
Abstract
In this study, we investigated the metabolic alterations associated with clinical response to chemotherapy in patients with ovarian cancer. Pre- and post-neoadjuvant chemotherapy (NACT) tissues from patients with high-grade serous ovarian cancer (HGSC) who had poor response (PR) or excellent response (ER) to NACT were examined. Desorption electrospray ionization mass spectrometry (DESI-MS) was performed on sections of HGSC tissues collected according to a rigorous laparoscopic triage algorithm. Quantitative MS-based proteomics and phosphoproteomics were performed on a subgroup of pre-NACT samples. Highly abundant metabolites in the pre-NACT PR tumors were related to pyrimidine metabolism in the epithelial regions and oxygen-dependent proline hydroxylation of hypoxia-inducible factor alpha in the stromal regions. Metabolites more abundant in the epithelial regions of post-NACT PR tumors were involved in the metabolism of nucleotides, and metabolites more abundant in the stromal regions of post-NACT PR tumors were related to aspartate and asparagine metabolism, phenylalanine and tyrosine metabolism, nucleotide biosynthesis, and the urea cycle. A predictive model built on ions with differential abundances allowed the classification of patients’ tumor responses as ER or PR with 75% accuracy (10-fold cross-validation ridge regression model). These findings offer new insights related to differential responses to chemotherapy and could lead to novel actionable targets.
Similar content being viewed by others


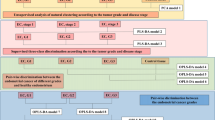
Introduction
The standard first-line chemotherapy approach in high-grade serous ovarian cancer (HGSC) has been a combination of taxanes and platinum for over two decades1. High overall mortality2 from HGSC is related to an advanced stage at diagnosis and the rapid emergence of chemotherapy resistance. Mechanisms of resistance, including metabolic changes and adaptation, are not fully understood. Here, we used an innovative strategy to characterize spatially resolved metabolic changes in a highly clinically annotated set of HGSC samples collected before and after neoadjuvant chemotherapy (NACT) from patients treated consistently according to a surgical algorithm3.
Mass spectrometry (MS) is a powerful technology to spatially characterize the molecular composition of tissues within the tumor microenvironment (TME)4,5,6,7,8. We employed desorption electrospray ionization (DESI)-MS, which allows the simultaneous detection of diverse metabolites and lipid species directly from native tissues under ambient conditions9,10. The use of histologically compatible spray solvents allows for the same tissue sections to be stained with hematoxylin and eosin (H&E) to visualize tissue morphology11. To corroborate metabolic findings with molecular data, we performed global proteomics and phosphoproteomics using laser capture (LC)-MS. These results provide new insights into the metabolic alterations in the tumor and stromal compartments based on response to NACT.
Results
Metabolic profiling of pre-chemotherapy tumor tissues using DESI-MS
We first used DESI-MS on pre-NACT tissues from patients stratified as having excellent response (ER) or poor response (PR) to NACT (Supplementary Table 1). MS imaging data were extracted from the epithelial and stromal regions following manual segmentation (Fig. 1). A preliminary analysis on reproducibility was performed where two sections from the same tumor (one mouse xenograft and two human ovarian cancer samples) were analyzed with DESI-MS (Supplementary Fig. 1A) and a cosine similarity test was performed, yielding a score of 0.981 for mouse xenograft (Supplementary Fig. 1B) and 0.998 (Supplementary Fig. 1C) and 0.968 (not shown) for human tumors. In addition, four sections from 4 mouse xenografts were analyzed with an average cosine score of 0.972 (Supplementary Fig. 1D). Manual segmentation allowed us to distinguish between epithelial and stromal areas; the resolution of the technique does not allow for single cell-level segmentation. Stromal regions were mostly characterized by fibroblast-like cells and extracellular matrix (e.g., elastic fibers) that can be identified with H&E stain. Necrotic areas were excluded. Representative DESI-MS images of nine metabolites for two PR and ER tissues are shown in Fig. 2a. We employed negative ion mode DESI-MS imaging to detect small molecules, such as sugars, nucleotides, and amino acids, and a vast range of lipid classes. Multiple lipids, such as fatty acids, monoacylglycerols, ceramides, cardiolipins, and phospholipids, showed different relative abundances between ER and PR tissues. Significance analysis of microarrays (SAM) revealed that the epithelial regions of PR samples, as compared to those of ER samples, had significantly higher relative abundances of fatty acids, phosphatidic acids, ceramides, cardiolipins, and monoacylglycerols; the stromal regions of PR samples had higher relative abundances of fatty acids and phosphatidic acids (Table 1 and Fig. 2b) as compared to those of ER samples. A number of small molecules were also detected in both the ER and PR samples (Fig. 2c); SAM revealed that several molecules had significantly higher relative abundances in the epithelial PR samples than in the epithelial ER samples. In particular, hydroxybutyric acid and ubiquinone were detected at significantly higher relative abundances in the ER samples, while taurine and uridine were detected at significantly higher relative abundances in the PR samples. The stromal regions of the ER samples had significantly higher relative abundances of hydroxybutyric acid, hexose, and uridine, while metabolites with significantly higher abundances in the PR stroma samples included succinic acid and taurine (Table 1).

R0 absence of macroscopic residual at surgery, ER excellent responders, PR poor responders.

a DESI-MS imaging of tumor tissue sections obtained from 52 patients (30 ER and 22 PR) was performed. Negative ion mode DESI-MS ion images of two ER and two PR tumors showing the spatial distribution and relative abundances of nine metabolites and lipid species for each sample are represented. Within each column, the ion images were normalized to the same ion intensity (100% relative abundance, red) for ease of comparison among individual samples and response groups. Optical images of the H&E-stained tissue sections are shown for each sample, with regions of tumor epithelium outlined in black and regions of stroma outlined in red. b Distribution of lipid classes representing higher relative abundances in the stroma and epithelium of ER and PR pre-chemotherapy tissues, from DESI-MS. c Histograms representing relative abundances of small metabolites in the epithelium and stroma of ER and PR tumors. d Plots of ridge regression coefficients for the predictive model. The analysis was restricted to the primary tumor sites (adnexa and ovaries, N = 16), and samples from metastatic sites (omentum or abdominal organs) were excluded (N = 36). A ridge regression model was used to estimate the probability of every mass spectrum belonging to either the ER or PR group. ER excellent responders, PR poor responders.
