
Abstract
Adverse pregnancy outcomes (APOs) affect a large proportion of pregnancies and represent an important cause of morbidity and mortality worldwide. Yet the pathophysiology of APOs is poorly understood, limiting our ability to prevent and treat these conditions. To search for genetic markers of maternal risk for four APOs, we performed multi-ancestry genome-wide association studies (GWAS) for pregnancy loss, gestational length, gestational diabetes, and preeclampsia. We clustered participants by their genetic ancestry and focused our analyses on three sub-cohorts with the largest sample sizes: European, African, and Admixed American. Association tests were carried out separately for each sub-cohort and then meta-analyzed together. Two novel loci were significantly associated with an increased risk of pregnancy loss: a cluster of SNPs located downstream of the TRMU gene (top SNP: rs142795512), and the SNP rs62021480 near RGMA. In the GWAS of gestational length we identified two new variants, rs2550487 and rs58548906 near WFDC1 and AC005052.1, respectively. Lastly, three new loci were significantly associated with gestational diabetes (top SNPs: rs72956265, rs10890563, rs79596863), located on or near ZBTB20, GUCY1A2, and RPL7P20, respectively. Fourteen loci previously correlated with preterm birth, gestational diabetes, and preeclampsia were found to be associated with these outcomes as well.
Similar content being viewed by others

Introduction
Adverse pregnancy outcomes (APOs) are a serious threat to the health of pregnant persons and children. APOs affect a significant fraction of pregnancies across the globe and are among the leading causes of morbidity and mortality worldwide1. Among the most common APOs are preterm birth (which occurs in over 10% of pregnancies in the United States (US)2), preeclampsia (which develops in 5–10% of pregnancies3), gestational diabetes (occurring in roughly 6% of pregnancies in the US4), and pregnancy loss (estimated to occur in about 7–14% of pregnancies5,6). APOs are also highly correlated with future disease in birthing parents. For example, gestational diabetes carries a lifetime 50% risk of type 2 diabetes (T2D) in the mother7, while preeclampsia is associated with a 2–threefold increase of cardiovascular disease later in life8. Yet, factors driving these diseases remain poorly understood, hindering efforts in prevention and treatment.
To better understand the mechanisms and improve prediction of APOs in nulliparous individuals, the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be (nuMoM2b) consortium9,10 recruited and prospectively followed a large cohort of nulliparous people beginning in their first trimester of pregnancy. Participants underwent several assessments over the course of their pregnancies, resulting in a comprehensive profile that included biospecimens, clinical measurements, ultrasounds, behavior (through interviews and questionnaires), physical activity assessment, and dietary content.
By precisely characterizing different aspects of over 10,000 pregnancies, the nuMoM2b cohort has already yielded valuable insights into factors that contribute to APOs11,12. Additionally, the availability of biospecimens provides a unique opportunity to study the genetic underpinnings of APOs. The objective of this study was to test for association between common variants across the maternal genome and four APOs in the nuMoM2b cohort: gestational length (as a proxy for preterm birth), preeclampsia, gestational diabetes mellitus (GDM), and pregnancy loss. Our investigation leverages ancestrally diverse populations to further isolate potential genetic factors involved in these APOs.
Methods
Participants
The participants of the analysis were enrolled in the nuMoM2b cohort (https://www.nichd.nih.gov/research/supported/nuMoM2b), a longitudinal, multiethnic cohort study of nulliparous individuals. All participating centers, documented in Haas et al.10, obtained approval by the local Institutional Review Boards (IRBs) of their corresponding recruitment institutions. The genotype analysis is covered by Indiana University’s IRB, which was Protocol Study number 1008–08, approved on 9/28/2010. This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. All participants included in this study provided informed consent. The study enrolled 10,038 nulliparous people from the first trimester of their pregnancy to participate in three study visits during pregnancy. Collection of health status and biomarkers were conducted at regular intervals, and documentation of pregnancy outcomes was performed by medical record abstraction using a priori definitions (details of this process were described by Haas et al.9,10).
Phenotype definitions
Pregnancy loss: All subjects who had a pregnancy loss, regardless of gestational age, were considered as cases (Table 1). A pregnancy loss occurs when the fetus dies during gestation. This categorization includes all cases of fetal demise occurring under 20 weeks of gestation, and all stillbirths (defined as a fetal death occurring from 20 weeks of gestation onward). Individuals who underwent termination of pregnancy were excluded from the pregnancy loss analysis. Subjects who had a live birth were treated as controls.
