCorrel_LLL_25HUgl, GLCM_invVar_LLL_25HUgl and FOS_RMS_LLL). The best prognosis model uses eight (SNS_max3d, FD_max_LLL_25HUgl, FD_max_LLH_25HUgl, GLCM_invVar_25HUgl, FOS_RMS_LLL, GLCM_invVar_LLL_25HUgl, GLCM_IDN_LLH_25HUgl and NGTDM_Coarse_LLL_25HUgl). Where SNS = size and shape features, FD = fractal dimensions, GLCM = Grey-level co-occurrence matrix, GLSZM = Grey-level size zone matrix, FOS = first order statistics, GLRLM = Grey-Level Run Length Matrix, NGTDM = Neighborhood grey- tone difference matrix, GLCM = Grey-level co-occurrence matrix).

a TMR-CT (image embeddings space ya) (b) metabolomic embeddings yb and (c) image and metabolomic shared latent space zab.

a F1-score for classification of AC and SCC using TMR-CT for different classification models. b F1-score for classification of AC and SCC using CT_emb for different classification models. c F1-score for classification of AC and SCC using radiomic features, with the x-axis being the predictive models and the y-axis corresponding to feature selection techniques. d C-index for a prognosis for different models using TMR-CT. e C-index for a prognosis for different models using CT_emb. f C-index using radiomic features, with the x-axis being the prognosis models and the y-axis corresponding to feature selection techniques.
From Fig. 7, it is evident how RF using TMR-CT features significantly outperforms the CT_emb and traditional radiomics features extracted using TextLab 2.0 for histology classification and prognosis without performing any feature selection. This finding is important as it shows that the quality of the TMR-CT features is sufficiently high. Thus, no feature selection is required. This can be further seen in Tables 3 and 4 where we chose the best performing feature selection and model, respectively, from Fig. 7 to report the results on the four external datasets and the ROC curves are plotted in Supplementary Fig. 5.
The three external test sets of Kaplan Meier curves are shown in Fig. 8 and demonstrate good separation between high and low-risk groups with log-rank tests confirming a statistically significant difference a 5% level in the GSTT and 1% for the ICHT and RMH.

Dichotomized predicted probabilities using k-means clustering of the RSF with TMR-CT on the external validation dataset: (a) ICHT, (b) GSTT and (c) RMH (P-values are from log-rank tests. Plots demonstrate good separation between high and low risk groups with log-rank tests confirming the statistical significance of 5% in the GSTT and 1% in ICHT and RMH.
To understand the importance of different features in our best prognosis model noted in Table 4, we reported the hazard ratio that our model calculated together with the P-value for the log-rank test for each feature modality in Table 5. We observe in a multivariable model that TMR-CT exhibited the highest hazard values in both the validation and external test datasets showing the high importance of TMR-CT for prognosis. This finding is notably more significant than clinical features, including age, gender, N-stage and gross tumor volume (GTV).
Discussion
In this study, we have shown that a deep learning framework - DPCCA - can model a connection between CT scans of lung nodules and their tissue metabolomics profiles against the premise that certain metabolites and/or their intensities, representing tumor growth and/or tumor microenvironment factors maximally co-vary together, linearly or non-linearly, with CT image features. Furthermore, we have shown the usefulness of such models, embodied within TMR-CT, for histology subtype classification and prognosis of NSCLC patients non-invasively, thus asserting clinical relevance. Notably, the DPCCA-generated learned representations could be used for downstream classification or prognosis tasks even when we only have CT scans available. Such metabolomic profile-correlated features are more interpretable biologically. This methodology would be useful in guiding treatment decisions, particularly in the context of patients that are unfit for biopsy.
The generated metabolite pairs are not inherently intuitive. For example, the most important metabolites (in our DPCCA model) differ from our previously published top metabolites from metabolomics-only analysis, in the study by Moreno et al. (2018). One of the main reasons for this is that, in the metabolomics-only study, the top metabolites are chosen as those most discriminative between tumor and non-tumor cases for AC and SCC separately, obtaining 20 different metabolites19. In the current study, however, the top ten metabolites chosen are those most correlated with our TMR-CT, whose purpose is to reconstruct the two data modalities as a composite phenotype of both the CT features of the lesion and the metabolomics profile. Regardless, the most correlated metabolites appear to regulate cell growth and membrane activity through glycolysis, pentose-phosphate, DNA synthesis and fatty acid metabolism.