To investigate if the small metabolites whose relative abundances significantly differed between ER and PR tumors were associated with specific metabolic pathways, we used two publicly available software programs. These programs analyzed the metabolites with higher or lower relative abundances in the different cohorts and tissues and provided the metabolic pathways in which such metabolites are particularly enriched. Taurine and uridine (Table 1B), which were detected at higher relative abundances in the epithelium of PR samples, mapped mainly to the “recycle of bile acids and salts” pathway (FDR-adjusted p < 0.05) and the “pyrimidine salvage and catabolism” pathway (FDR-adjusted p < 0.05) (Supplementary Table 2A, B). In contrast, the metabolites hydroxybutyric acid and ubiquinone (Table 1A), which had higher relative abundance in the epithelial regions of ER samples, showed a correlation with the “respiratory electron transport” and “metabolism of amino acids and derivatives” pathways (FDR-adjusted p < 0.05) (Supplementary Table 3A).
In the stromal regions of the pre-chemotherapy tissues, hydroxybutyric acid, hexose, asp-his, and uridine, which were detected at higher relative abundances in ER samples (Table 1C), were associated with several pathways, including the “pyrimidine salvage” pathway (Supplementary Table 4A, B, Table 2A). The metabolites succinic acid and taurine (Table 1D), which had higher abundances in PR tumors, were related to the “transport of bile salts and organic acids metal ions and amine compounds” and ”oxygen-dependent proline hydroxylation of hypoxia-inducible factor alpha” pathways (FDR-adjusted p < 0.05 for both) (Supplementary Table 5A, B, Table 2A). Table 2A summarizes the pathways related to the detected metabolites that may be altered in pre-chemotherapy ER and PR tissue samples.
Next, we built a predictive model based on the DESI-MS data extracted from the pre-NACT samples from the ER and PR groups acquired from tumor primary sites (N = 16). This model predicted response to chemotherapy using cross-validation with a per-pixel sensitivity of 83%, specificity of 69%, and total accuracy of 76%, with a positive predictive value of 92% (Fig. 2d). When the predictive performance per patient was analyzed, sensitivity, specificity, and accuracy values of 75% were achieved.
Metabolic profiling of post-chemotherapy tumor tissues
Next, we analyzed the matched post-chemotherapy tissues from ER and PR tumors and examined the metabolic changes occurring in response to chemotherapy. Discriminant analysis using the sparse partial least squares algorithm was used to identify and plot the most discriminative features12. When the epithelial areas of matched pre- and post-NACT tissues were analyzed, the number of metabolic species with lower relative abundances after chemotherapy (as compared to pre-chemotherapy) was higher in ER tumors than in PR tumors. Specifically, 113 metabolites (small molecules and lipids) had lower relative abundances in the epithelial areas of post-NACT tissues of ER tumors, while 65 metabolic species had higher relative abundances in the epithelial areas of post-NACT tissues of ER tumors (SAM, FDR p < 0.01) (Table 3A, B). In the epithelial areas of PR tumors, 60 metabolic species showed lower relative abundances in post-NACT tissues, while 45 metabolic species showed higher relative abundances in post-NACT tissues compared to pre-chemotherapy ones (Table 3C, D). Small metabolites (but not lipids) with lower relative abundances in the epithelial regions of post-NACT ER tissues included glycerophosphoethanolamine, citrate, glutamic acid, hypoxanthine, aspartate, pyroglutamate, fumarate, and uracil. Conversely, hydroxyglutaric acid was the only small metabolite with a lower relative abundance in the epithelial areas of post-NACT PR tissues.
In the epithelial regions, fatty acid species had a lower relative abundance in post-NACT tissues for both ER and PR tumors, while the relative abundance of phosphatidic acids was higher in the epithelial areas of post-NACT tissues for both ER and PR tumors. Indeed, phosphatidic acids were detected at higher relative abundances after chemotherapy in both ER and PR tumors (Fig. 3a). Interestingly, ceramides were highly abundant in the epithelial areas of post-NACT ER tumors. In the stromal areas, glycerophosphoinositol species were highly abundant in post-NACT tissues in both ER and PR tumors, while glycerophosphoserine and glycerophosphoethanolamine species were particularly abundant in post-NACT PR tumors. Heatmaps show distinct distributions of the normalized ion intensities of lipid species with different abundances in the epithelial areas (Fig. 4a, b) and stromal areas (Fig. 4c, d) in both pre- and post-NACT tissues from ER and PR tumors.

a, b Pie charts summarize the number of lipids in each lipid class, with higher and lower relative abundance in the epithelium (a) and stroma (b) of ER and PR post- versus pre-chemotherapy tissues identified using DESI-MS analysis. c, d Histograms representing the relative abundances of small metabolites in the epithelium and stroma of ER (c) and PR (d) tumors of post- versus pre-NACT tissues identified using DESI-MS. The data shown in the pie charts were obtained from DESI-MS analysis of tumor tissue sections from pre-chemotherapy samples from 52 patients (30 ER and 22 PR) and post-chemotherapy samples from 37 patients (20 ER and 17 PR). ER excellent responders, PR poor responders.

a, b Heatmaps representing the relative abundances of lipids in the epithelial areas of ER tumors (a); epithelial areas of PR tumors (b); stromal areas of ER tumors (c); and stromal areas of PR tumors (d). e, f Plots for sparse partial least squares discriminant analysis (sPLS-DA) in tri-dimensional (e) and bi-dimensional (f) settings. post post-NACT; pre pre-NACT; ER excellent responders, PR poor responders. For heatmap abbreviations see Tables 1 and 3. The data shown in the heatmaps were obtained from DESI-MS analysis of tumor tissue sections from pre-chemotherapy samples from 52 patients (30 ER and 22 PR) and post-chemotherapy samples from 37 patients (20 ER and 17 PR).
