
Abstract
According to the World Health Organization, Chagas disease (CD) is the most prevalent poverty-promoting neglected tropical disease. Alarmingly, climate change is accelerating the geographical spreading of CD causative parasite, Trypanosoma cruzi, which additionally increases infection rates. Still, CD treatment remains challenging due to a lack of safe and efficient drugs. In this work, we analyze the viability of T. cruzi Akt-like kinase (TcAkt) as drug target against CD including primary structural and functional information about a parasitic Akt protein. Nuclear Magnetic Resonance derived information in combination with Molecular Dynamics simulations offer detailed insights into structural properties of the pleckstrin homology (PH) domain of TcAkt and its binding to phosphatidylinositol phosphate ligands (PIP). Experimental data combined with Alpha Fold proposes a model for the mechanism of action of TcAkt involving a PIP-induced disruption of the intramolecular interface between the kinase and the PH domain resulting in an open conformation enabling TcAkt kinase activity. Further docking experiments reveal that TcAkt is recognized by human inhibitors PIT-1 and capivasertib, and TcAkt inhibition by UBMC-4 and UBMC-6 is achieved via binding to TcAkt kinase domain. Our in-depth structural analysis of TcAkt reveals potential sites for drug development against CD, located at activity essential regions.
Similar content being viewed by others


Introduction
CD is a potentially chronic and life-threatening disease caused by the protozoan parasite Trypanosoma cruzi, which is classified by the World Health Organization (WHO) as the most prevalent of poverty promoting neglected tropical diseases1. CD is endemic in Latin America and is gradually turning into a global health problem due to globalization and climate change2,3,4,5,6.
Parasite transmission from T. cruzi to the human host is primarily vector-borne via direct interaction with infected Triatominae bugs. Other transmission routes include oral ingestion of contaminated products, transplacental passage from mother to fetus, blood transfusions and organ transplantations from infected donors7,8. The clinical spectrum of CD is broad and potentially causes fatal chronic illness like cardiomyopathy, gastrointestinal or neurological diseases1,7,8.
The current treatment relies on two drugs developed more than five decades ago: nifurtimox and benznidazole2,9. These nitroheterocyclic drugs require long treatment periods, are ineffective in chronic stages of CD and are associated with a high prevalence of toxic side-effects leading to discontinued treatments10. Emerging resistant T. cruzi strains against both drugs also raise the demand for new molecular targets and treatment options against CD11.
Over the past few decades, many T. cruzi targets have been analyzed for drug development purposes12,13,14,15,16,17,18,19,20,21. However, until now none of those targets have led to new candidates in clinical trials yet9,22. In fact, the drug development pipeline for CD remains limited, even compared to other neglected diseases, like leishmaniasis22.
TcAkt was previously proposed as a promising target for the development of drugs against CD23,24. Akt kinases play a central role in all organisms as they are key regulators involved in the balance between growth, proliferation and apoptosis25,26. TcAkt inhibition causes apoptosis-like events in T. cruzi, confirming its essential role for the survival of the parasite23,24,27. Furthermore, TcAkt is present in all stages of the parasite life cycle—epimastigotes, trypomastigotes and amastigotes—which is crucial for effective treatment of CD. Considerable effort has been made to inhibit TcAkt based on bioinformatics and molecular docking studies23,24. However, the three-dimensional structure of the Akt-like kinase in trypanosomatids has not been determined yet, resulting in a lack of detailed information and understanding of its mechanism of action27.
Akt kinases, also called protein kinase B (PKB), contain an N-terminal pleckstrin homology (PH) domain, a catalytic (kinase) domain and a disordered, regulatory, C-terminal tail (C-tail)28. Regulation of human Akt (HsAkt), comprising of the isoforms Akt1, Akt2 and Akt3, involves the interaction of the PH and the kinase domain through the so-called ‘autoinhibitory intramolecular interface’25,26,28,29,30. In this autoinhibitory state, the PH domain is packed against the kinase domain, resulting in a closed conformation of HsAkt. In its autoinhibitory state, HsAkt is constitutively phosphorylated at T450 and primed with adenosine triphosphate (ATP) bound in a deep cleft between the N-terminal (N-lobe) and C-terminal lobe (C-lobe) of the kinase domain26,30. Upon recruitment of HsAkt to the plasma membrane, the PH domain binds to membrane-associated phosphatidylinositol-3,4,5-trisphosphate (PIP3) or phosphatidylinositol-3,4-bisphosphate (PI(3,4)P2), thereby inducing a disruption of the intramolecular interface26,31 and forming an open conformation of HsAkt30. Dislodgement of the PH domain from the kinase domain, triggered by conformational changes in loop regions of the PH domain28,32, leads to subsequent phosphorylation of S473 and T308 and induces a re-orientation of the activation loop enabling high-affinity substrate binding and, in turn, full activation of HsAkt26,30.
The active site of Akt kinases contains several structural elements that are necessary for substrate phosphorylation: the glycine-rich G-loop involved in ATP-binding, the activation loop binding the peptide substrate and the catalytic loop33,34. Precise interactions between these elements create an environment for phospho-transfer to substrates that contain the characteristic motif RXRXX(S/T)f, with f representing a large hydrophobic residue35,36,37,38,39,40,41. Phosphorylation of these targets on serine or threonine residues leads to activation or inhibition of the given protein35,36.
A crucial step in the activation represents the binding of the PH domain to membrane-associated PIPs to induce specific cellular processes42,43,44,45,46. T. cruzi expresses all necessary enzymes to synthesize PI(3,4,5)P3 as well as PI(3,4)P2, PI(3,5)P2 and PI(4,5)P244,47,48. Each of those PIP residues is associated with distinctive molecular functions: PI(3,4,5)P3 is involved in actin polymerization, cell survival and cell growth, PI(3,5)P2 plays a crucial role in homeostasis and stimuli response, whereas PI(4,5)P2 is important for cell migration, gene expression and endo-/exocytosis44. Still, the understanding of the signaling mechanisms of PIPs and involved enzymes remains elusive and needs to be investigated further44,47,48,49.
Two putative phosphorylation sites (p-sites) could be identified in T. cruzi by multiple sequence alignment (MSA), T290 in the activation loop and S450 in the hydrophobic motif (h-motif)27. While these sites have not been experimentally confirmed in T. cruzi, the phosphorylation of an equivalent threonine in Leishmania panamensis—a closely related trypanosomatid parasite—could be experimentally detected50.