Gestational length: We opted to use a quantitative phenotype, gestational length, instead of a binary preterm/full term outcome to gain additional information and statistical power by using a more granular phenotype. Gestational length was determined from an estimated due date established by a first-trimester ultrasound crown-rump length measurement and was recorded in weeks10. Preterm birth was defined as any live birth that occurred before 37 weeks gestational age. Cases of stillbirth, fetal demise, and termination (elective and indicated) were all excluded from this phenotype group (Table 1). Cases of preterm birth that were medically indicated (e.g. for preeclampsia), were excluded. Term births that had labor induced or were by planned cesarean delivery were included as they occurred at term and because gestational age at delivery is a right-bordered outcome. Thus, we did not believe that their inclusion would alter this result and excluding these cases would limit the sample size of deliveries > 37 weeks, potentially skewing the data toward lower gestational age means.
Gestational diabetes (GDM): GDM was diagnosed through clinical evaluation, from fasting blood sugar, sequential 1-h glucose challenge test followed by a 3-h glucose tolerance test (GTT), or a single step 2-h 75-g GTT13. We excluded individuals diagnosed with pregestational diabetes from GDM analyses, and all other individuals were treated as controls.
Preeclampsia: Cases are individuals with a diagnosis of preeclampsia (with and without severe features), eclampsia, and chronic hypertension with superimposed preeclampsia. A detailed description of nuMoM2b study definitions of hypertensive disorders of pregnancy was published in the supplement to the paper by Facco et al.13. All other individuals were treated as controls. We did not exclude individuals with hypertension antepartum, as we are interested in unearthing markers specific to gestational disease, not general hypertension.
Genotyping
We genotyped all participants who had adequate samples and agreed to be genotyped (n = 9,757). We used the Infinium Multi-Ethnic Global Array (MEGA; Illumina, USA), which is designed to adequately query individuals of multiple genetic ancestries14 (a known issue in genotyping studies), enriched for variants of clinical importance15, and has been successfully used to study recent admixed populations14,16. The MEGA allowed us to genotype > 1 million variants that are on average 1.4 Kb apart, effectively covering the entire genome. DNA extractions from whole blood were carried out on a QIAsymphony instrument (from Qiagen; extraction kit DSP DNA Midi Kit #937,355, protocol Blood_1000_V7_DSP) at the Center for Genomics and Bioinformatics (Indiana University, Bloomington), and genotyping was completed at the Van Andel Institute (Grand Rapids, MI, USA). We imposed standard quality control filters at this stage, all involving technical measurements of the raw intensity data (cluster separation < 0.3, normalized R-value mean < 0.2 for all genotypes, 10th percentile of the GenCall scores < 0.3) using GenomeStudio v2.4 (Illumina). Genotype calls for the ~ 1.7 million loci that passed initial quality control (98.3% of all markers in the array) were made with Beeline autoconvert (Ilumina).
Quality control pipeline
We leveraged a multi-step quality control (QC) pipeline, depicted in Fig. 1, to adequately address the heterogeneous nature of the dataset. The pipeline is broken into five separate modules that integrate current best practices in GWAS QC, which are further described in the text below. Unless otherwise stated, both the quality control steps and analyses were carried out using PLINK1.917.

Overview of the quality control (QC) pipeline used to preprocess nuMoM2b subjects.
Module 1: preprocessing and QC
The initial preprocessing of the dataset removed poorly genotyped individuals and SNPs according to the following criteria: (1) minor allele frequency (MAF) < 1%, (2) missingness of genotyping per individual and per marker > 5%, (3) Hardy–Weinberg Equilibrium (HWE) test p-value < 5 × 10–6, and (4) heterozygosity F-statistic within 3 standard deviations from the mean heterozygosity across all subjects using autosomes (Supplementary Figure S1). Significantly reduced heterozygosity may be indicative of high levels of consanguinity and subjects with excessive heterozygosity are suggestive of sample contamination, thus we excluded these subjects from the downstream dataset.
Module 2: cryptic relatedness
The self-reported racial and ethnic diversity of the nuMoM2b cohort necessitates a careful approach in assessing relatedness. As the pairwise identity-by-descent (IBD) estimation implemented in PLINK assumes a homogeneous subset, we used the KING-Robust algorithm, a pairwise kinship estimator for GWAS that is robust to the presence of unknown population substructure18. We inferred kinship estimates between all pairs of subjects in the cohort (Supplementary Figure S2), randomly removing one subject from the pairs of subjects with first- or second-degree relatedness (kinship coefficient > 0.0884) such that we minimized the number of subjects removed.