To investigate if the metabolites that had higher or lower relative abundances in post- versus pre-NACT tissues of ER and PR tumors were related to specific metabolic pathways, we analyzed the non-lipid metabolites identified with DESI-MS. The histograms in Fig. 3c, d represent the higher and lower relative abundances of metabolites in ER and PR tumors of post- versus pre-NACT samples. The epithelial regions of post-NACT samples from ER tumors showed lower relative abundances of uracil, fumarate, pyroglutamate, aspartate, hypoxanthine, glutamic acid, citrate, and galactosylglycerol (Table 3A), which are involved in the “urea cycle,” “phenylalanine and tyrosine metabolism,” and “nucleotide metabolism” pathways (FDR-adjusted p < 0.01) (Supplementary Table 6A, B), whereas several metabolites, including hydroxy valeric acid, taurine, leucinic acid, hydroxyniconitic acid, and glutamine (Table 3B), which had higher relative abundances in the epithelial regions of ER tumors, are involved in the “TP53-regulated metabolic genes” and “metabolism of nucleotides” pathways (FDR-adjusted p < 0.05) (Supplementary Table 7A, B). Metabolites with lower abundances in the post-NACT stromal regions of ER tumors, including uracil, hypoxanthine, glutamic acid, xanthine, and inosine (Table 3E), were associated with the pathways “metabolism of nucleotides” (FDR-adjusted < 0.01), “nucleotide salvage” (FDR-adjusted p < 0.01), and “purine catabolism” (FDR-adjusted p < 0.01) (Supplementary Table 8A, B), while metabolites such as valeric acid, fumaric acid, taurine, glutarate semialdeyde, and succininc acid (Table 3F), which had high relative abundances in the stromal regions, were mostly involved in the “urea cycle” and “citric acid cycle” pathways (FDR-adjusted p < 0.05) (Supplementary Table 9A, B).
In the epithelial regions of post-NACT PR tumors, taurine, glutamine, xanthine, aconitic acid, ascorbic acid, hexose, asp-his, inosine, and glutathione (Table 3D) had higher relative abundances and were mostly associated with the “metabolism of nucleotides” and “TP53 regulated-metabolic genes” pathways (FDR-adjusted p < 0.05) (Supplementary Table 10A, B, Table 2B). In the stromal regions of post-NACT PR tumors, the metabolites with higher abundances included fumaric acid, taurine, pyroglutamic acid, aspartic acid, and aconitic acid (Table 3G), which were related to the “aspartate and asparagine metabolism,” “phenylalanine and tyrosine metabolism,” and “nucleotide biosynthesis” and “urea cycle” pathways (Supplementary Table 11A, B, Table 2B), whereas less abundant metabolites, such as valeric acid, glutarate semialdehyde, succinic acid, hydroxyvaleric acid, malic acid, glutamic acid, and hydroxyglutaric acid, were associated with the “GABA degradation and synthesis” pathway (Supplementary Table 12A, B, Table 2B). Table 2B summarizes the deregulated pathways in post-NACT ER and PR tissues compared with pre-treatment tissues.
Sparse partial least squares discriminant analysis (sPLS-DA) of the data acquired from pre- and post-NACT samples in both ER and PR tumors showed a clear separation of the two tissue groups in the tri- or bi-dimensional score plots (Fig. 4e, f). These results indicate that different adaptive metabolic changes occur in tissues based on response to NACT.
Quantitative proteomic and phosphoproteomic analyses of pre-chemotherapy samples from ER and PR tumors
To identify differentially expressed enzymes and phosphoproteins in PR versus ER tumors, we generated global proteomic and phosphoproteomic data for whole-tumor equivalent collections of pre-chemotherapy samples, as described previously13. A total of 7148 proteins and more than 1075 phosphosites were co-quantified across cases (Supplemental Tables 13–19). We selected proteins and phosphosites with significantly different expressions based on clinical response and metabolic pathways previously identified by DESI-MS. Pathways with the highest number of proteins quantified included the “metabolism of amino acids and derivatives,” “metabolism of nucleotides,” and “respiratory electron transport and related” pathways. Differential analysis revealed that most proteins and phosphosites that differed significantly (LIMMA p < 0.05, ±1.5-fold change) between PR and ER cases mapped to the “metabolism of amino acids and derivatives” and “metabolism of nucleotides” pathways (z-score = 0.728 p-value 1.69E-13, derived from Ingenuity Pathway Analysis) (Fig. 5a). Principal component analysis of these proteins by case revealed a distinct separation of the pre-NACT PR and ER tumors (Fig. 5b).

a Unsupervised clustering of protein expression in ER and PR tissues. b PCA plot of the same features by case. c Proposed mechanism for the metabolic interactions between stroma and cancer cells. PCA principal component analysis, Ser serine, gly glycine, GLDC glycine decarboxylase. The results from the proteomics analyses shown in this figure were obtained from analysis performed on tumor tissue sections from pre-chemotherapy samples from 15 patients (7 ER and 8 PR). Differential analyses of global proteome or transcriptome matrixes were performed using LIMMA.
The quantitative proteomic analysis confirmed that phosphosites, which are related to the metabolism of nucleotides and particular pyrimidines, were significantly elevated in the PR tumors, which was concordant with the DESI-MS data (Supplementary Tables 14, 16, and 18). Interestingly, uridine (which DESI-MS revealed to have a high relative abundance in the tumor epithelium of pre-NACT PR samples), which serves as a substrate for cytidine 5-prime triphosphate synthetase (CTPS1), was highly abundant in pre-NACT PR samples (logFC 0.97, LIMMA p = 0.008) (Supplementary Table 18). CTPS1 catalyzes the conversion of uridine triphosphate into cytidine triphosphate and regulates intracellular rates of RNA, DNA, and phospholipid synthesis14,15; its phosphorylation is inhibitory, which could explain the higher abundance of uridine. Lastly, the increase of ornithine aminotransferase in PR tumors, measured by proteomic analysis, correlated well with the high relative abundances of molecules detected by DESI-MS that are associated with aspartate and asparagine pathways in the stroma (e.g., fumaric acid, taurine, aspartic acid, and glucose)16.