The phosphorylation motif RXRXX(S/T)f is a common consensus sequence recognized by protein kinases and was detected with a 1.74% abundance in the T. cruzi phosphoproteome at the epimastigotes, a replicative form of the parasite that colonize the digestive tract of the vector37. A short version of this motif, the RXXS sequence, represents a phosphorylation motif during the differentiation process from trypomastigotes to amastigotes. It could be identified in phosphoproteins associated with processes that are highly relevant to the differentiation stimulus–response such as cell communication, cellular organization, and biogenesis51. Analysis of the T. cruzi genome exhibits several homologs to identified human Akt substrates38,39,40, thus presenting putative TcAkt targets: glycogen synthase kinase 3 GSK-3 (TritrypDB ID: TcCLB.507993.80), ribosomal protein S6 (ID: TcCLB.508277.120), Rab11 (ID: TcCLB.511407.60) and glyceraldehyde-3-phosphate dehydrogenase TcGAPDH (ID: TcCLB.506885.413)41. However, further in vitro and in vivo experiments are needed to identify and confirm putative TcAkt substrates.
In this work, we present the first experimentally determined structure of an Akt-like protein domain of a protozoan parasite. Our NMR solution structure of TcAkt-PH reveals a typical PH domain fold including a basic charged cleft. By using NMR Chemical Shift Perturbation (CSP) experiments52,53 we confirm the binding of inositol-1,3,4,5-tetraphosphate (Ins(1,3,4,5)P4), the soluble headgroup of PIP3, to TcAkt-PH inducing local conformational changes of loop regions β1-β2, β3-β4 and β6-β7, affecting its dynamics. The presented experiments reveal that phosphorylation patterns of inositol headgroups induce distinctive conformational changes either stabilizing or destabilizing loop-to-helix transitions suggesting that dynamics of loop regions of TcAkt-PH play a crucial role in regulating versatile Akt-like functionality. By combining experimental information with Alpha Fold (AF) calculations we present a model for the mechanism of action of TcAkt involving a PIP-induced disruption of the autoinhibitory intramolecular interface between the TcAkt kinase and PH domain, via bending of TcAkt-PH loop β1-β2 upon ligand interaction. The molecular docking derived structure of ATP-bound TcAkt reveals strong similarities to HsAkt unravelling activity essential residues. Further docking experiments show that human Akt inhibitors PIT-132,54 and capivasertib55,56 recognize TcAkt via similar binding modes compared to HsAkt. Additionally, based on the AF structure of TcAkt, the exact localization of the binding site of previously described TcAkt inhibitors UBMC-423 and UBMC-624 could be determined at the kinase domain of TcAkt, thus suggesting kinase-involved inhibition modes.
The presented experiments offer primary insights into the structure and function of the central protein Akt-like of a protozoan parasite and furthermore reveal potential drug target regions crucial for TcAkt function but exhibiting significant structural differences to HsAkt. The detailed understanding of the mechanism of action of TcAkt forms the basis for the development of effective drugs against the expanding CD.
Results
N-terminal TcAkt-PH forms a positively charged flexible cleft
The presented NMR solution structure of the membrane binding domain TcAkt represents to date the only experimentally determined structure of an Akt-like protein domain of a protozoan parasite. The structural determination of TcAkt-PH (11.7 kDa, 100 aa) was achieved by a combination of NMR experimental data including long range NOEs and CS-Rosetta57 (Figs. 1a, S1a, Tables S1, S2, and S3). The following structural analysis is based on the lowest energy structure of TcAkt-PH (PDB: 8OZZ) (Fig. 1b).

NMR derived structure of TcAkt-PH (PDB: 8OZZ) (a) CS-Rosetta57,58 plot: All atom energies of TcAkt-PH models with respect to their Cα-RMSD values relative to the lowest-energy model. 10 000 structures were calculated. For each structure the Cα-RMSD to the lowest-energy structure (S_07667) was calculated and plotted against the all-atom energy of each model. The run is designated as converged due to the shape of the plot and the averaged Cα-RMSD value of 1.7 Å of the final 10 structures to the lowest-energy model (S_07667). (b) Lowest energy structure as cartoon representation: TcAkt-PH forms a typical PH domain fold, built of two antiparallel β-sheets shown in blue (β1-β4, β5-β7) and a C-terminal α-helix α1 (red). Loops are shown in grey. (c) Topology and amino acid sequence of TcAkt-PH (aa 2–105).
TcAkt-PH has a typical PH domain fold46,59 consisting of a C-terminal α-helix and two antiparallel β-sheets, formed by four and three β-strands, respectively (Fig. 1b,c). All structural elements (strands β1-β7, helix α1) contribute to the formation of the hydrophobic core (Fig. S1) resulting in a stable structure with an averaged rotational correlation time of 8.68 ns (Eq. 2), as observed by NMR relaxation experiments (Fig. S2a–d). Additionally, TcAkt-PH exposes an intense network of seven surface-exposed intramolecular salt bridges, providing further stabilization of the fold (Fig. S3). Expression constructs of C-terminal truncated TcAkt-PH (aa 1–95), results in a destabilization of the structure, probably due to missing helix-stabilizing residues S96 and K97.
Calculation of the electrostatic surface potential (see Eq. 1) in combination with NMR relaxation analysis (Figs. 2b and S2a) reveals a dynamic, positively charged cleft of TcAkt-PH (Fig. 2a) gated by three flanking loops β1-β2, β3-β4 and β6-β7, which include nine basic charged residues (K11, R23, K15, K18, K19, K36, R37, R38, K76) (zoomed details in Fig. 2a).

Surface representation of electrostatic potential and flexibility of TcAkt-PH. (a) Electrostatic surface of TcAkt-PH apo-form (PDB: 8OZZ) with zoomed details of nine basic amino acids: K11 (β1); R23 (β2); K15, K18, K19 (loop β1-β2); K36, R37, R38 (loop β3-β4); K76 (loop β6-β7). Coulombic electrostatic potential (kcal/(mol.ε) at 298 K) was calculated using ChimeraX60,61,62 (see Eq. 1). Negative electrostatic potential is shown in red (min: − 14.51), positive electrostatic potential in blue (max: 23.87). (b) Transparent surface with display of hetNOE values (see Eq. 3) of TcAkt-PH apo-form: low hetNOE values are colored in cyan, high hetNOE values in purple, not assigned (n.a.) residues in white. Orientation is consistent with Fig. 1b.