A fundamental point of consideration in this research is whether optimization of CT features, detached from metabolomics (using a radiomics pipeline; current state of the art in clinical setting) compares favourably with TMR-CT (generated by the DPPCA pipeline). Thus, we compared feature optimization employing a matrix of combinatorial feature reduction and machine learning model for prediction, detached from metabolomics information, with the DPPCA pipeline informed by tissue metabolomics. When comparing the two machine learning pipelines - TMR-CT versus radiomics optimised feature combinations - we observed that TMR-CT performed significantly better in two clinically distinct tasks: histology classification and prognosis. This suggests that TMR-CT optimized vector contains more relevant features than radiomics vectors, even though there are significantly fewer features in the former. This difference was observed regardless of whether TMR-CT was used alone or combined with patient information, meaning that TMR-CT exhibits overall superior performance compared to radiomic features for NSCLC histology classification and prognosis determination.
To our knowledge, over 16 different models have attempted to integrate multiomics using deep learning to gain a better understanding of the complex biological process of cancer17. However, most of these models aim to fuse multiomics of the same modality, making it a significantly easier process relying on the availability of both data modalities during the test time. Various studies have successfully integrated CT features with other biomarkers for lung cancer diagnosis and prognosis; however, a direct comparison is challenging due to differences in the datasets30,31. The said methods only work when both modalities are present32. Thus, the benefit of using DPCCA over other models is that it can be applied during test time even without information about the tissue metabolomic profiles (with only the CT available). We show that TMR-CT derived from DPCCA was superior to conventional radiomics for histology classification and prognosis in patients who only had the CT scan available.
Our current study exhibits a number of limitations. First, a potential limitation of the study is the sample size used for training. Of note, however, the DPCCA is particularly attractive for medical applications with a small sample size but a large feature space, as it explicitly models uncertainty18. In that study, DPCCA utilized two inputs: gene expression (18,659 features) and 2D tissue histology images of size (128 × 128) with three colour channels, so a total of 49,152 features. In our study, we used the 174 metabolomic features that were consistently present in all patients and CT images of size 32 × 32 × 32 with one colour channel and a total of 32,768 features. Although our dataset was slightly smaller with fewer features, this was compensated through data augmentations techniques, ultimately enabling our model to perform efficiently. Secondly, regardless of the aim of the approach is to permit future use of the more routine method (in this case CT), it is clear that we only validated the performance of the TMR-CT on external CT datasets but not metabolomics. In theory, one could inversely predict the values of metabolomics counterpart from CT data. This generative aspect of the model could be investigated when independent test tissue metabolomics data become available Future analysis on this independent cohort with paired CT and metabolomics data would be required to validate the stability of the correlations identified in our study. Lastly, a wider range of histology could have been used. In our study, we only examined patients with AC and SCC, but our technique could easily be extended to incorporate other lung cancer histology with minimal adaptation when those data become available.
In the future, two primary directions could be explored by researchers. The first is to validate the TMR-CT features on an external dataset of paired CT scans and metabolomics features. The second is to increase the number of patients in the paired CT and metabolomics dataset to contain a larger number of patients that are more representative of the wider population by including small cell lung cancer patients. Unfortunately, such datasets don’t currently exist, so we could not incorporate these ideas into our study. Nonetheless, by showcasing the efficacy of a niche algorithm in a specific context, we establish a foundation for future studies that aim to extend its performance and validation to diverse settings. By conducting a prospective study that combines TMR-CT, radiomics, and body fluid metabolomic analysis, it may be possible to improve prognostic capabilities when tissue metabolomics is unavailable. This is particularly relevant for patients who are deemed unsuitable for surgery or face obstacles in accessing tumor material for histology classification and prognostication prior to making a decision about surgery.
Our study investigates the feasibility of using deep learning to combine patients’ paired CT and steady-state metabolomics information to find a shared representation that can allow the reconstruction of both modalities. One benefit of using our two-step deep learning model is the ability to independently extract deep features from a single modality without needing another modality. This is of specific importance in the clinical setting, where it is often the case that a single data modality is more readily accessible than the other. Enhancing the features obtained from lesions on CT images, we are improving the usefulness of CT scans, which are more readily available when evaluating NSCLC patients for early diagnosis and tumor prognostication33.