For the four enzymes that were upregulated in the pre-NACT PR samples (Supplementary Tables 13, 15, and 17), we analyzed the correlation between mRNA expression and progression-free survival (PFS) in patients with HGSC from publicly available databases (Supplementary Fig. 2). Interestingly, higher expression of glycine decarboxylase (GLDC) was positively correlated with worse prognosis (HR 1.16, CI 1.00–1.35, p = 0.046) (KMplotter.com). We also investigated the expression of GLDC mRNA in several organs (gTEX.org) and the expression of its protein (Protein Atlas) and mRNA (TCGA) in several cancer types (Supplementary Fig. 3). GLDC mRNA expression is low in normal ovarian and fallopian tube samples, but it is detected at high protein levels in ovarian cancer. A possible mechanism explaining the metabolic interaction between stroma and cancer cells involving GLDC activity is proposed in Fig. 5c. We further explored the possible impact of GLDC expression on the response of ovarian cancer cells to chemotherapy. After consulting the public database DepMap Public 22Q4 at Cancer Cell Line Encyclopedia (CCLE), we identified the ovarian cancer cell lines with higher GLDC expression based on average [log2(TPM + 1)] expression (Supplementary Fig. 4A). IGROV1 cells are among the top two ovarian cancer cell lines listed in CCLE with the highest GLDC expression. We then measured GLDC mRNA expression in IGROV1 and other ovarian cancer cell lines and in the HIO 180 non-transformed epithelial ovarian cancer cells to confirm that IGROV1 had the highest GLDC expression among them (Supplementary Fig. 4B). Next, the level of GLDC mRNA in IGROV1 was evaluated after transient transfection with three silencing RNAs (siRNA29, siRNA35, siRNA03). SiRNA35 resulted in the lowest mRNA levels of GLDC (Supplementary Fig. 4C). Cell viability was then evaluated in IGROV1 cells transfected with siRNA35 or siRNA control and treated for 72 h with carboplatin. The IC50 level of carboplatin in IGROV1 cells transfected with siRNA35 was 3.3 times lower than the IC50 in IGROV1 cells transfected with siRNA control, 3.7 µM versus 12.3 µM (P = 0.0003, hypothesis test, alpha 0.05) (Supplementary Fig. 4D).
Discussion
Relapse represents a major challenge in the treatment of patients with ovarian cancer, and studying the molecular changes related to therapy response is essential to identifying novel actionable targets. Nutrient availability inside the TME and paracrine communication influence the metabolic reprogramming of cancer cells, generating a complex metabolic profile17. In particular, reprogramming of nucleotide metabolism towards increased levels of nucleotide precursors and nucleotides has been found in recurrent tumor cells, including several cancer models14,18. Also, the metabolic dependency of ovarian cancer cells on neighboring stroma cells plays an important role in fueling tumor cell growth15. Many studies have investigated the metabolic interactions between the TME and cancer cells in inducing a permissive environment for tumor growth. The increased use of glucose and glutamine by cancer cells results in lactate accumulation, which decreases the activation of dendritic and T cells while stimulating the polarization of macrophages towards an M2-like phenotype16,19. Moreover, lactate stimulates angiogenesis20 and promotes acidification of the TME. This stimulates the proteolytic activity of metalloproteinases21, which in turn enhances extracellular matrix degradation and tumor invasion. Therefore, while much is known about how the metabolic interactions between stroma and cancer cells induce tumor cell proliferation and invasion, less is known about how these interactions promote resistance to chemotherapy. A few recent studies identified some important metabolic vulnerabilities in ovarian cancers that can be exploited to increase treatment response; these vulnerabilities include glutamine and serine metabolism22,23. To thoroughly study the metabolic heterogeneity of the HGSC TME in relation to therapy response, we performed a comparative analysis of metabolic species (nucleotides, proteins, sugars, and lipids) present in pre- and post-NACT tissues. We used DESI-MS imaging to obtain spatially resolved metabolomic information about the epithelial and stromal regions, and we used global proteomics and phosphoproteomics to corroborate the metabolic findings.
The use of highly clinically annotated samples, as presented here, is important for obtaining reliable results. When comparing the relative abundances of metabolites detected within the stromal regions of post- versus pre-NACT tissues, we found that PR tumors had higher abundances of fumaric acid, taurine, and aspartic acid, which are related to aspartate and asparagine metabolism. Interestingly, proliferating cells with impending glutamine depletion often adapt by utilizing asparagine, which is structurally similar to glutamine and can be used to fuel the TCA cycle, as an energy source24,25. It is plausible that an elevated demand for glutathione to counteract chemotherapy-induced cell damage may result in the depletion of glutamine in proliferating cells; in this scenario, stromal cells may support cancer cells by fueling them with asparagine to sustain their proliferation in tissues that respond less to chemotherapy. Moreover, the stroma of post-chemotherapy PR tissues showed elevated abundances of glycerophosphoserines (PS 36:1, PS 34:1, PS 36:2), which might be substrates for serine synthesis26. We believe that the increased nucleotide metabolism in PR cancer cells might be sustained by an increased influx of glycine and serine from the tumor stroma, leading to the increased activity of GLDC. GLDC fuels one-carbon metabolism via glycine breakdown to form CO2, NH3, and 5,10-methylene-tetrahydrofolate (CH2-THF)27; in particular, CH2-THF has been shown to be crucial for nucleotide synthesis28,29. As shown in other cancer types, GLDC may sustain nucleotide synthesis during cell proliferation in HGSC tumorigenesis30,31. However, an increase in nucleotide synthesis does not necessarily translate into a higher sensitivity to carboplatin-based chemotherapy (the main type of neoadjuvant chemotherapy used for ovarian cancer in our cohort and in general), since carboplatin does not show cell-cycle specificity32.
Our findings suggest several strategies to overcome chemotherapy resistance in HGSC, including targeting glycerophosphoserine and interfering with the metabolism of aspartate and glycine. While the antibody-based targeting of phosphatidylserine has been shown in pre-clinical studies to overcome resistance to radiation and chemotherapy33,34, the targeting of glycerophosphoserine has yet to be investigated. Interfering with aspartate and glycine metabolism could be done by blocking GLDC. GLDC, a mitochondrial enzyme, is part of a complex that oxidatively decarboxylates glycine35; high GLDC activity is strongly correlated with high rates of glutaminolysis and the synthesis of acetyl-CoA and fatty acids36. Notably, patients with HGSC with elevated GLDC levels have significantly worse PFS37, a finding similar to that in patients with non-small cell lung cancer30, in whom GLDC inhibition has been investigated in vitro and in vivo38. Combined treatment with GLDC inhibitors and platinum-based compounds, a completely novel strategy, might enhance sensitivity to chemotherapy in ovarian cancer. As mentioned above, and based on prior publications, glutaminolysis, and glutamine metabolism are part of the basis of reprogramming ovarian cancer cells towards increased proliferation and invasiveness39. Our study provides additional data on how the heterogeneous metabolism inside ovarian cancers might affect response to chemotherapy by promoting glycine-dependent nucleotide synthesis in cancer cells. However, additional work is needed to further test the biological importance of GLDC in ovarian and other cancers.