Phosphoinositides bind into the basic cleft of TcAkt-PH
To analyze if TcAkt-PH binds to PIP ligands, we performed NMR CSP experiments using the soluble headgroup Ins(1,3,4,5)P4 of the proposed ligand PI(3,4,5)P3 (Figs. S4 and S5). The experiments confirmed an interaction of Ins(1,3,4,5)P4 with TcAkt-PH exhibiting a dissociation constant of 40 ± 14 µM (Table S4). NMR 1H-15N HSQC spectra of TcAkt-PH were recorded before and after the addition of increasing amounts of the ligand (Fig. S5). NMR chemical shifts are sensitive to their local chemical environment. Thereby, residues involved in direct interaction with a ligand can be detected via the degree of change of their chemical shift compared to the apo-form52. Residues directly located in the binding pocket generally exhibit a strong change of their chemical shift and/or peak intensity. Residues that do not bind to the ligand directly but experience ligand-induced conformational changes, can also be detected via CSP. For each amino acid of TcAkt-PH, the Euclidean distance or d-value was calculated (Eq. 4) (Fig. 3a,b), defined as combined value of 15N- and 1H-shifts, thus representing the degree of change of the chemical shift of a specific residue upon ligand addition and its involvement in the ligand interaction (Fig. 3a,b).

TcAkt-PH PIP binding site evaluation by NMR and MD simulations. (a) Chemical shift perturbations of TcAkt-PH. The graph includes calculated d-values (see Eq. 4) for each amino acid of TcAkt-PH. (b) Surface display of TcAkt-PH colored by determined d-values according to a gradient. Residues exhibiting high d-values are shown in red (max: 0.69), low d-values in light yellow (min: 0.01), not affected residues below threshold (< 0.008) in white, n.a. residues (due to overlap or missing peaks) are shown in grey. (c) MD derived structure of TcAkt-PH interacting with Ins(1,3,4,5)P4 (a frame was carefully chosen from the last 1 µs MD simulation to showcase the interacting residues adequately): Interacting residues are shown in sticks, contacts in dashed yellow lines, phosphate groups in red (ball representation). (d) H-bonds, ionic interactions, and water bridges plotted as interaction fractions for each interacting residue: Bar charts are normalized over the course of the trajectory. A value of 1 indicates that the respective interaction type persists throughout 100% of the simulation time, while a value greater than 1 signifies multiple instances of the same interaction type between corresponding residue and ligand over the simulation duration (e.g., multiple hydrogen bonds between arginine and the ligand). Additional details on the computed interactions (H-bonds, ionic interactions, and water bridges) can be found in the supplementary information (SI Section 1 ‘Detailed description for computed protein–ligand interactions of TcAkt-PH and Ins(1,3,4,5)P4 shown in Fig. 3d’).
The presented experimental data reveals that ligand-induced conformational changes affect mainly residues in or at close proximity to the positively charged cavity of TcAkt-PH including loops β1‑β2, β3‑β4, and β6‑β7, thus facilitating the required opposite charge for interactions with phosphate groups of Ins(1,3,4,5)P4 (Figs. 2a and 3a,b). In detail, high d-values could be determined for strands β1, β2 and connecting β1-β2 loop involving residues: T12, G13, G14, F16, Y17, N20, Q21 and T22 (Fig. 3a). Significant chemical shift changes could be furthermore detected for residues located at strands β3, β4 and loop β3-β4 (Y34, R38, T47) as well as strands β6, β7 and loop β6-β7 (S66-T81). Residues of loop β4-β5 (A55, R56) are probably affected indirectly by the ligand interaction due to conformational changes since their location is spatially distant from the binding site. Chemical shift changes of basic residues K11, K15, K18, K19 and R23, located in strands β1, β2 and loop β1-β2, could not be analyzed in the 1H-15N HSQC spectrum due to missing signals or spectral overlap. Nevertheless, surrounding residues are highly influenced by ligand binding (Fig. 3a), thus suggesting that the mentioned basic amino acids play a crucial role in the interaction with Ins(1,3,4,5)P4.
Subsequent Molecular Dynamics (MD) simulations in combination with NMR experimental data reveal that the nature of the protein–ligand interactions between TcAkt-PH and Ins(1,3,4,5)P4 is strictly polar including H-bonds, ionic interactions and water bridges (Fig. 3d, see SI Section 1, Fig. S7). Except for R38 and K19, all basic charged residues located in the positively charged cleft are interacting with the ligand (Figs. 2a and S6). Additionally, N20 has profound interactions with Ins(1,3,4,5)P4, as well as Y34 and T47, which form water-mediated interactions (Fig. 3c,d). Interestingly, N20 faces away from the binding site in the apo-form of TcAkt but when bound to PIP3, N20 rotates towards the ligand, enabling interactions with PIP phosphate groups (Fig. 3b,c).
TcAkt PH domain undergoes local conformational changes while hosting Ins(1,3,4,5)P4
For analyzing the impact of ligand interaction on TcAkt-PH dynamics, {1H}-15N heteronuclear NOE (hetNOE) experiments were recorded before and after Ins(1,3,4,5)P4 addition (Fig. 4a). As shown in Fig. 4b, ligand binding induces a slight increase of rigidity of residues located at the binding site (loop regions β1-β2 and β6-β7) or close to the binding cleft of TcAkt-PH (strand β3 and loop β3-β4), whereas the dynamics of the rest of the protein are not affected (Fig. 4b,c).

Conformational and dynamic changes of TcAkt-PH initiated by Ins(1,3,4,5)P4 binding. (a) Dynamic analysis of apo TcAkt-PH and Ins(1,3,4,5)P4 bound TcAkt-PH. The hetNOE values (see Eq. 3) of residues of apo TcAkt-PH are shown as black spheres. The hetNOE values of residues of TcAkt-PH bound to Ins(1,3,4,5)P4 are shown as white spheres. TcAkt-PH secondary structure elements are shown above the graph: β-strand forming residues are marked as blue squares, helix forming residues are represented as red spheres. (b) Structure of TcAkt-PH colored according to determined hetNOE values of TcAkt-PH bound to Ins(1,3,4,5)P4: low hetNOE values are shown in cyan (min: 0.28), high hetNOE values in purple (max: 0.90), n.a. in white. Orientation of TcAkt-PH is consistent with Fig. 1b. (c) Structural superimposition of TcAkt-PH apo-form (yellow loops) and TcAkt-PH bound to Ins(1,3,4,5)P4 (green loops, light green 310 helix).
As shown by MD simulations the increase of rigidity observed by NMR can be explained by ligand-induced loop-to-helix transitions. The apo-form of TcAkt-PH already reveals a tendency for loop-to-helix transitions (Figs. 5a, S8a, and S9). However, binding of Ins(1,3,4,5)P4 further stabilizes mentioned conformational changes for a more extended period (Figs. 5a, S8b, and S9).