Module 3: subpopulation stratification
With the goal of minimizing spurious genetic associations in downstream analyses driven by population stratification and ancestry-based allele frequency differences, we clustered the cohort by genetic ancestry (Supplementary Figure S3). We determined the ancestry of each subject using SNPweights v.2.1 and a set of approximately 40,000 ancestry-informative markers curated by the 1000 Genomes Consortium19. Samples were clustered into five ancestry groups concordant with the 1000 Genomes Consortium19 subpopulations: African (AFR, n = 1425), Admixed American (AMR; n = 846), East Asian (EAS; n = 323), European (EUR, n = 6082), and South Asian (SAS; n = 112). Membership in each ancestry subpopulation was assigned based on having ≥ 80% ancestry in the specific subpopulation (Supplementary Figure S4). We observed a large fraction of highly admixed individuals; thus, we also established a sixth group (ADM, n = 891) of subjects who have no percent ancestry > 50% in any single ancestry group. Not all ancestry groups contained enough subjects to power a genome-wide association. Accordingly, we only proceeded with imputing the European, African, and Admixed American ancestry sub-cohorts. From this module onward, all QC steps are performed at the sub-cohort level.
Next, we checked the consistency of reported sex with sex assignments imputed from X chromosome breeding coefficients. This step was performed after subjects were grouped into subpopulations because F-statistic approximation for the X-chromosome relies on accurate MAF estimates, which vary at the subpopulation level. The sex check consists of four steps: (1) unambiguously re-coding the pseudo-autosomal region of the X-chromosome, (2) performing LD-based pruning on the set of markers used for the F-estimate, (3) confirming that all F estimates yield female calls using the threshold F < 0.6 (PLINK suggests a cutoff of F > 0.8 for a male call), and (4) removing any subjects with discordant results. Lastly, population structure was determined in PLINK using pruned SNPs from the data (linkage disequilibrium pruning r2 < 0.1). The top ten principal components were computed for each of the three subpopulations (EUR, AFR, AMR).
Module 4: phasing and imputation
Subjects in these sub-cohorts were phased with Eagle2 and imputed by Minimac320 using the TOPMED Imputation Server21,22. Prior to phasing, TOPMED partitions the data into 20 megabase length chunks and removes SNPs that are: (1) duplicates, (2) indels, (3) strand ambiguous (C/G and A/T), (4) not included in the Haplotype Reference Consortium (HRC) panel, and (5) mismatched between the reference panel and study. Data were then imputed using version R2 of the TOPMED panel, currently the largest panel of sequenced human genomes, and containing representation from the ancestry groups observed in nuMoM2b.
Module 5: imputation postprocessing
Following imputation, we excluded SNPs with an r2 quality score < 0.7, MAF < 1%, and a Hardy–Weinberg Equilibrium (HWE) P < 5 × 10–6 within each imputed group.
Genome-wide associations
Association testing was carried out using regression models implemented in PLINK v1.917. The model was adjusted for each subject’s rank-transformed age, body mass index (BMI), and the first ten principal components from population structure analysis. As maternal age has a nonlinear effect on pregnancy23,24, we transformed each subject’s age into the distance from the median percentile in all subjects. The suggestive association threshold was P < 1 × 10–5 and the threshold for genome-wide significance was P < 5 × 10–8. Any subjects with missing age or phenotype information were excluded from analysis. The multi-ancestry results from the sub-cohorts were fixed-effect meta-analyzed using GWAMA25 and result plots were displayed using R libraries. As the majority of the subjects in nuMoM2b are of European ancestry, we only considered variants shared by this group, as the African and Admixed American sub-cohorts were underpowered in detecting signal in a standalone GWAS. Due to the smaller cohort size and large class imbalance (7 cases, 768 controls) observed in the Admixed American cohort for pregnancy loss outcomes, we did not perform a pregnancy loss GWAS for this sub-cohort.
Using the results of the genome-wide associations for each APO, we inferred SNP-based heritability of these traits using LDscore26. We scanned for putative regulatory effects by carrying out a transcriptome-wide association study (TWAS) as implemented in the Fusion software, and using the available expression reference weights for whole blood and adipose tissue27, as well as liver, pancreas, vagina, and uterus, and whole blood from the GTEx v7 multi-tissue RNA-seq data set. For TWAS, P-values were Bonferroni-corrected by the number of genes in each panel. Conditional expression analyses were performed using R scripts from the Fusion software. As both SNP-based heritability and TWAS inferences rely on reference panels, and relevant panels for these were developed with EUR cohorts, we used only EUR individuals in these two analyses.
Variant fine mapping and annotation
We utilized the previously defined fine-mapping approach28 to delineate the 95% credible set for each significant meta-analysis locus containing more than one SNP of interest. In this approach, the posterior probability that each variant is causal is computed using approximations of the necessary Bayes factors. Only variants with a cumulative posterior probability above 95% are included in the credible set.