In summary, we were able to show that there is a connection between the metabolomic and CT features of NSCLCs. Furthermore, it is possible to exploit the learned representation within CT images of patients with NSCLC that co-vary with tissue metabolomic profiles and demonstrate their usefulness clinically for histology subtype classification and prognosis on external datasets when only a CT scan is present.
Methods
Pre-processing of CT images
All image pre-processing was done using TorchIO, a package allowing for effective pre-processing of CT images34. To ensure comparability, the CT scans from all datasets were resampled to isotropic voxels of 1 × 1 × 1 mm. This was performed using linear and nearest neighbor interpolation for the image and segmentation, respectively35.
Pre-processing data for training and testing DPCCA
We had 48 paired samples from the UHRS, each with two data views: CT scans and the metabolomic profiles of tumor/normal tissue. We performed a stratified split of the dataset into 32 paired samples for training, validation and 16 paired samples for testing whilst keeping the balance of AC/SCC consistent across the splits.
To ensure that the model was clinically relevant and matched the target population as much as possible we further considered the significance of the representative test data to ensure that our predictions hold clinical relevance. While assessing clinically-relevant predictions, we considered that the test data should match the target population rather than be a random subset of the same data pool as the train data36. To achieve this, we employed a multi-step stratification process that extended beyond the histological subtype: Split the dataset by histology, TNM8 Stage (due to clinical significance of staging), then gender (to account for potential variation in disease presentation between gender), and then age.
The stratification order was chosen based on its importance. We rationalized that ensuring the model could represent all stages and the frequency of occurrences (fewer females) was important, as such implemented this order in the test set. The following pre-processing steps were implemented on the training and testing data split separately.
Given the 3D segmentations, we calculated the center of mass (COM) and bounding box of the tumor. A 3D isotropic patch of 50 × 50 × 50, around the COM of tumor volume, was extracted, resulting in 48 3D tumor patches. We then created 3D patches of 32 × 32 × 32 randomly and ensuring that at least 65% of tumor was captured by the bounding box. The 3D patches were normalized to a range of 0–1 and lower upper boing of −1024 and 302135.
For the metabolomics features, we only included those that were profiled in all patients for both tumor and non-tumor adjacent tissue, such that we had a total of 174 metabolites. The reason for this was to increase the reproducibility of the chosen features. Subsequently, we normalized the values of the metabolomic features to have a mean of 0 and a standard deviation of 1.
Data augmentation makes it possible to increase the data available for training without actually collecting new samples by applying a range of techniques. In this study, an augmentation factor of 186,624 was applied to the patches resulting in a training dataset of approximately nine million 3D patches. These augmentations were chosen based on other similar studies and consisted of ±18 pixels in three axes, random rotations at 90° intervals along the longitudinal axes, and random flipping along three axes35. The augmentations were applied in real-time during training, and simultaneously, we applied Gaussian noise with a standard deviation of 0.1 to the image patches and metabolomic features35. No augmentation was applied during validation or testing.
Building DPCCA model
The DPCCA model is a deep generative model that fits the probabilistic canonical correlation analysis (PCCA) into two autoencoders, one for the CT image and the other for metabolomic. Figure 1 shows a detailed image of this model and where it fits the PCCA to the embeddings of two autoencoders. The code for this model was adapted from https://github.com/gwgundersen/dpcca. Specifically, we optimized the image autoencoder to enable studies with 3D images instead of 2D and used the 3D-DCGAN developed specifically for medical images37,38. The model was trained end-to-end using the mean squared error (MSE) for regression model fitting of paired CT image and metabolomics data; and also, for the reconstruction of the loss function for the modalities separately. The following section details the DPCCA method and its adaptation to our task.
Given a paired sample (xa, xb), the linear and convolution encoder embedded the CT images and metabolomics, respectively. These embedded vectors ya and yb are then fitted by the PCCA and incorporate an l1 penalty on the PCCA metabolomic weights, thus, encouraging sparsity in the metabolomic profiles and resulting in shared and view-specific latent variables z = [zab za zb]T.