Conformational changes of TcAkt-PH loop regions induced by binding of different InsP ligands. (a) Table of loop-to-helix transitions of TcAkt-PH interacting with Ins(1,3,4,5)P4, Ins(1,3,4)P3, Ins(1,4,5)P3 or Ins(1,3,5)P3. As determined by NMR experiments, strongest binder Ins(1,3,4,5)P4, stabilizes loop-to-π-helix transitions in stretches 15–17 and 46–48 and simultaneously destabilizes region 41–43. Ins(1,3,5)P3 reduces the π-helix tendency of stretch 15–17 but induces π-helix formation in regions 41–43 and 52–54. Ligands Ins(1,3,4)P3 and Ins(1,4,5)P3 induce less conformational changes compared to Ins(1,3,4,5)P4 and Ins(1,3,5)P3, showing a significant impact mainly in region 15–17 of TcAkt-PH. Ins(1,4,5)P3 stabilizes a π-helix in 13–15, similar to Ins(1,3,4,5)P4, whereas Ins(1,3,4)P3 represents the only ligand that induces a 310-helix formation in stretch 15–17. (b-d) MD determined protein–ligand structures of TcAkt-PH interacting with (b) Ins(1,3,4)P3, (c) Ins(1,4,5)P3 and (d) Ins(1,3,5)P3: Interacting residues are shown in sticks, contacts in dashed yellow lines, phosphate groups in red (ball representation). The presented frames were carefully chosen from the last 1 µs MD simulation to showcase the interacting residues adequately.
Although regions 41–43 (SGP) and 46–48 (PTG) are both located in loop β3‑β4, Ins(1,3,4,5)P4 interaction seems to have different effects on the dynamics and conformations of the stretches. Stretch 41–34 is more solvent exposed and experiences an increase of flexibility upon ligand interaction (Figs. 4c and S9) whereas stretch 46–48 is closer to strand β4 and its helical tendency is stabilized when Ins(1,3,4,5)P4 is bound (Fig. S9).
Eventually, Ins(1,3,4,5)P4 pulls the loops β3‑β4 and β6‑β7 towards the binding site compared to their respective conformations in the apo-form (Fig. 4c). Consequently, loop β1‑β2 changes into a more open conformation upon Ins(1,3,4,5)P4 binding, compared to the compact, apo-form (Fig. 4c).
Phosphorylation patterns of PIP ligands induce different conformational changes of TcAkt-PH
As previously stated, PIPs act as signaling molecules involved in numerous cellular pathways depending on the position of phosphate groups on their inositol ring guiding broad Akt functionality42,43,44.
To monitor the binding behavior of different PIP ligands to TcAkt-PH, MD simulations were performed using inositol phosphates (InsP): Ins(1,3,4,5)P4, Ins(1,3,4)P3, Ins(1,3,5)P3 and Ins(1,4,5)P3 (Fig. S4). According to MD data, all InsP ligands bind in the same cavity of TcAkt-PH and remain bound throughout the 2 µs MD run (Figs. 3c and 5b–d), exhibiting RMSD values less than 1.5 Å (Fig. S10b) which indicates a stable interaction63. Ins(1,3,4,5)P4 shows the strongest binding due to an additional phosphate group. Similar interaction energies IE (Fig. S10a) could be calculated for other InsP ligands.
Although tested ligands bind into the same cavity of TcAkt-PH, ligand-induced conformational changes of TcAkt-PH loop regions are versatile and complex as shown in detail in Fig. 5a (Figs. S8 and S9). Phosphorylation patterns of PIP headgroups impact distinctive conformational changes of TcAkt loops β1-β2, β3-β4 and β4-β5, resulting in ligand-specific structural rearrangements of the PH domain, which subsequently could influence the versatile functionalities of TcAkt43,44. Still, all of the tested ligands induce a similar bending of loop β1-β2 when binding to TcAkt-PH, independent on their phosphorylation pattern (Fig. S11).
TcAkt-PH prefers PIP ligands with P3 and P5 phosphorylations
In humans, a binding preference of Akt1 PH domain to PI(3,4,5)P3 and PI(3,4)P226,32,42,64 was determined by monitoring its intrinsic tryptophane fluorescence upon binding to different inositol phosphates65, as well as by competitive HPLC binding experiments of 32P-labeled phosphoinositides66. To analyze TcAkt binding preferences, we calculated the solvent-accessible surface area (SASA) and the buried surface area (BSA) of each of the tested ligands when bound to TcAkt-PH (Fig. S10d). The more buried a ligand is in the binding site, the larger its BSA will be. The extent of BSA increase is an important descriptor of ligand binding67,68 and can therefore be used for analyzing the impact of different phosphorylation patterns of PIP ligands on the interaction with TcAkt-PH.
The calculated averaged BSA (Fig. S10d) shows no significant differences between all InsPs, reflecting that the depth of the binding pocket remains consistent and is equally accessible to all InsPs irrespective of the number and relative positions of phosphate groups on the inositol ring. However, when analyzing each phosphate position individually, different BSA values could be observed (Fig. S10c). Irrespective of the type of InsP, the P1 phosphate group has the lowest BSA, meaning that it is more exposed to the solvent relative to other phosphate positions. In the membrane-bound PIP ligand, the fatty acid tail is attached on phosphate group P1 and is therefore oriented towards the membrane rather than the binding site and does not seem to form specific interactions with TcAkt.
In comparison, the P3 phosphorylation has the highest BSA values in all InsPs. For Ins(1,4,5)P3, which lacks the P3 phosphate group, P5 seems to compensate for P3 with comparable BSA values. Thus, we propose a preference of TcAkt-PH for PIP ligands containing P3 and/or P5 phosphorylations.
As previously shown, MD data reveals that all PIP ligands induce a bending of loop β1-β2 (Fig. S11). Nevertheless, different phosphorylation patterns of PIP ligands initiate different local conformational changes of TcAkt and also vary in binding behavior, thus supporting a ligand specificity of TcAkt. In contrast to HsAkt1, which favors P3 and P4 phosphorylations of PIP26,32,42,64, TcAkt shows a preference for P3 and P5 phosphate groups.
The kinase domain of TcAkt contains conserved motifs essential for Akt activity
Since structure determination of full-length TcAkt by X-ray crystallography was not successful, we used AF69 for calculating the structure of TcAkt (Figs. 6 and S12). According to internal quality parameters, the calculation runs were designated as successful (see Materials and Methods section ‘AF calculations’).