In characterizing the putative causal role of our variants of interest, we further annotated each SNP using Variant Effect Predictor29 (VEP) to identify the corresponding gene function, and RegulomeDB30 to understand the regulatory context. VEP enables genome interpretation by providing the gene and transcript level context, while also describing the regulatory location within the gene and type of point mutation for each variant. RegulomeDB heuristically scores variants on a scale of 1a (high confidence) to 7 (low confidence) given the presence of regulatory function as assessed by various functional screens, including annotations from ENCODE31 and other sources.
Results
Pregnancy loss
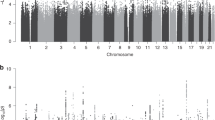
The multi-ancestry meta-analysis revealed a set of 12 novel SNP associations across two loci associated with pregnancy loss (Table 2 and Supplementary Table S1). Significant SNPs were located within or near the genes TRMU and RGMA (Table S1, Supplementary Figures S10-S12). While most of the associated SNPs appeared in both the European and African cohorts, two SNPs (rs143149726 and rs142372194) were only found in the European cohort (Supplementary Table S2). Additionally, fine-mapping the eleven SNPs identified on or near TRMU eliminated rs147382049 from the 95% credible set for the locus. For the nine remaining multi-ancestry variants, the direction of effect was concordant across the two ancestry cohorts and suggestive of an increased odds of pregnancy loss in carriers of the alternative allele. The genomic inflation factor was suggestive of minimal confounding effects in the meta-analysis (Supplementary Figure S5). This trait showed the highest SNP-based heritability (h2 = 0.30, SE = 0.075) of the four APOs studied.
Further investigation of the variants using annotation tools pinpoints potential functional effects. RegulomeDB scores all the associated variants as “regulatory” (scoring < 5 on the RegulomeDB scale). rs143149726, a downstream gene variant of TRMU, (TRNA 5-Methylaminomethyl-2-Thiouridylate Methyltransferase), scored the lowest score across all associated variants (indicating the greatest amount of evidence for the variant to be in a functional region). The variant had a RegulomeDB rank of 1f. with a probability of 1 (Table S1), indicating that it was “likely to affect binding and linked to expression of a gene target” given evidence of an expression quantitative trait locus (eQTL) and transcription factor binding or DNase peak. rs62021480, the top SNP association at the RGMA locus, encodes a 3 prime UTR variant for the gene, which is a glycoprotein that guides developing axons and may act as a tumor suppressor.
Through TWAS, we found a correlation between pregnancy loss and gene expression levels of TTC38 (Tetratricopeptide Repeat Domain 38) in uterine (Z = 5.17, P = 2.39 × 10–7), liver (Z = 4.31, P = 1.62 × 10–5), and ovarian tissue (Z = 4.61, P = 3.91 × 10–6; Supplementary Figure S20, Supplementary Table S3). We were able to replicate previously reported associations (with P < 0.05) in three SNPs (out of 32; Table 3 and Supplementary Table S4), all near the LINC01717 (Long Intergenic Non-Protein Coding RNA 1717) locus.
Gestational length
One multi-ancestry and one European-specific variant were implicated in the quantitative meta-analysis of gestational length (Supplementary Figures S13-S14). rs58548906 is an intergenic variant located on the X chromosome near AC005052.1 and appears to be associated with a reduced gestational length (beta = − 1.43, CI = [− 1.94, − 0.921]) across African, Admixed American, and European ancestries. rs2550487, located on the 3 prime UTR of WFDC1, was found in the European sub-cohort only, and individuals carrying the minor allele are associated with lower gestational length (beta = − 1.28, CI = [0.215, − 1.70] ). Both SNPs show “minimal binding evidence” in their RegulomeDB ranking, with evidence of transcription factor binding or a DNase peak.
The genomic inflations of each sub-cohort GWAS ranged between λ = 0.99 and λ = 1.02, while the GWA meta-analysis across sub-cohorts did not show genomic inflation (Supplementary Figure S6). SNP-based heritability for this trait was h2 = 0.23 (SE = 0.13). We found evidence of association (P < 0.05) in 6 out of 79 SNPs previously reported to be associated with gestational length or similar phenotypes (Table 3, Table S4). These markers were clustered around four genes: ADAMST14 (ADAM Metallopeptidase with Thrombospondin Type 1 Motif 14), EBF1 (EBF transcription factor 1), MSI2 (Musashi RNA Binding Protein 2), and KAZN (Kazrin, Periplakin Interacting Protein).
Gestational diabetes mellitus
Three significant loci were identified in the GWAS meta-analysis for GDM (Table 2, Table S2, Supplementary Figures S15-S19). A set of four SNPs appear in the intergenic region near RPL7P20, Ribosomal Protein L7 Pseudogene 20. The most significantly associated SNP in this set is the intergenic variant, rs79596863 (Table 2). The second significant locus includes intronic variants rs61167087 and rs72956265 in ZBTB20, a Zinc Finger and BTB Domain Containing 20. Lastly, rs10890563, a 3’UTR variant on the gene GUCY1A2, Guanylate Cyclase 1 Soluble Subunit Alpha 2, had a significant association with GDM as well. Several loci show suggestive associations in the meta-analysis, which we report in the supplementary text (Fig. 2; Table S2). We did not find genomic inflation in any of the ancestry groups or in the meta-analysis (λ = 1.001, Supplementary Figure S7), and we estimated SNP-based heritability to be h2 = 0.13 (SE = 0.085).