Mathematically the PCCA can be expressed by Eq. 1 as follows:
Where \({B}^{j}\in {{\mathbb{R}}}^{{p}^{j}\times k}\), \({\Lambda }^{j}\in {{\mathbb{R}}}^{{p}^{j}\times k}\) and \({\Psi }^{j}\in {{\mathbb{R}}}^{{p}^{j}\times {p}^{j}}\). This can be reformulated as a factor analysis problem18, thus, suggesting that inference in the PCCA can be performed using expectation-maximization (EM), where the parameters are updated using the following tilling as seen in Eq. 2:
Once the shared and view-specific latent variables \(z={[{z}^{{ab}}{z}^{a}{z}^{b}]}^{{\rm{{\rm T}}}}\) are derived, the next step is to use the reparameterization trick to sample from the PCCA representation \({\hat{y}}^{j}{\mathscr{ \sim }}{\mathscr{N}}{\mathscr{(}}{\Lambda }^{{j}^{* }}{z}^{{ab}}+{{\rm{{\rm B}}}}^{{j}^{* }}{z}^{j};{\Psi }^{{j}^{* }})\) and obtain embedding samples \({\hat{y}}^{j}\). This step ensures that the Monte Carlo estimate of the expectation is distinct with respect to the encoder parameters and, thus, the model can be trained in an end-to-end fashion by defining the following loss function in Eq. 3:
In the formulation described in this section, there are five hyperparameters (pa,pb,kab,ka and kb) determining the dimensions of the modality embeddings and latent space. In this case: \(y\in {{\mathbb{R}}}^{p}\) such that p = pa + pb, where pa represents the dimensionality of CT embedding, and pb represents the dimensionality of the metabolomics embedding. The latent space is \(z\in {{\mathbb{R}}}^{k}\) where = kab + ka + kb, \(\Lambda \in {{\mathbb{R}}}^{p\times k}\) and \(\Psi \in {{\mathbb{R}}}^{p\times p}\). To identify the best set of hyperparameters we did a grid search \(p\in \{\mathrm{5,10,25,50}\}\) and k\(\in \left\{\mathrm{2,3,5,10}\right\}\) such that k ≤ p was always satisfied and we selected the smallest number that resulted in a high image and metabolomics reconstruction. This was found to be pa = pb = 10 and kab = ka = kb = 3, such that p = 20 and k = 9, through the loss function defined in Eq. 3 and the reconstruction of both modalities as seen in Fig. 4.
Building image autoencoder
The image autoencoder was based on the 3D-DCGAN similar to the image autoencoder in the DPCCA to make them directly comparable and trained using the mean squared error loss function to reconstruct the image. The same image preprocessing techniques described in “Pre-processing data for training and testing DPCCA” were used for this section and we performed image augmentation. A grid search was performed to identify the best hyperparameter, specifically the hyperparameter determining the size of the image embedding \({p}^{a}\in \{\mathrm{5,10,25,50}\}\). This was found to be pa = 10, through the mean squared error loss function.
Analysis of dataset for testing image embeddings for histology classification and survival
For this section, we trained our model on the TCIA cohort of (n = 203) and then performed the test on the test section of TCIA and three different datasets from the (n = 320) Royal Marsden Hospital (RMH), (n = 128) Guy’s and St Thomas’ Hospital (GSTT) and (n = 101) Imperial College Healthcare Trust (ICHT)25. We first filtered the datasets only to have patients with AC and SCC histology.
As a baseline for feature quality, we used TextLab 2.0 software to extract 438 features from the lesion. The methods in Table 6 were applied to features extracted using the DPCCA, image autoencoder and TextLab 2.0, the latter for radiomics analysis.
There exists a wide range of feature selection and machine learning techniques. Identifying the feature selection and machine learning algorithm is task-dependent and critical when developing clinically applicable models. Therefore, we combined different feature reduction techniques for the classification and survival tasks39. To find the best combination, we performed a ten-fold cross-validation using the training split of the TCIA data. Then, we used the average accuracy to select the best feature reduction and machine learning algorithms. The acronyms of each feature section, classification and survival method are defined in Table 6.
For the histology classification task on radiomic features, we selected 15 feature selection methods and combined them with 12 machine-learning classifiers based on previous related research39,40. The filter selection methods consisted of univariate and multivariate filter methods, which are classifier-independent and embedded methods such as penalty and tree-based methods, which incorporated the feature selection in the training process. For the classification task, we selected a broad range of methods as suggested by previous studies. We used a cross-combination strategy to select the method with the best mean F1 score across the ten-fold validation41. The feature selection and classification task were performed using the scikit-learn package in Python42.