AF derived model of full-length TcAkt. (a) Schematic representation of TcAkt domains: TcAkt consists of an N-terminal PH-domain (aa 1–100) (turquoise), a flexible linker (aa 100–134) (grey), a kinase domain divided in a N-lobe and C-lobe (aa 134–395) (sand) and a C-tail (aa 395–458) (lavender). Two putative p-sites are shown in red: T290, S450. (b) Cartoon representation of full-length TcAkt model. Same color scheme as in (a).
Crucial residues in regions associated with Akt activation are typically highly conserved among kinases and are also present in the AF TcAkt structure (Figs. 6 and S14): The glycine-rich G-loop in the kinase C-lobe is crucial for ATP binding and was stated to have the consensus sequence GXGXΦG, with Φ as hydrophobic residue33,34,56. While this is consistent with the G-loop in HsAkt1, in TcAkt the third glycine within this sequence is replaced by a serine (Fig. S14), an exception that is also found in other protein kinases70,71. The catalytic loop, also present in the C-lobe, is responsible for the phospho-transfer from ATP to the substrate and carries a conserved aspartic acid (in TcAkt D257) that interacts directly with the target S/T p-site (Hs: T308, Tc: T290) (Figs. S13 and S14)27,34,56. The activation loop starts with the highly conserved DFG motif and ends with the APE motif33,34, whereas the crucial threonine p-site (in T. cruzi T290) is located in between (Fig. S14). The h-motif is part of the C-tail and usually contains the sequence FXXF(S/T)(Y/F)27 and a p-site. TcAkt shows a shortened version of the h-motif (FSF) including the putative p-site S450 (Figs. S13 and S14). Other p-sites in the C-tail that were linked to HsAkt1 activation (Hs: S477, T479) are absent in TcAkt25,28,29.

ATP bound to TcAkt and HsAkt2 kinase domains. TcAkt kinase domain (dark grey) bound to ATP (orange sticks) was calculated with RoseTTAFold72. HsAkt2 kinase domain (light grey) was complexed with ATP analog, AMP-PnP (not shown) (PDB: 1O6K)73,74 (all-atom RMSD of aligned structures 0.883 Å). Zoomed details show ATP binding site. TcAkt residues are shown in dark sticks and HsAkt residues are shown in light sticks. Colors refer to specific regions of the kinase domain: Catalytic spine residues (turquoise), shell residues (red), DFG motif (blue), C-tail residues (yellow), G-loop (green), other residues interacting with ATP (grey).
Full-length TcAkt retains kinase activity and possesses a similar ADP/ATP binding pocket compared to human Akt
To evaluate functional properties in a full-length context, we expressed and purified recombinant TcAkt (Fig. S15a) and tested its enzymatic activity in vitro using a polyclonal antibody that recognizes its phosphorylated form. The synthetic peptide RPRAATF was used as a substrate. As shown in Fig. S15b, TcAkt efficiently phosphorylates the threonine residue of the peptide substrate as did HsAkt3, which was used as a positive control. This indicates that TcAkt is capable of recognizing the consensus recognition motif RXRXX(S/T)f associated with Akt-mediated phosphorylation35,36,37.
Based on the presented AF structure, an ATP bound model of TcAkt was calculated using the recent release of RoseTTAFold All-Atom72. The protein–ligand model proposes that TcAkt binds ATP in a similar manner compared to HsAkt2 in complex with AMP-PnP, an ATP analog, and Mn2+ (PDB: 1O6K) (Fig. 7), thus emphasizing a strong structural and sequential conservation in this region. Figure 7 shows the ATP binding site of TcAkt kinase domain with involvement of residues from the G-loop, the C-tail, the DFG motif, catalytic spine residues and shell residues56,75,76. According to Kornev et al.76 catalytic spine residues of protein kinases, as well as the DFG motif, are essential for positioning ATP and Mn2+, while shell residues were identified to have a stabilizing effect77 (Fig. 7).
Differential scanning fluorimetry (DSF) experiments revealed an increased melting temperature (Tm) upon ADP and ATP binding only in the presence of Mn2+ (Figs. S16 and S17). These findings match observations reported by Pascuccelli et al.71, showing that TcAkt requires Mn2+ to phosphorylate substrates in vitro, but is not dependent on Mg2+ (Figs. S16 and S17). The Mn2+ binding residues in the crystal structure of HsAkt2 (N293 and D280) align with the corresponding residues in TcAkt (N262 and D275), confirming a conserved binding mechanism of TcAkt and HsAkt (see SI Section 2).
Interdomain interface of TcAkt is established via hydrophobic and aromatic interactions
The interface between the kinase and the PH domain represents a favorable target for the inactivation of HsAkt1 due to its autoinhibitory functionality25,26,28,29,30. To analyze TcAkt potential as drug target against CD, a detailed examination of this region is therefore of high interest.
The AF model of full-length TcAkt is present in a closed conformation forming interdomain interactions between the TcAkt-PH and the kinase domain (Figs. 6 and 8c). The interface between loop β1-β2 of TcAkt-PH and the C-lobe of the TcAkt- kinase domain is constructed from a network of non-bonded interactions (33 contacts between 20 residues) (Figs. 9d and S18) and has a surface area of 412–460 Å2. The tip of loop β1-β2 of TcAkt-PH carries two aromatic residues, F16 and Y17, which contribute to the hydrophobic aromatic cluster of the kinase domain built of F289, F291, F302 and Y340 (Figs. 8a, S14, and S19). F16 interacts directly with F291 and Y340, and additionally forms a weak π-stacking interaction with Y340 (ring-to-ring distance 4.2 Å) (Fig. S19). The involvement of aromatic residues in the formation of the interdomain interface between loop β1-β2 of the PH domain and the C-lobe of the kinase domain is also observed in HsAkt1 (Figs. 9c and S19).

Proposed model of TcAkt activation via disruption of its autoinhibitory interface upon PIP-binding. TcAkt PH domain is shown in turquoise, TcAkt kinase domain in sand and the C-tail in lavender. (a) Interdomain contacts between the hydrophobic tip of the PH domain (sticks colored in turquoise) and the kinase domain (sticks colored in sand) are shown as zoomed detail. Phosphorylation site T290 is colored in red. Conserved residues are marked with an asterisk (*). (b) Ligand induced bending of TcAkt loop β1-β2. Superposition of TcAkt apo-form (a) and PIP-bound TcAkt determined by MD simulations (pink). Red arrow indicates conformational changes upon binding to PIPs. (c) TcAkt is present in a closed conformation (inactive) and gets recruited to the membrane. The PH domain and the kinase domain are interacting via an autoinhibitory interface. (d) The binding of the TcAkt PH domain to PIP molecules in the membrane induces conformational changes of the hydrophobic tip (F16, Y17) of the PH domain loop β1-β2, leading to an opening of the structure and the disruption of the intramolecular autoinhibitory interface including a surface exposure of otherwise buried phosphorylation site T290.