Manhattan Plot of GWAS Results: GWA meta-analysis was carried out using the inverse variance–weighted method implemented in GWAMA. The dotted line indicates the threshold for genome-wide significance (P < 5 × 10–8). (A) Gestational length; (B) GDM; (C) Pregnancy Loss.
We also found a positive association with gestational diabetes for four previously reported variants (P < 0.05; Table 3). These SNPs were mapped near four genes: Hexokinase HKDC1, transcription factor TCF7L2, melatonin receptor MTNR1B, and major histocompatibility complex HLADQB1.
Preeclampsia
We found no SNPs associated with preeclampsia (Supplementary Figure S8). Again, genomic inflation was low (λ = 1.005, Supplementary Figure S9), and SNP-based estimate of heritability was h2 = 0.02 (SE = 0.076). We confirmed 1 of 30 previously linked variants: rs4655789 (near gene WLS; Table S2).
Discussion
We present a set of multi-ancestry GWAS of four of the most ubiquitous APOs. Overall, we identified variants at six novel loci associated with our APOs of interest, while also replicating 14 signals from previous studies. Results from the European-ancestry analyses also reveal two secondary signals at the TRMU locus, and the discovery of a new locus association with gestational length on WFDC1. Using a Bayesian fine-mapping approach, we further narrowed the list of putative causal variants to those within a credible posterior probability of at least 95%. The candidate genetic markers identified in the present study provide a starting point for molecular exploration of each APO of interest.
The cluster of 11 significant variants at the TRMU locus in the pregnancy loss GWAS are characterized as having a significant regulatory role in binding and gene expression given high-throughput experimental data from RegulomeDB. Indeed, the expression quantitative trait locus (eQTL) annotation from RegulomeDB and VEP indicate that the SNPs in the aforementioned cluster (Table S1) mediate gene expression across several two genes located on chromosome 22q13.31: TRMU and CELSR1. Additionally, TWAS analysis implicates the gene TTC38 in pregnancy loss across three tissue types: liver, ovary, and uterus. A top SNP in this locus, rs114058457 (Table S1), was previously found by Jansen et al. as an eQTL for TTC38 in blood in a cohort of 4,896 subjects of European ancestry (FDR-corrected P < 1.34 × 10–5)32. Overall, there appears to be consistent evidence mapping the top SNPs from the chromosome 22q13.31 locus to biologically meaningful roles.
While the preliminary annotations of the chromosome 22q13.3 SNP cluster seem to be promising, further work is necessary to pinpoint the mechanistic contribution of the four target genes. Previous work has shown that TRMU encodes a mitochondria-specific tRNA-modifying enzyme and plays a key part in mitochondrial translation33. Aberrant expression of TRMU is likely pathogenic to humans early in life, as variants in TRMU have been linked to several disease phenotypes, including infantile liver failure33, and infantile reversible cytochrome c oxidase deficiency34. Next, TTC38, implicated through the TWAS, has been posited as a factor associated with kidney development35, while CELSR1 is highly prevalent in embryonic tissues36,37 and linchpins embryonic development across humans and other vertebrates38,39. The genes we have outlined here span a diverse set of functions, however we speculate that an underlying commonality is that they act as important contributors to early development.
Rs62021480, located near RGMA and associated with greater risk of pregnancy loss, plays a role in transcription factor binding and lies in an open chromatin region, however the evidence for a regulatory role is minimal. Still, rs62021480 lies within the 3’ UTR of RGMA, and therefore may have post-transcriptional regulatory function. RGMA is a repulsive guidance molecule for axons of the retina, which is a pivotal step in the developing brain41.
Interpretation of the results from the remaining two GWAS is less straightforward as the associated SNPs have greater ambiguity from a functional perspective. The top GDM SNPs identified localize to three genes: RPL7P20, ZBTB20, and GUCY1A2. While RPL7P20 codes for a pseudogene, several studies have made associations between SNPs at this locus and increased heart rate42 as well as general cognition43,44. The zinc finger ZBTB20 has been linked to Primrose syndrome45, which may include insulin-resistant diabetes and other metabolic disruptions. This transcription factor regulates beta-cell function in mice46 and has been hypothesized to be involved in the control of glucose metabolism. GUCY1A2 (a guanylate cyclase, GTP-binding protein) has shown high expression levels in placenta and was differentially expressed in rat placentas responding to hyperglycemia47. In addition, GUCY1A2 has been inferred to be associated with diabetes through exposure to several toxins (reported in the Comparative Toxicogenomics Database48).