This study provides evidence that tumors with low sensitivity to NACT are characterized by different metabolic profiles, a finding that can be leveraged to stratify patients for treatment purposes. Additional research is needed to examine the therapeutic efficacy of targeting these differences. The availability of highly annotated tissues from patients undergoing standardized treatment and follow-up makes our results particularly relevant and translatable to the clinic. Future work will focus on the analysis of metabolomic and proteomics/phosphoproteomics profiles within an expanded cohort of tissues from patients with HGSC.
Our subgroup analysis was limited by the small sample size; therefore, a larger cohort and additional validation studies are needed. Tissue segmentation in stroma and epithelium was based on the morphological analysis of H&E-stained slides; additional subclassification of the TME with the identification of vessels, immune cell clusters, and different fibroblast subtypes is needed to further elucidate the metabolic changes in the stroma. Although MS imaging data provide spatially resolved molecular information, they are not quantitative; thus, the pathway analyses based on these data were exploratory in nature. Moreover, it should be noted that the predictive model was built from data extracted from tumor regions annotated by pathologic evaluation within the primary tissue types. As such, the model is limited to these tissue types and needs to be further expanded and validated for use in tissues in which higher degrees of cellular heterogeneity may influence the metabolic profiles.
Methods
Patients
A total of 112 frozen samples from 59 patients were collected and analyzed with DESI-MS; these included pre-chemotherapy samples from 52 patients (30 ER and 22 PR) and post-chemotherapy samples from 37 patients (20 ER and 17 PR). Among these, frozen tumor sections were retrieved from 48 patients from the MD Anderson Department of Gynecologic Oncology, 7 patients from the Gynecologic Cancer Translational Research Center of Excellence (GYN-COE) Program, and 4 patients from Washington University, St. Louis, as part of a collaborative study with the University of Iowa and MD Anderson Cancer Center. When available, two pre- or post-chemotherapy samples (one from adnexa and the other from a metastatic site such as the omentum, uterus, or abdominal organs) for each patient were collected and analyzed.
The collection of tissues from patients diagnosed and treated at the MD Anderson Cancer Center followed a specific algorithm: patients with suspected advanced primary ovarian cancer underwent surgical laparoscopy, during which their metastatic burden was assigned a modified Fagotti score40,41 and their tissues obtained and stored. Following laparoscopy, patients with a predictive index value < 8 underwent primary reductive surgery, and patients with a predictive index value ≥ 8 underwent NACT followed by interval reduction surgery. After three to four cycles of carboplatin-based NACT (generally a paclitaxel- and carboplatin-based regimen), patients were considered “excellent responders” (ER) if there was a complete response or only microscopic disease left at time of interval surgery, or they were considered “poor responders” (PR) if they presented stable or progressive disease on radiologic evaluation and/or suboptimal interval cytoreduction after NACT, according to Response Evaluation Criteria in Solid Tumors version 1.1. At interval surgery, post-chemotherapy tissues were collected and stored. The study was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center, and all samples were collected after obtaining written informed consent from patients.
For the collection of tissues from GYN-COE, frozen tumors and clinical data were collected before and after neoadjuvant paclitaxel and carboplatin chemotherapy from patients with histologically confirmed advanced-stage, high-grade serous ovarian or tubal carcinoma and banked at the Women’s Health Integrated Research Center in Annandale, VA. These patients provided broad consent for their tissues to be used in future research under WCG IRB Protocol #20110222, Tissue and Data Acquisition Activity for the Study of Gynecologic Disease. The paired tumor specimens and clinical data were collaboratively evaluated under WCG IRB Protocol #14-1679, an Integrated Molecular Analysis of Endometrial Cancer, Ovarian Cancer, and Other Medical Conditions to Identify and Validate Clinically Informative Biomarkers and Factors, and the fully executed Material Transfer Agreement #205-20.
For the collection from Washington University, frozen tumors and clinical data were collected before neoadjuvant paclitaxel and carboplatin chemotherapy from patients with histologically confirmed advanced-stage, high-grade serous ovarian or tubal carcinoma and banked at the University of Iowa as part of a collaborative study; these patients gave informed consent as part of our Washington University Tumor Tissue Banking IRB 201105400 or our collaborative R01 with Iowa: IRB 201104242 and 20511102. The study was approved by the Institutional Review Board of the University of Iowa (protocol #201507805).
Unidentified frozen blocks from HGSC of two different patients were obtained from the Cooperative Human Tissue Network (CHTN) and used to test the reproducibility of DESI-MS on multiple sections (Supplementary Fig. 1).
In vivo models of ovarian cancer
Animal protocols were approved by the MD Anderson Institutional Animal Care and Use Committee and experiments were performed with 6- to 8-week-old female athymic nude mice (NCr-nude) obtained from Taconic Biosciences. Luciferase-labeled SKOV3ip1 ovarian cancer cells were used to establish xenograft models for all mouse experiments as described before42. Cells were cultured to 70–90% confluence and then trypsinized, washed twice with phosphate-buffered saline, and resuspended in ice-cold Hank’s Balanced Salt Solution (cat. #21-021-CV; Cellgro). The mice were then inoculated with 1 × 106 SKOV3ip1 cells via intraperitoneal injection to the right side of the abdomen. Tumor establishment was subsequently confirmed after injection of 200 μL of 14.3 mg/mL luciferin (cat. #LUCK-1G; GoldBio) using a Xenogen IVIS in vivo imaging system. Following tumor establishments, mice were randomly assigned to treatment groups as described in {Glassman, 2023 #119}. For the purpose of this study, tumors from mice treated with vehicle control were considered. Investigators sacrificed the mice via carbon dioxide euthanasia followed by cervical dislocation once the mice were moribund. At the time of gross necropsy, mouse tumor weights, nodule numbers, distribution of metastasis, and presence of ascites were recorded. All tumor tissues were dissected, and samples were snap-frozen, fixed in formalin for paraffin embedding, or snap-frozen in optimal cutting temperature compound (Mercedes Scientific) for frozen slide preparation.
DESI-MS
A 2-dimensional Omni Spray (Prosolia Inc, Indianapolis, IN) was used for tissue imaging with an LTQ-Orbitrap Elite mass spectrometer (Thermo Scientific, San Juan, CA). DESI-MS imaging was performed in the negative ion mode from m/z 100 to 1500 with a hybrid mass spectrometer, which allows tandem MS experiments to be performed with high mass accuracy (<5 ppm mass error) and high mass resolution (60,000 resolving power). Imaging was performed using a spatial resolution of 200 µm. Ion images were assembled using Biomap (Novartis) software. For negative ion mode analyses, the histologically compatible spray solvent dimethylformamide:acetonitrile (DMF:ACN) 1:1 (v/v) was used to perform the imaging analyses at a flow rate of 1.2 µL/min38. DESI-MS data were deposited at https://data.mendeley.com/datasets/zzr5rk7vj5/1. For many cases we analyzed multiple sections of the same tumor; prior studies have evaluated the reproducibility of DESI-MS imaging on serial tissue sections43.