In the survival analysis, we used a different set of feature selection methods and models as suggested by the literature and included the Cox proportional hazard model as a benchmark43. These specifics are capable of handling censored, heterogenous and high-dimensional data. The machine learning algorithms selected in this section can be divided into four categories: penalized Cox regression, boosted Cox regression (GLM), boosted based on trees and random forests. To select the best combination, we used a cross combination strategy to select the method with the best mean C-index across the ten-fold validation. The feature selection and classification task were performed using the ‘survival’ package in R44.
The best feature selection and machine learning model combination were then selected to perform histology subtype classification and survival analysis using the TCIA validation cohort and the three external datasets, as reported in Tables 3 and 4.
Ethics
This study used retrospective human data and complied with all relevant ethical regulation except where this was waived. Specifically three types of retrospective data were used: (a) Dataset obtained from the University Hospital Reina Sofia (UHRS), Spain. The research study was conducted in accordance with the Helsinki Declaration and was approved by the Cordoba Clinical Research Ethics Committee; all patients provided a signed written informed consent for participation in the study. In addition to CT scans. Correlated tissue Tissue samples were stored by the Andalusian Health Services Biobank, and the metabolomic profiling was performed under contract by Metabolon as reported previously19. (b) OCTAPUS-AI dataset (UK). OCTAPUS-AI represents a study from multiple UK cancer centers including Guy’s and St Thomas’ NHS Foundation Trust, Imperial College Healthcare NHS Trust and the Royal Marsden NHS Foundation Trust, collected for the explicit purpose of developing robust predictive lung cancer algorithms as previously reported25. This study was approved by the UK Health Research Authority (reference number: 20/HRA/3051); ClinicalTrials.gov identifier, NCT04721444. As the data used in the study were de-identified, patient consent was not required for this type of study and as per the respective Health Research Authority and Research Ethics Council approvals. (c) TCIA dataset. The Cancer Imaging Archive (TCIA) provides the cancer research community with an open-source repository of de-identified and highly curated radiology and histopathology imaging data (www.cancerimagingarchive.net). In keeping with TCIA’s grant-funded mandate from United States National Institute of Health, the dataset is considered de-identified information as defined by the Health Insurance Portability and Accountability Act of 1996, as amended (“HIPAA”). Institutional Review Board approval for TCIA data was not required for use of the dataset.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The TCIA data is publicly available from https://wiki.cancerimagingarchive.net/display/Public/NSCLC-Radiomics. The UHRS dataset is not publicly available but can be request to the corresponding authors. The (GSTT, Imperial and RMH). data are not publicly available but can be requested to the corresponding authors and/or OCTAPUS-AI.
Code availability
The underlying code used to reproduce the key findings are publicly accessible in Mendeley Data with the identifier https://doi.org/10.17632/ft2f3xhjz7.138.
References
LU, H. et al. A mathematical-descriptor of tumor-mesoscopic-structure from computed-tomography images annotates prognostic-and molecular-phenotypes of epithelial ovarian cancer. Nat. Commun. 10, 764 (2019).
Hunter, B. et al. A radiomics-based decision support tool improves lung cancer diagnosis in combination with the Herder score in large lung nodules. EBioMedicine 86, 104344 (2023).
Bera, K., Braman, N., Gupta, A., Velcheti, V. & Madabhushi, A. Predicting cancer outcomes with radiomics and artificial intelligence in radiology. Nat. Rev. Clin. Oncol. 19, 132–146 (2022).
Mao, K., Tang, R., Wang, X., Zhang, W. & Wu, H. A comprehensive algorithm for evaluating node influences in social networks based on preference analysis and random walk. Complexity 2018, 3078374 (2018).
Kadir, T. & Gleeson, F. Lung cancer prediction using machine learning and advanced imaging techniques. Transl. Lung Cancer Res. 7, 304–312 (2018).
Ardila, D. et al. End-to-end lung cancer screening with three-dimensional deep learning on low-dose chest computed tomography. Nat. Med. 25, 954–961 (2019).
Kumar, D., Wong, A., Clausi, D. A. Lung nodule classification using deep features in CT images. Conference on Computer and Robot Vision. 12, 133–138 (2015).
Boubnovski, M. M. et al. Development of a multi-task learning V-Net for pulmonary lobar segmentation on CT and application to diseased lungs. Clin. Radiol. 77, e620–e627 (2022).