Two significant SNPs map to two separate loci in the GWAS of gestational length: AC005052.1 and WFDC1. While the gene profile of AC005052.1 is relatively unknown, WFDC1 modulates inflammatory response49. Inflammation has a clearly established link to preterm birth50 and dysregulation of the immune system through inflammation may lead to harmful effects on pregnancy51. These initial findings suggest potential mechanisms through which the SNP findings may shorten gestational length during pregnancy.
In conjunction with the GWAS findings, nuMoM2b offers a uniquely valuable resource for genetic studies on two levels. First, the dataset spans over 4,600 features per subject12, representing a multimodal array of clinical, biological, and environmental factors influencing pregnancy. The deep, longitudinal phenotyping of the cohort presents varied opportunities for additional GWAS and downstream analyses. Second, another advantage of this study and any future genetic studies performed using nuMoM2b’s multi-ancestry population is the potential for better genetic signal triangulation in the face of confounds. As nuMoM2b contains several distant ancestral populations, meta-analyses of GWAS from each ancestry group takes advantage of naturally occurring differences in linkage disequilibrium between SNPs to disentangle false positives from the most probable causal variant. In particular, having an African ancestry sub-cohort is not only inherently beneficial to study, but also helpful in that African populations contain a more diverse set of haplotypes52 that can hone in on causal variant signals from a more homogeneous population. The preprocessing pipeline established in this study can be used in future studies to study additional variants in an ancestry-stratified manner.
There are still important limitations. When considering many of the disease phenotypes in nuMoM2b, there is an imbalanced breakdown of subjects in terms of both case–control status and genetic ancestry. This imbalance persists in the analyses presented here—indeed, the pregnancy GWAS meta-analysis excluded the Admixed American sub-cohort due to extreme class imbalance, and the meta-analysis results contain > 70% of individuals of European ancestry, despite the inclusion of diverse ancestry populations. Our results would therefore benefit from further validation cohorts, particularly of individuals of non-European genetic ancestry, to improve with broad applicability of the findings. Future studies may want to study quantitative traits to improve statistical power given modest sample sizes.
This study confirmed previously reported associations between several SNPs and APOs but failed to do so for most markers queried. This result is not unexpected, however, since low levels of replication in GWAS are not uncommon53,54. While we investigated the functional role of our variants of interest through fine-mapping approaches and annotation tools, the APOs studied in this paper are ultimately defined by complex genetic architectures. The effect sizes observed for individual SNPs suggest that the APOs studied are highly polygenic. These findings imply that, rather than aiming for genetic screening at one or a few markers, better prediction of these APOs will come through the modeling of genetic signals genome-wide (e.g., through polygenic risk scoring) together with non-genetic factors. Examination of gene-by-environment interactions, which disentangle the nonlinear interplay of variants and external factors on phenotype, may further illuminate drivers of APOs55. Such efforts are already underway, with auspicious results for the prediction of GDM through the combination of polygenic risk scores and behavioral data56. It is also possible that these complex phenotypes are largely controlled by rare genetic variants57 for which GWAS are not adequate. Identifying rare causative variants would require sequencing efforts including whole-genome and whole-exome sequencing, as well as larger study populations of diverse genetic ancestry.
The results of our multi-ancestry study highlight the role of previously unknown loci across several APOs: gestational length, GDM, pregnancy loss, and preeclampsia. We unearth potential contributors to preterm birth at multiple levels of granularity—SNP, gene, and tissue. We also confirm previously identified genetic associations for preterm birth, GDM, and preeclampsia. These findings broaden our understanding of the complex polygenic nature of the APOs studied, informing further research directions and enabling downstream genetic analyses.
Data availability
Summary statistics of the GWA meta-analysis and corresponding genetic data will be available through dbGaP under study accession phs002808.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002808.v1.p1).
References
Lawn, J. E. & Kinney, M. Preterm birth: now the leading cause of child death worldwide. Sci. Transl. Med. 6, 263ed21 (2014).
Martin, J. A., Hamilton, B. E., Osterman, M. J. K. & Driscoll, A. K. Births: Final data for 2019. Natl. Vital Stat. Rep. 70, 1–51 (2021).
Sibai, B., Dekker, G. & Kupferminc, M. Pre-eclampsia. Lancet 365, 785–799 (2005).
Deputy, N. P., Kim, S. Y., Conrey, E. J. & Bullard, K. M. Prevalence and changes in preexisting diabetes and gestational diabetes among women who had a live birth - United States, 2012–2016. MMWR Morb. Mortal. Wkly. Rep. 67, 1201–1207 (2018).