Histopathology and light microscopy
The same tissue sections analyzed by DESI-MS were then subjected to standard H&E staining. Pathologic diagnosis was made by Dr. Jinsong Liu using light microscopy. Light microscopy images were obtained and subjected to manual tissue segmentation into the two regions of interest, epithelium, and stroma, based on morphologic assessment.
DESI-MS reproducibility
In order to test the reproducibility of DESI-MS on several sections from the same tumor block, we used sections from frozen tumor blocks derived from two patients with ovarian cancer and 5 total ovarian cancer xenograft mice. Two sections from each human tumor and four sections from four of the five xenografts were used for the analysis. For one of the five xenografts two sections were analyzed. After DESI-MS, mass spectra were identified, and a Cosine Similarity analysis was performed. (Supplementary Fig. 1).
Global proteomics and phosphoproteomics analysis
Global proteomics and phosphoproteomics analysis of pre-chemotherapy samples from 7 ER and 8 PR patients, including tumors from primary and metastatic disease sites for a subset of cases, was performed as described previously13. Briefly, laser microdissection was used to collect whole tumor samples (cancer and stromal cells combined), which underwent pressure-assisted digestion employing a barocycler (2320EXT Pressure BioSciences, Inc.) and a heat-stable form of trypsin (SMART Trypsin, ThermoFisher Scientific, Inc.). Peptide digestion was labeled per tandem mass tag channel (TMT-11, ThermoFisher Scientific, Inc.). Sample multiplexes were separated offline using basic reversed-phase liquid chromatographic fractionation on a 1260 Infinity II liquid chromatograph (Agilent) into 96 fractions using a linear gradient of acetonitrile (0.69% min) followed by concatenation (36 total fractions for global proteomics and 12 fractions for phosphopeptides serially enriched by TiO2 and Fe-IMAC). Each pooled fraction was resuspended in 100 mM NH4HCO3 and analyzed by LC-MS/MS employing a nanoflow LC system (EASY-nLC 1200, ThermoFisher Scientific) coupled online with an Orbitrap Fusion Lumos Tribrid mass spectrometer (ThermoFisher Scientific). In brief, each fraction was loaded onto a nanoflow HPLC system fitted with a reversed-phase trap column (Acclaim PepMap100 C18, 20 mm, nanoViper, Thermo Scientific) and a heated (50 °C) reversed-phase analytical column (Acclaim PepMap RSLC C18, 2 µm, 100 Å, 75 µm × 500 mm, nanoViper, Thermo Fisher Scientific) coupled online with the mass spectrometer. Peptides were eluted using a linear gradient of 2% mobile phase B (95% acetonitrile, 0.1% formic acid) to 32% mobile phase B over 120 min at a constant flow rate of 250 nL/min. High-resolution (R = 60,000 at m/z 200) broadband (m/z 400-1600) mass spectra were acquired, followed by the selection of the 12 most intense molecular ions in each MS scan for high-energy collisional dissociation. Global protein-level and phosphosite identifications were generated by searching .raw data files with a publicly available, non-redundant human proteome database (Swiss-Prot, Homo sapiens [http://www.uniprot.org/]) using Mascot (Matrix Science), Proteome Discoverer (Thermo Fisher Scientific), and in-house tools using identical parameters as described previously34. Differential analyses of global proteome or transcriptome matrixes were performed using the LIMMA package (version 3.8) in R (version 3.5.2), and candidates mapping to metabolomic pathways of interest identified from the Reactome databases were prioritized for downstream analysis. A more detailed description of this method can be found in13. A total of 7148 proteins and >1075 phosphosites were co-quantified across cases (Supplemental Tables 13–18). Protein and phosphosite mapping to the Reactome pathways altered between ER and PR cases observed in the metabolomic analysis were selected for further analysis. Pathways with the highest number of proteins quantified included the metabolism of amino acids and derivatives, metabolism of nucleotides, and respiratory electron transport and related pathways. Differential analysis revealed that most proteins and phosphosites were significantly altered (LIMMA p < 0.05, ±1.5-fold change) between the PR and ER cases mapped to the metabolism of amino acids and derivatives and metabolism of nucleotide pathways. A heatmap for the eight proteins significantly altered in PR versus ER pre-chemotherapy tumors, mostly mapping to the metabolism of amino acids and derivatives pathway (z-score = 0.728 p-value 1.69E-13, derived from Ingenuity Pathway Analysis), is shown in Fig. 5a. A principal component analysis plot of these proteins by case (Fig. 5b) illustrates a distinct separation of the pre-chemotherapy PR and ER tumors. For the phosphoproteome analysis, the comparison reflects the relative abundance of a given phosphosite, i.e., phosphorylated residue, of interest in one condition versus the other. The mass spectrometry proteomics data are available at the ProteomeXchange Consortium via the PRIDE (https://doi.org/10.1093/nar/gky1106) partner repository with the dataset identifier PXD014980.
Quantification
MS data were extracted from the regions of interest using MSiReader software. Significance analysis of microarrays was used to identify ions with significantly different abundances in ER samples compared with PR samples and in pre-chemotherapy samples compared with post-chemotherapy samples. Features below 10% FDR were selected. Selective analyses were carried out separately for data extracted from epithelial and stromal regions. The identified metabolites were divided for analytical purposes into non-lipid (such as uracil, fumarate, hypoxanthine, glutamic acid, and citrate) and lipid (such as fatty acids, glycolipids, ceramides, and cardiolipins) categories.
Pathway analysis
To study the non-lipid metabolic species, we carried out pathway analyses using REACTOME (https://reactome.org/) and confirmed the findings in Pathway Studio (https://www.pathwaystudio.com/). Enriched REACTOME pathways were ordered according to a probability score corrected for FDR using the Benjamini-Hochberg method. We selected the 10 most relevant pathways in REACTOME sorted by p-value, and pathways with a common hit in Pathway Studio were considered. Pathway analysis was performed only when at least two metabolites (small molecules) were recognized by the software. In cases in which one software was not able to recognize more than two metabolites, the results from the other software were considered.