Structural analysis of activity essential regions of TcAkt and HsAkt1. Due to high differences in the residual arrangements the structures are not superimposed. HsAkt PH domain is shown in green, HsAkt kinase domain in blue and the C-tail in grey. TcAkt PH domain is shown in turquoise, TcAkt kinase domain in sand and the C-tail in lavender. Flexible interdomain linkers are shown in light grey. PIP binding site: (a) PIP binding site of HsAkt1: PIP interacting residues are shown as green sticks (derived from the crystal structure of HsAkt1-PH in complex with Ins(1,3,4,5)P4 ;PDB: 1UNQ) and presented on the apo structure (PDB: 1UNP) for comparative reasons. (b) PIP binding site of TcAkt: PIP interacting residues are shown as turquoise sticks (derived from NMR analysis and MD simulations) and presented on the apo structure (PDB: 8OZZ). Intramolecular interface: (c) Intramolecular interface of HsAkt1 as zoomed detail from full-length HsAkt1 (PDB: 7APJ): Involved residues of the kinase (HsAkt1-K: blue) and the PH domain (HsAkt1-PH: green) are shown as sticks. (d) Intramolecular interface of TcAkt as zoomed detail from full-length TcAkt (AF model): Involved residues of the kinase (HsAkt1-K: sand) and the PH domain (HsAkt1-PH: turquoise) are shown as sticks. Conserved residues are marked with an asterisk (*).
Model of PH domain-mediated Akt autoinhibition in T. cruzi
In HsAkt1 PIPs recruit the PH domain to the membrane by direct interactions, thus disrupting the autoinhibitory interface between the kinase and the PH domain25,26,28,29,30. This conformational rearrangement exposes the activation loop and the h-motif of the kinase domain, thereby enabling phosphorylation of T308 and S473 and activation of Akt26,30. In the absence of PIPs, HsAkt1 stays in its inactive closed conformation, maintaining the autoinhibitory interface between the kinase and the PH domain78.
We propose a similar mechanism of action for TcAkt. As shown in the presented AF model, hydrophobic interdomain interactions between the activation loop of the kinase domain and loop β1-β2 of the PH domain, cause a closed conformation of TcAkt (Fig. 8a,c). In this conformation, the putative phosphorylation site of TcAkt T29027 is not accessible, as it is shielded by the PH domain (Figs. 8a and S13). Binding of the TcAkt-PH domain to a PIP headgroup induces a bending of loop β1-β2, as observed by MD simulations (Fig. S11). An opening of loop β1-β2 involves a change of the position of residue F16, which is thus pulled away from the interdomain interface as shown in Fig. 8b. As a consequence, the hydrophobic cluster of the kinase domain needs to be rearranged to shield the non-polar residues from the solvent exposure. This rearrangement of the region around the activation loop, including T290, could lead to an exposed position of T290, thereby enabling its phosphorylation and consequently the activity enhancement of TcAkt (Fig. 8b). The described disruption of the interface is accelerated as residues K76, T47, K15 and K36, previously involved in the interface, are switching interaction partners upon PIP contact (Fig. 8b,d). The disruption of the autoinhibitory interface consequently leads to an open conformation (Fig. 8d) that is associated with activation of Akt.
The interdomain interface of TcAkt differs from HsAkt
For evaluation of TcAkt’s potential as drug target against CD, it is inevitable to characterize similarities and differences to its human ortholog in order to highlight putative regions for the inhibition of TcAkt. The following analysis concentrates on two activity essential regions: the autoinhibitory interface and the PIP binding site, both revealing clear structural and sequential differences between TcAkt and HsAkt1 as shown in Fig. 9.
Overall, the presented AF model of TcAkt is in good alignment with the crystal structure of HsAkt1 (PDB: 7APJ) and the HsAkt1 AF model (AF-DB: AF-P31749-F1), where loop regions of the kinase domain are visible25 (Fig. S12a,b). In contrast to the kinase domains which usually share a high conservation, the sequences of PH domains are generally more diverse among different species45. The sequence similarity (SS) of full-length sequences of TcAkt and HsAkt1 is 52.6%, while kinase domains share a 64.4% SS and PH domains have a SS of only 36.2% (Fig. S21).
As previously described, the interdomain interface of TcAkt represents a promising target for Akt inhibition. As shown in Fig. 9, the structural built of the interdomain interface differs significantly between HsAkt and TcAkt. Despite the distinctive structural arrangements, the interdomain interface of TcAkt and HsAkt1 is basically constituted via a hydrophobic interface, including an aromatic cluster (Figs. 9c,d and S19). In both organisms, the hydrophobic tip of loop β1-β2 of the PH domain interacts with hydrophobic residues of the kinase domain (Fig. 9c,d). In TcAkt, the hydrophobic tip of β1-β2 in TcAkt is formed by residues F16 and Y17, compared to residues Y18 and I19 in HsAkt1. In HsAkt1, Y18 was stated to form a π-stacking interaction with F309 of the kinase domain25,29. In the presented AF model of TcAkt, a weak π-stacking interaction is established between F16 and Y340. In HsAkt1, functional relevant residues D323 and D325, as well as E17, have been described, which are involved in the interdomain interface29. Mentioned residues are not present in TcAkt. In HsAkt1 D323 and D325 form interfacial contacts to the PH domain (loop β6-β7, loop β3-β4, strands β1 and β2) and mutations result in HsAkt1 hyperphosphorylation29. HsAkt1 E17 forms a salt bridge with R86 stabilizing the autoinhibitory interface29. Oncogenic HsAkt1 mutant E17K results in enhanced membrane binding29. In TcAkt, the interdomain interface is not stabilized by salt bridges. TcAkt contains K15, at a similar position to HsAkt1 E17, but forming a hydrogen bond with F291.