Zhao, W. et al. 3D deep learning from CT scans predicts tumor invasiveness of subcentimeter pulmonary adenocarcinomas. Cancer Res. 78, 6881–6889 (2018).
Yang, J. et al. Medical Image Computing and Computer Assisted Intervention–MICCAI 2020: 23rd International Conference, Lima, Peru, 2020, Proceedings, Part VI 23. Springer International Publishing, (2020).
Fornacon-Wood, I., Faivre-Finn, C., O’Connor, J. P., Price, G. J. Radiomics as a personalized medicine tool in lung cancer: Separating the hope from the hype. Lung Cancer. 146, 197–208 (2020).
Ibrahim, A. et al. Radiomics for precision medicine: current challenges, future prospects, and the proposal of a new framework. Methods 188, 20–29 (2021).
Frix, A. N. et al. Radiomics in lung diseases imaging: state-of-the-art for clinicians. J. Pers. Med. 11, 602 (2021).
Shiri, I. et al. Impact of feature harmonization on radiogenomics analysis: Prediction of EGFR and KRAS mutations from non-small cell lung cancer PET/CT images. Comput. Biol. Med. 142, 105230 (2022).
Carrillo-Perez, F. et al. Machine-learning-based late fusion on multi-omics and multi-scale data for non-small-cell lung cancer diagnosis. J. Pers. Med. 12, 601 (2022).
Takahashi, S. et al. Predicting deep learning based multi-omics parallel integration survival subtypes in lung cancer using reverse phase protein array data. Biomolecules 10, 1460 (2020).
Lee, T. Y., Huang, K. Y., Chuang, C. H., Lee, C. Y. & Chang, T. H. The fractal dimension as a measure for characterizing genetic variation of the human genome. Comput. Biol. Chem. 87, 107277 (2020).
Gundersen, G., Dumitrascu, B., Ash, J. T., Engelhardt, B. E. End-to-end training of deep probabilistic CCA on paired biomedical observations. In Proceedings of The 35th Uncertainty in Artificial Intelligence Conference (2020).
Moreno, P. et al. Metabolomic profiling of human lung tumor tissues--nucleotide metabolism as a candidate for therapeutic interventions and biomarkers. Mol. Oncol. 12, 1778–1796 (2018).
Li, Y., Wu, X., Yang, P., Jiang, G., Luo, Y. Machine Learning for Lung Cancer Diagnosis, Treatment, and Prognosis Genomics. Proteomics & Bioinformatics 20, 850–866 (2022).
Kumar, A. & Misra, B. B. RNF4—a paradigm for SUMOylation‐mediated ubiquitination. Proteomics 19, 1900042 (2019).
Armato III, S. G. et al. The Reference Image Database to Evaluate Response to therapy in lung cancer (RIDER) project: A resource for the development of change-analysis software. Clin. Pharmacol. Ther. 84, 448–456. (2008).
Zhao, B. et al. Evaluating variability in tumor measurements from same-day repeat CT scans of patients with non-small cell lung cancer. Radiology 252, 263–272 (2009).
Aerts, H. J. W. L. et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 5, 4006 (2014).
Hindocha, S. et al. Gross tumour volume radiomics for prognostication of recurrence & death following radical radiotherapy for NSCLC. NPJ Precis. Oncol. 6, 77 (2022).
Langley, P., Bowers, E. J., Murray, A. Principal component analysis as a tool for analyzing beat-to-beat changes in ECG features: application to ECG-derived respiration. IEEE transactions on biomedical engineering 57, 821–829 (2009).
Rosato, A. et al. From correlation to causation: analysis of metabolomics data using systems biology approaches. Metabolomics 14, 1–20 (2018).
Van Timmeren, J. E., Cester, D., Alkadhi, H., Baessler, B. Radiomics in medical imaging—“how-to” guide and critical reflection. Insights into Imaging 11, 1–16 (2020).
Teng, X. et al. Improving radiomic model reliability using robust features from perturbations for head-and-neck carcinoma. Front. Oncol. 12, 974467 (2022).
Yang, Y. et al. A multi-omics-based serial deep learning approach to predict clinical outcomes of single-agent anti-PD-1/PD-L1 immunotherapy in advanced stage non-small-cell lung cancer. Am. J Transl. Res. 13, 743 (2021).