Prediction model
The glmnet package44 in R version 3.6.3 was used to create a ridge regression model for the classification of treatment response. Prior to analysis, data were pre-processed by removing m/z values that were present in less than 10% of all spectra. Intensities were median-normalized by dividing the intensity of each individual ion in a spectrum by the median intensity for the same spectrum. Ten-fold cross-validation was used to create a ridge regression model for the classification of ER and PR samples from the epithelial regions of pre-chemotherapy tumor tissues. The analysis was restricted to the primary sites (adnexa and ovaries), and samples from metastatic sites (omentum or abdominal organs) were excluded. The ridge regression model used 78 features for class distinction, represented by a variety of small molecules and lipid species between 100 and 1000 m/z. The ridge regression model was used to estimate the probability of every mass spectrum belonging to either the ER or PR group. If more than 50% of the spectra were correctly predicted for a single sample, we considered the sample to be correctly classified in our per-sample prediction results. Pixel-based and sample-based accuracies, sensitivities, and specificities were calculated.
Sparse partial least squares discriminant analysis
Metaboanalyst 5.0 was used for sparse partial least squares discriminant analysis (sPLS-DA). Prior to sPLS-DA, data were TIC-normalized and mean-centered. sPLS-DA plots were used to visualize the distinct separation between pre- and post-chemotherapy samples in both the ER samples and PR samples (Fig. 3b).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contacts, Anil K. Sood (asood@mdanderson.org) and Livia S. Eberlin (Livia.Eberlin@bcm.edu). The MS proteomics data are available at the ProteomeXchange Consortium via the PRIDE (https://doi.org/10.1093/nar/gky1106) partner repository with the dataset identifier PXD014980. DESI-MS data were deposited at https://data.mendeley.com/datasets/zzr5rk7vj5/1.
Code availability
The glmnet package in R44 was used to create a ridge regression model for the classification of treatment response. Metaboanalyst 5.0 was used for sparse partial least squares discriminant analysis (sPLS-DA).
References
Piccart, M. J. et al. Long-term follow-up confirm’s a survival advantage of the paclitaxel-cisplatin regimen over the cyclophosphamide-cisplatin combination in advanced ovarian cancer. Int J. Gynecol. Cancer 13, 144–148 (2003).
Siegel, R. L., Sahar, L., Portier, K. M., Ward, E. M. & Jemal, A. Cancer death rates in US congressional districts. Ca-Cancer J. Clin. 65, 339–344 (2015).
Fleming, N. D. et al. Laparoscopic surgical algorithm to triage the timing of tumor reductive surgery in advanced ovarian cancer. Obstet. Gynecol. 132, 545–554 (2018).
Chughtai, S. et al. A multimodal mass spectrometry imaging approach for the study of musculoskeletal tissues. Int J. Mass Spectrom. 325, 150–160 (2012).
Norris, J. L. & Caprioli, R. M. Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem. Rev. 113, 2309–2342 (2013).
Eberlin, L. S. et al. Classifying human brain tumors by lipid imaging with mass spectrometry. Cancer Res 72, 645–654 (2012).
Eberlin, L. S. et al. Molecular assessment of surgical-resection margins of gastric cancer by mass-spectrometric imaging. P Natl Acad. Sci. USA 111, 2436–2441 (2014).
Guenther, S. et al. Spatially resolved metabolic phenotyping of breast cancer by desorption electrospray ionization mass spectrometry. Cancer Res 75, 1828–1837 (2015).
Venter, A., Sojka, P. E. & Cooks, R. G. Droplet dynamics and ionization mechanisms in desorption electrospray ionization mass spectrometry. Anal. Chem. 78, 8549–8555 (2006).
Wiseman, J. M., Ifa, D. R., Song, Q. Y. & Cooks, R. G. Tissue imaging at atmospheric pressure using desorption electrospray ionization (DESI) mass spectrometry. Angew. Chem. Int Ed. 45, 7188–7192 (2006).
Sans, M. et al. Metabolic markers and statistical prediction of serous ovarian cancer aggressiveness by ambient ionization mass spectrometry imaging. Cancer Res 77, 2903–2913 (2017).
Cao, K. A. L., Boitard, S. & Besse, P. Sparse PLS discriminant analysis: biologically relevant feature selection and graphical displays for multiclass problems. Bmc Bioinformatics. 12, https://doi.org/10.1186/1471-2105-12-253 (2011).
Lee, S. et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Rep. 31, https://doi.org/10.1016/j.celrep.2020.03.066 (2020).
van Gastel, N. et al. Induction of a timed metabolic collapse to overcome cancer chemoresistance. Cell Metab. 32, 391 (2020).
Nieman, K. M. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med 17, 1498–U1207 (2011).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559 (2014).
Desbats, M. A., Giacomini, I., Prayer-Galetti, T. & Montopoli, M. Metabolic plasticity in chemotherapy resistance. Front Oncol. 10, https://doi.org/10.3389/fonc.2020.00281 (2020).
Fox, D. B. et al. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2, 318 (2020).
Goetze, K., Walenta, S., Ksiazkiewicz, M., Kunz-Schughart, L. A. & Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J. Oncol. 39, 453–463 (2011).
Vegran, F., Boidot, R., Michiels, C., Sonveaux, P. & Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappa B/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 71, 2550–2560 (2011).
Rothberg, J. M. et al. Acid-mediated tumor proteolysis: contribution of cysteine cathepsins. Neoplasia 15, 1111–1123 (2013).
Nyen, T. V. et al. Serine metabolism remodeling after platinum-based chemotherapy identifies vulnerabilities in a subgroup of resistant ovarian cancers. Nat. Commun. 13, https://doi.org/10.1038/s41467-022-32272-6 (2022).
Yang, L. F. et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 24, 685–700 (2016).
Pavlova, N. N. et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 27, 428 (2018). -+.
Zhang, J. et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 (2014).
Kopp, F. et al. The glycerophospho metabolome and its influence on amino acid homeostasis revealed by brain metabolomics of GDE1(−/−) Mice. Chem. Biol. 17, 831–840 (2010).
Kume, A. et al. The glycine cleavage system - molecular-cloning of the chicken and human glycine decarboxylase Cdnas and some characteristics involved in the deduced protein structures. J. Biol. Chem. 266, 3323–3329 (1991).