Wang, X. et al. Conception, early pregnancy loss, and time to clinical pregnancy: a population-based prospective study. Fertil. Steril. 79, 577–584 (2003).
Zinaman, M. J., Clegg, E. D., Brown, C. C., O’Connor, J. & Selevan, S. G. Estimates of human fertility and pregnancy loss. Fertil. Steril. 65, 503–509 (1996).
Kim, C., Newton, K. M. & Knopp, R. H. Gestational diabetes and the incidence of Type 2 Diabetes: A systematic review. Diabetes Care 25, 1862–1868 (2002).
Mongraw-Chaffin, M. L., Cirillo, P. M. & Cohn, B. A. Preeclampsia and cardiovascular disease death: prospective evidence from the child health and development studies cohort. Hypertension 56, 166–171 (2010).
Haas, D. M. et al. Pregnancy as a window to future cardiovascular health: Design and implementation of the nuMoM2b heart health study. Am. J. Epidemiol. 183, 519–530 (2016).
Haas, D. M. et al. A description of the methods of the Nulliparous pregnancy outcomes study: Monitoring mothers-to-be (nuMoM2b). Am. J. Obstet. Gynecol. 212(539), e1-539.e24 (2015).
Catov, J. M. et al. Patterns of leisure-time physical activity across pregnancy and adverse pregnancy outcomes. Int. J. Behav. Nutr. Phys. Act. 15, 68 (2018).
Goretsky, A. et al. Data preparation of the nuMoM2b dataset. https://doi.org/10.1101/2021.08.24.21262142.
Facco, F. L. et al. Association between sleep-disordered breathing and hypertensive disorders of pregnancy and gestational diabetes mellitus. Obstet. Gynecol. 129, 31–41 (2017).
Galanter, J. M. et al. Genome-wide association study and admixture mapping identify different asthma-associated loci in Latinos: the Genes-environments & Admixture in Latino Americans study. J. Allergy Clin. Immunol. 134, 295–305 (2014).
Bien, S. A. et al. Strategies for enriching variant coverage in candidate disease loci on a multiethnic genotyping array. PLoS One 11, e0167758 (2016).
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518 (2019).
Rentería, M. E., Cortes, A. & Medland, S. E. Using PLINK for genome-wide association studies (GWAS) and data analysis. Methods Mol Biol https://doi.org/10.1007/978-1-62703-447-0_8 (2013).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Fuchsberger, C., Abecasis, G. R. & Hinds, D. A. minimac2: Faster genotype imputation. Bioinformatics 31, 782–784 (2015).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Loh, P.-R. et al. Reference-based phasing using the haplotype reference consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Cnattingius, S., Forman, M. R., Berendes, H. W. & Isotalo, L. Delayed childbearing and risk of adverse perinatal outcome A population-based study. JAMA 268, 886–890 (1992).
Fuchs, F., Monet, B., Ducruet, T., Chaillet, N. & Audibert, F. Effect of maternal age on the risk of preterm birth: A large cohort study. PLoS One 13, e0191002 (2018).
Mägi, R. & Morris, A. P. GWAMA: Software for genome-wide association meta-analysis. BMC Bioinformatics 11, 288 (2010).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Wellcome Trust Case Control Consortium et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 44, 1294–1301 (2012).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Dong, S. et al. Annotating and prioritizing human non-coding variants with RegulomeDB vol 2. Nat. Genet. 55, 724–726 (2023).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Jansen, R. et al. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum. Mol. Genet. 26, 1444–1451 (2017).
Zeharia, A. et al. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 85, 401–407 (2009).
Uusimaa, J. et al. Reversible infantile respiratory chain deficiency is a unique, genetically heterogenous mitochondrial disease. J. Med. Genet. 48, 660–668 (2011).
Zhao, J., Zou, W. & Hu, T. Novel genes associated with folic acid-mediated metabolism in mouse: A bioinformatics study. PLoS One 15, e0238940 (2020).
Tissir, F., De-Backer, O., Goffinet, A. M. & Lambert de Rouvroit, C. Developmental expression profiles of Celsr (Flamingo) genes in the mouse. Mech. Dev. 112, 157–160 (2002).
Shima, Y. et al. Differential expression of the seven-pass transmembrane cadherin genes Celsr1-3 and distribution of the Celsr2 protein during mouse development. Dev. Dyn. 223, 321–332 (2002).
Feng, J., Han, Q. & Zhou, L. Planar cell polarity genes, Celsr1-3, in neural development. Neurosci. Bull. 28, 309–315 (2012).
Tissir, F. & Goffinet, A. M. Atypical cadherins Celsr1-3 and planar cell polarity in vertebrates. Prog. Mol. Biol. Transl. Sci. 116, 193–214 (2013).