Ding, Y. et al. Improving the efficiency of identifying malignant pulmonary nodules before surgery via a combination of artificial intelligence CT image recognition and serum autoantibodies. Eur. Radiol. 33, 3092–3102 (2023).
Leng, D. et al. A benchmark study of deep learning-based multi-omics data fusion methods for cancer. Genome Biol. 23, 1–32 (2022).
Hunter, B. et al. A radiomics-based decision support tool improves lung cancer diagnosis in combination with the Herder score in large lung nodules. EBioMedicine 86, 104344 (2022).
Pérez-García, F., Sparks, R. & Ourselin, S. TorchIO: a python library for efficient loading, preprocessing, augmentation and patch-based sampling of medical images in deep learning. Comput. Methods Prog. Biomed. 208, 106236 (2021).
Hosny, A. et al. Deep learning for lung cancer prognostication: a retrospective multi-cohort radiomics study. PLoS Med. 15, e1002711 (2018).
Varoquaux, G. & Cheplygina, V. Machine learning for medical imaging: methodological failures and recommendations for the future. NPJ Digit. Med. 5, 4848 (2022).
Nie, D. et al. Medical image synthesis with deep convolutional adversarial networks. IEEE Transactions on Biomedical Engineering 65, 2720–2730 (2018).
Boubnovski, M. et al. 3D-DPCCA, Mendeley Data, V1 (2023).
Sun, P., Wang, D., Mok, V. C. & Shi, L. Comparison of feature selection methods and machine learning classifiers for radiomics analysis in glioma grading. IEEE Access 7, 102010–102020 (2019).
Yin, P. et al. Comparison of radiomics machine-learning classifiers and feature selection for differentiation of sacral chordoma and sacral giant cell tumour based on 3D computed tomography features. Eur. Radiol. 29, 1841–1847 (2019).
Destito, M. et al. Radiomics-based machine learning model for predicting overall and progression-free survival in rare cancer: a case study for primary CNS lymphoma patients. Bioengineering 10, 285 (2023).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. 12, 2825–2830 (2011).
Spooner, A. et al. A comparison of machine learning methods for survival analysis of high-dimensional clinical data for dementia prediction. Sci. Rep. 10, 20410 (2020).
Therneau, T. M., Lumley, T. Package ‘survival’. R Top Doc 128, 28–33 (2015).
Patra, K. C., Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 39, 347–354 (2014).
Krall, A. S., Xu, S., Graeber, T. G., Braas, D. & Christofk, H. R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 (2016).
Acknowledgements
The authors would like to thank the following organizations for providing data for the work reported: University Hospital Reina Sofia, Spain (metabolomics and CT scans), The Cancer Imaging Archive, USA (CT scans), and OCTAPUS-AI (Optimizing Cancer Treatment Surveillance and Ascertaining cause of Pneumonitis Using Artificial Intelligence); a full list of OCTAPUS-AI members are located in the Supplementary information file. This article is independent research funded by the Medical Research Council and Imperial STRATiGRAD PhD programme. The authors acknowledge support from Medical Research Council Grant (MR/M015858/1), Imperial College NIHR Biomedical Research Centre award (WSCC_P62585), Imperial College Experimental Cancer Medicines award (C1312/A25149) and National Cancer Imaging Translational Accelerator (C2536/A28680). This work was funded by the Spanish Ministerio de Ciencia e Innovación (MICINN, PID2021-124314OB-I00), and Junta de Andalucía-Consejería de Conocimiento, Investigación y Universidad (P20_00470) grants to M.A.C. The funding source did not have a role in the study design, data collection, analysis, or interpretation of data.
Author information
Authors and Affiliations
Contributions
E.O.A. and M.A.C. conceived the project. M.B.M. designed the project and provided computational analysis. K.L.-R. provided bioinformatics analysis. P.M., M.Á.‐B. and Á.S collected UHRS dataset. M.C. performed nodule segmentations of UHRS dataset. S.H., R.L., J.M.P., E.O.A. and M.B.M. contributed to the interpretation of results. M.B.M. and E.O.A. wrote the manuscript. All authors edited and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boubnovski Martell, M., Linton-Reid, K., Hindocha, S. et al. Deep representation learning of tissue metabolome and computed tomography annotates NSCLC classification and prognosis. npj Precis. Onc. 8, 28 (2024). https://doi.org/10.1038/s41698-024-00502-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00502-3