Bhatia, M. et al. Allosteric inhibition of MTHFR prevents futile SAM cycling and maintains nucleotide pools in one-carbon metabolism. J. Biol. Chem. 295, 16037–16057 (2020).
Tibbetts, A. S. & Appling, D. R. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 (2010).
Zhang, W. C. et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis (vol 148, pg 259, 2012). Cell 148, 1066–1066 (2012).
Liu, R. et al. mTORC1 activity regulates post-translational modifications of glycine decarboxylase to modulate glycine metabolism and tumorigenesis. Nat. Commun. 12, https://doi.org/10.1038/s41467-021-24321-3 (2021).
Knox, R. J., Friedlos, F., Lydall, D. A. & Roberts, J. J. Mechanism of cytotoxicity of anticancer platinum drugs - evidence that Cis-Diamminedichloroplatinum(Ii) and Cis-Diammine-(1,1-Cyclobutanedicarboxylato)Platinum(Ii) differ only in the kinetics of their interaction with DNA. Cancer Res. 46, 1972–1979 (1986).
Budhu, S. et al. Targeting phosphatidylserine enhances the anti-tumor response to tumor-directed radiation therapy in a preclinical model of melanoma. Cell Rep. 34, 108620 (2021).
Schad, S. et al. Phosphatidylserine targeting antibody enhances anti-tumor activity of adoptive cell therapies in a mouse melanoma model. J. Immunol. 204 (2020).
Hiraga, K. & Kikuchi, G. The mitochondrial glycine cleavage system - functional association of glycine decarboxylase and aminomethyl carrier protein. J. Biol. Chem. 255, 1671–1676 (1980).
Alptekin, A. et al. Glycine decarboxylase is a transcriptional target of MYCN required for neuroblastoma cell proliferation and tumorigenicity. Oncogene 38, 7504–7520 (2019).
Gyorffy, B., Lanczky, A. & Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr.-Relat. Cancer 19, 197–208 (2012).
Woo, C. C., Kaur, K., Chan, W. X., Teo, X. Q. & Lee, T. H. P. Inhibiting glycine decarboxylase suppresses pyruvate-to-lactate metabolism in lung cancer cells. Front. Oncol. 8, https://doi.org/10.3389/fonc.2018.00196 (2018).
Han, C., Lu, X. B. & Nagrath, D. Regulation of protein metabolism in cancer. Mol Cell Oncol. 5, https://doi.org/10.1080/23723556.2017.1285384 (2018).
Fagotti, A. et al. A laparoscopy-based score to predict surgical outcome in patients with advanced ovarian carcinoma: a pilot study. Ann. Surg. Oncol. 13, 1156–1161 (2006).
Hansen, J. M. et al. Concordance of a laparoscopic scoring algorithm with primary surgery findings in advanced stage ovarian cancer. Gynecol. Oncol. 151, 428–432 (2018).
Glassman, D. et al. Exploiting metabolic vulnerabilities after anti-VEGF antibody therapy in ovarian cancer. Iscience. 26, https://doi.org/10.1016/j.isci.2023.106020 (2023).
Dill, A. L., Eberlin, L. S., Costa, A. B., Ifa, D. R. & Cooks, R. G. Data quality in tissue analysis using desorption electrospray ionization. Anal. Bioanal. Chem. 401, 1949–1961 (2011).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Acknowledgements
This work was supported in part by the MD Anderson Ovarian Cancer Moon Shot; CPRIT (RP180381); the National Institutes of Health (CA217685 [Ovarian Cancer SPORE], CA193249, CA209904, and CA193249-S1); the Ovarian Cancer Research Alliance; the American Cancer Society; the Dunwoody Fund; the Le Bert Suess Family Endowment for Ovarian Cancer Research; the Frank McGraw Memorial Chair in Cancer Research; the Foundation for Women’s Cancer; the Amy Krouse Rosenthal Foundation; and Judy’s Mission to End Ovarian Cancer Foundation (Research Grant for Early Detection of Ovarian Cancer). Y.W is supported in part by Department of Defense Ovarian Cancer Research Program (W81XWH-20-1-0335). We acknowledge the Research Medical Library at MD Anderson Cancer Center for editing the text. For the GYN-COE collection, the collection and banking of these specimens and data were funded by awards HU0001-16-2-0006, HU0001-19-2-0031, HU0001-20-2-0033, and HU0001-21-2-0027 from the Uniformed Services University of the Health Sciences from the Defense Health Program to the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., Gynecologic Cancer Center of Excellence Program (PI: Yovanni Casablanca, Co-PI: G. Larry Maxwell). We acknowledge BioRender for figure composition.
Author information
Authors and Affiliations
Contributions
S.C.: Project administration, writing original draft and review/editing, supervision. P.L.L.: Software suggestion. J.L.: Validation. S.B., M.S., M.K., I.P., Y.W., N.W.B., W.B., T.P.C., L.G.M.: Formal analysis and data curation. S.B., M.S., M.K., I.P., N.W.B., W.B., T.P.C., L.G.M., P.L.L., S.K.L., P.H.T., M.J.G., J.L., S.L., L.S.E., A.K.S., K.M.D.: Review/editing of the text. K.I.F., E.S., E.B., Y.W.: Revision/editing of the text. S.C., S.L., L.S.E., A.K.S.: Investigation. S.C., L.S.E., A.K.S.: Conceptualization. S.C., E.S., E.B.: Visualization. S.C., K.I.F.: Data curation. K.M.D., S.K.L., P.H.T., M.J.G.: Provision of study material (patients/laboratory samples). N.W.B., T.P.C., L.G.M.: Funding aquisition. L.S.E., A.K.S.: Methodology, resources, funding.
Corresponding authors
Ethics declarations
Competing interests
N.F. declares that she is a consultant for GSK and Immunogen. L.S.E. declares that she receives funding from Thermo Fisher, Merck & Co, Waters corporation, and Eli Lilly and has stocks in MS Pen Technologies. A.K.S. declares that he is a shareholder of BioPath, and is a consultant for Merck, AstraZeneca, Onxeo, ImmunoGen, Ivlon, GSK, and Kiyatec. The remaining authors declare no competing financial or non-financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Corvigno, S., Badal, S., Spradlin, M.L. et al. In situ profiling reveals metabolic alterations in the tumor microenvironment of ovarian cancer after chemotherapy. npj Precis. Onc. 7, 115 (2023). https://doi.org/10.1038/s41698-023-00454-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00454-0