Matsunaga, E., Nakamura, H. & Chédotal, A. Repulsive guidance molecule plays multiple roles in neuronal differentiation and axon guidance. J. Neurosci. 26, 6082–6088 (2006).
Ramírez, J. et al. Thirty loci identified for heart rate response to exercise and recovery implicate autonomic nervous system. Nat. Commun. 9, 1947 (2018).
Hill, W. D. et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol. Psychiat. 24, 169–181 (2019).
Savage, J. E. et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 50, 912–919 (2018).
Cordeddu, V. et al. Mutations in ZBTB20 cause Primrose syndrome. Nat. Genet. 46, 815–817 (2014).
Zhang, Y. et al. The zinc finger protein ZBTB20 regulates transcription of fructose-1,6-bisphosphatase 1 and β cell function in mice. Gastroenterology 142, 1571-1580.e6 (2012).
Meng, R. et al. Changes in gene expression in rat placenta at gestational day 16.5 in response to hyperglycemia. Gen. Comp. Endocrinol. 320, 113999 (2022).
Davis, A. P. et al. Comparative toxicogenomics database (CTD): Update 2023. Nucleic Acids Res. 51, D1257–D1262 (2023).
Ressler, S. J. et al. WFDC1 is a key modulator of inflammatory and wound repair responses. Am. J. Pathol. 184, 2951–2964 (2014).
Cappelletti, M., Della Bella, S., Ferrazzi, E., Mavilio, D. & Divanovic, S. Inflammation and preterm birth. J. Leukoc. Biol. 99, 67–78 (2016).
Denney, J. M. et al. Longitudinal modulation of immune system cytokine profile during pregnancy. Cytokine 53, 170–177 (2011).
Rotimi, C. N. et al. The genomic landscape of African populations in health and disease. Hum. Mol. Genet. 26, R225–R236 (2017).
Liu, Y.-J., Papasian, C. J., Liu, J.-F., Hamilton, J. & Deng, H.-W. Is replication the gold standard for validating genome-wide association findings?. PLoS One 3, e4037 (2008).
Jiang, W., Xue, J.-H. & Yu, W. What is the probability of replicating a statistically significant association in genome-wide association studies?. Brief. Bioinform. 18, 928–939 (2017).
Virolainen, S. J., VonHandorf, A., Viel, K. C. M. F., Weirauch, M. T. & Kottyan, L. C. Gene-environment interactions and their impact on human health. Genes Immun. 24, 1–11 (2023).
Pagel, K. A. et al. The influence of genetic predisposition and physical activity on risk of gestational diabetes mellitus in the nuMoM2b cohort. https://doi.org/10.1101/2022.03.08.22271868.
Momozawa, Y. & Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 66, 11–23 (2021).
Acknowledgements
We thank Tatiana Foroud and Kymberleigh Pagel for helpful discussion in the planning stage of this work, and Jelena Radivojac for contributing to the design and logistics of DNA extraction and genotyping.
Funding
Precision Health Initiative of Indiana University, National Institutes of Health award R01HD101246 to DMH and PR. Cooperative agreement funding from the National Heart, Lung, and Blood Institute and the Eunice Kennedy Shriver National Institute of Child Health and Human Development: grant U10-HL119991 to RTI International; grant U10-HL119989 to Case Western Reserve University; grants U10-HL120034 and R01LM013327 to Columbia University; grant U10-HL119990 to Indiana University; grant U10-HL120006 to the University of Pittsburgh; grant U10- HL119992 to Northwestern University; grant U10- HL120019 to the University of California, Irvine; grant U10-HL119993 to University of Pennsylvania; and grant U10-HL120018 to the University of Utah. National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health to Clinical and Translational Science Institutes at Indiana University (grant UL1TR001108) and University of California, Irvine (grant UL1TR000153).
Author information
Authors and Affiliations
Contributions
R.R.K and R.F.G. led the analytical design, analyzed data and wrote the manuscript. R.F.G., D.M.H. M.W.H, and P.R. designed and coordinated the genotyping study. D.M.H. M.W.H, I.P., and P.R. secured funding for genotyping and analysis and supervised the study. R.J.W, W.A.G., H.S., R.S., J.H.C., U.M.R., D.M.H. conceptualized the original numom2b study, secured its funding, and led the work in clinics. A.R. and A.S. contributed to initial clinical data preprocessing and provided valuable feedback on the analysis and manuscript. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, R.R., Guerrero, R.F., Wapner, R.J. et al. Genetic polymorphisms associated with adverse pregnancy outcomes in nulliparas. Sci Rep 14, 10514 (2024). https://doi.org/10.1038/s41598-024-61218-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61218-9
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