The PIP3 binding site of TcAkt is structurally different to HsAkt but is recognized by human PIP3 competitor PIT-1 due to its basic charge
A putative region for TcAkt inhibition represents the PIP binding site which regulates its activity and guides Akt function42,43,44. Despite two conserved basic residues (Tc: K11, R23; Hs: K14, R25), the binding clefts of HsAkt1-PH and TcAkt-PH are structurally and sequentially diverse as described in Fig. 9. The proposed consensus sequence KXn(K/R)XR located in loop β1-β2 and strand β2 to predict interactions with PIPs phosphorylated at position 3, matches with HsAkt1 but not with TcAkt42,46 (Fig. S14). Despite the differences in the amino acid composition of the binding site, the binding mode to PIP ligands is also established in distinctive manners. In mammalian PH domains mainly strands β1 and β2 (including loop β1‑β2) provide primary contact sites to PIP ligands45,46, involving also hydrophobic residues (loop β1-β2: Y18, I19) (PDB: 1UNQ) (Fig. 9a). In contrast, NMR and MD experiments performed with TcAkt-PH reveal strictly polar protein–ligand interactions and furthermore show that besides strands β1 and β2, also loops β3‑β4 and β6‑β7 are involved in PIP interaction (Figs. 3, 4, and 5). Interestingly, according to MSA analysis, the PIP binding site also differs among closely related Trypanosoma and Leishmania species (see SI Section 3, Figs. 10, S22, and S23) proposing that PIP binding mechanisms are species-specific.

MSA derived conserved regions of Akt-like proteins in Trypanosoma and Leishmania species highlighted on full-length TcAkt (MSA: Figs. S22 and S23). Conserved residues are shown in red (in MSA residues are marked with an asterisk (*)), residues which have similar properties are colored in sand (in MSA residues marked with dots (:,.)), non-conserved residues are shown in grey. Residues involved in PIP interaction, are shown as sticks. (a) Conserved regions of Akt-like proteins among Trypanosoma spp. (Fig. S22) (b) Conserved residues in PIP binding site amongTrypanosoma spp. (c) Conserved regions of Akt-like proteins among Trypanosoma and Leishmania spp. (Fig. S23) (d) Conserved residues in PIP binding sites of Trypanosoma spp. and Leishmania spp. Further details are described in SI Section 3.
PIT-1 is a non-phosphoinositide small molecule antagonist of PIP3 that shows inhibition of Akt without affecting other PIP2-selective PH domains32,54. The PIT-1 binding site of human Akt overlaps with the PIP interaction area, involving residues W22, Y26, and N54 (N54 interacts with the phenyl group of PIT-1, W22 and Y26 interact with nitro-group of PIT-1)54. Using the presented AF model of TcAkt, we initiated docking experiments with human PIP3 competitor PIT-1 revealing that the inhibitor binds to human and trypanosomal Akt in a similar manner (Fig. 11, see SI Section 4 ‘Docking studies of TcAkt and human Akt inhibitors capivasertib and PIT-1’). The overall basic charge of the PIP3 binding pocket seems to compensate for the sequential and structural differences of the PIP3 interaction sites of human and trypanosomal Akt.

Docking studies using human PIP3 competitor PIT-1 with HsAkt and TcAkt. Docking derived model of PIT-1 bound to (a) HsAkt-PH and (b) TcAkt-PH. 2D representation of interacting residues within 4 Å to PIT-1 of (c) HsAkt and (d) TcAkt. Basic charged residues enable interaction with PIT-1. In HsAkt residues K14, R23, and R86 are involved in ionic interactions with PIT-1. In TcAkt PIT-1 residues K11, K18 and K76 enable PIT-1 interaction.
General Akt inhibitor capivasertib binds to TcAkt
One of the best-studied examples of ATP-competitive Akt inhibitors is capivasertib55. Capivasertib is a pyrrolo[2,3-d]pyrimidine derivative that acts as a pan-Akt inhibitor, inhibiting all human Akt isoforms, by binding into the ATP binding site. Only recently, capivasertib was approved by the FDA for breast cancer treatment, being the first ATP-competitive Akt inhibitor on the market55,56. Based on the calculated AF structure of TcAkt, we generated protein–ligand models of capivasertib to human and trypanosomal Akt using molecular docking approaches (Fig. 12, see SI Section 4 ‘Docking studies of TcAkt and human Akt inhibitors capivasertib and PIT-1’). Although binding sites are similar between the proteins, TcAkt reveals a strong stereospecifity for (S)-capivasertib over (R)-capivasertib, which is not observed to that extent for HsAkt (see SI Section 4). Whereas in HsAkt E234, A230 and E228 are most profound residues for interaction with (S)-capivasertib and (R)-capivasertib, in TcAkt E218, L214 and D212 are responsible for the inhibitor’s binding (Fig. 12). Further experiments may be needed for evaluating the inhibitory potential of capivasertib on TcAkt activity.

Docking studies with human Akt inhibitor capivasertib and HsAkt and TcAkt. Docking derived model of capivasertib bound to (a) HsAkt-K and (b) TcAkt-K. 2D representation of interacting residues within 4 Å of capivasertib of (c) HsAkt-K and (d) TcAkt-K. (see SI Section 4 ‘Docking studies of TcAkt and human Akt inhibitors capivasertib and PIT-1’).
TcAkt inhibition by UBMC-4 and UBMC-6 involves the kinase domain
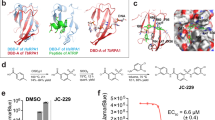
To date, two TcAkt inhibitors UBMC-423 and UBMC-624 have been defined, both detected via virtual screening and molecular docking approaches. Nevertheless, the lack of structural information restricts a detailed understanding of the inhibitory mechanisms. The presented AF-model enables the determination of the binding location of previously described TcAkt inhibitors, thus offering insights into their mechanism of action (Fig. 13).

TcAkt inhibitors UBMC-4 and UBMC-6 binding sites mapped on the AF model of TcAkt. TcAkt PH domain is shown in turquoise, the flexible linker in grey, the TcAkt kinase domain in sand and the C-tail (including the h-motif) in lavender. P-sites are colored in red and shown as sticks. Ligand interacting residues of TcAkt are shown in green and presented as sticks. (a) UBMC-4 binding site determined by Bustamante et al.23 (b) UBMC-6 binding site determined by Ochoa et al.24.
UBMC-4 has been recently described as a potent TcAkt inhibitor in T. cruzi cell structures, targeting vital cellular processes and resulting in various severe effects, including apoptosis-like events23. It furthermore reveals relatively low cytotoxicity on human cell lines (LC50 > 40 μM) and effective absorption in mice models23. TcAkt inhibitor UBMC-4 was assumed to bind to the PH domain according to published MD studies23. Nevertheless, combining published data with the presented AF model, localizes the UBMC-4 binding region at the linker region between the PH and the kinase domain (R103, T108) and at the kinase domain (L131, D132, T203, K204, F435) (Fig. 13). The binding position of UBMC-4 suggests that inhibition of TcAkt is achieved by restricting the flexibility of the interdomain linker region und thereby preventing PIP induced conformational changes as well as an opening of TcAkt.
UBMC-6 represents another putative TcAkt inhibitor, having an inhibitory effect on T. cruzi amastigotes and was also tested for its toxicity on human monocyte-derived macrophages24,79. According to the presented AF structure, UBMC-6 binds close to the h-motif of TcAkt and the p-sites T290 and S450 (K128, V129, S130, L131, D132, F201, T203, K205, P431, F435, E437), an important region to regulate Akt stability80. Likely, UBMC-6 binding blocks the activity essential h-motif and may lead to Akt protein degradation.
Discussion
The presented NMR structure, in combination with interaction experiments, MD simulations, Molecular Docking and AF-calculations, provides primary insights into the structure and function of a putative target against CD: TcAkt. The experiments reveal a PIP binding area, including ligand-induced versatile conformational changes of TcAkt regions, which likely guide the broad functionality of this central protein. The experimentally derived information combined with a full-length model of TcAkt and sequential analysis exposes activity essential regions of the proteins, leading to a proposed model of TcAkt activation via PIP-induced disruption of its interdomain interface. Detailed structural information reveil activity crucial regions of TcAkt with clear structural differences to the human ortholog HsAkt1 and thus highlights putative regions for targeting TcAkt inhibition.
Conformational changes induced by PIP ligands depend on phosphorylation patterns and play a crucial role in Akt activity
TcAkt binds PIP headgroups differing in phosphorylation of the inositol ring in the same binding pocket with comparable binding affinities but with clear preferences for P3 and P5 phosphorylations (Fig. S10). Phosporylation patterns of PIP headgroup inositol rings furthermore induce distinctive patterns of conformational changes, mainly regarding loop regions β1-β2, β3-β4 and β4-β5 of TcAkt-PH (Figs. 5a, S8, and S9). Nevertheless, all tested PIP headgroups result in an opening of loop β1-β2 upon binding to the PH domain, potentially inducing the disruption of the autoinhibitory interface (Fig. 8b).
In HsAkt1, activation is strictly limited to lipid second messenger molecules PI(3,4,5)P3 and PI(3,4)P2, revealing a different ligand specificity compared to TcAkt26,32,42,64,65,81,82. In HsAkt1, Ins(1,3,4,5)P4 induces a loop-to-helix transition of residues 44–46 (DVD) located in loop β3-β432,81,83. Destabilization of the helical formation by mutagenesis (DVD to GPG) consequently resulted in poor kinase activity of S473-phosphorylated Akt, suggesting that dynamics of the DVD motif are essential for the displacement of the PH domain from the kinase domain in response to pS47328.
Similarly, Ins(1,3,4,5)P4 also stabilizes a loop-to-helix transition of residues 41–43 (SGP) in loop β3-β4 of TcAkt (Figs. 5a, S8, and S9). Other tested ligands did not induce these conformational changes. Three of the four tested ligands result in helical conformations of region 15–17 (KFY) located at loop β1‑β2 at the direct binding site of PIP, whereas only Ins(1,3,5)P3 destabilizes the helix transition. Destabilizing effects of Ins(1,3,5)P3 lead to increased flexibility, which could enable interactions with other molecules or regulate functionality84. Nevertheless, further experiments are needed to determine the exact effect of local conformational changes on the activity of TcAkt.
The presented experiments show a complex pattern of diverse stabilizing or destabilizing effects of PIP ligands on TcAkt, proposing, similarly to HsAkt1, that the dynamics of loop regions of the PH domain are crucial for the functionality of TcAkt. Targeting the PIP-affected loop regions via binding of a ligand could offer another possibility for TcAkt inactivation by preventing subsequent structural changes essential for activity.
Potential strategies for TcAkt inhibition
HsAkt has undergone extensive investigation as a therapeutic target in oncology85 and more recently it has been studied as drug target for treatment of cardiovascular diseases86, metabolic syndrome54, Parkinson’s disease87 and schistosomiasis, caused by the parasite Schistosoma mansoni88. To inhibit Akt kinases, several inhibitors have been developed based on various approaches, which potentially guide the way for TcAkt inhibition.
ATP-competitive inhibitors target the active site in the open conformation of Akt kinases89,90,91,92. However, a high similarity of the ATP-binding site in TcAkt, HsAkt1 and potentially other AGC family kinases needs to be considered (Fig. 7)93,94. Presented docking studies could confirm that ATP-competitive inhibitor capivasertib recognizes human and trypanosomal Akt via similar binding areas (Fig. 12, SI Section 4).
Another target region for Akt inhibition represents the PIP-binding site of the PH domain. Small molecule antagonists of PIP3 showed activity against PIP3-dependent PI3K/Akt signaling54 supporting that this represents a promising region for Akt inhibition. Although the structure and sequence of the PIP binding region seems to be species-specific (Figs. 5, 9, 10, and 11), the general basic charge of this area may compensate for structural differences as shown by docking experiments with human PIP3 competitor PIT-1 and human Akt and TcAkt (Fig. 11, SI Section 4).
Allosteric inhibitors bind to the inactive closed conformation of Akt by stabilizing the PH-kinase domain interface thus preventing Akt recruitment to the membrane and consequent Akt activation89,95. By targeting both domains, allosteric inhibitors reveal a high selectivity also among Akt isoforms96,97,98. Exploiting the presented significant differences between HsAkt1 and TcAkt interdomain interfaces (Fig. 9), this class holds potential for the development of TcAkt-specific inhibitors.
Conclusion
In 2019 the WHO established a ‘World Chagas Disease Day’ on 14th of April in order to raise awareness for this neglected tropical disease and its global spread due to factors like climate change. The current lack of safe and efficient treatment in combination with the rising drug resistance of causative protozoan parasite T. cruzi, emphasizes the need for new strategies in order to fight this potentially chronic disease. This work offers a detailed analysis of a central protein of T. cruzi (Akt-like) as potential drug target against CD and includes atomic resolution data of activity essential regions, thereby forming the basis for structure-based rational drug design.
Materials and methods
Chemicals
All reagents were purchased in analytical grade. Ammonium chloride (15N, 99%) 98% (CAS 39466-62-1) was purchased from Eurisotop. D-Glucose (U-13C6, 99%) 98% (CAS 110187-42-3) was purchased from Cambridge Isotope Laboratories, Inc. Inositol 1,3,4,5-tetrakisphosphate > 95% (CAS 210488-61-2) was purchased from Echelon Biosciences Inc.
Constructs
The gene encoding TcAkt (TriTrypDB: TcCLB.509047.110, NCBI GeneID: Tc00.1047053509047.110) (1374 bp) originates from the T. cruzi CL Brener strain. The sequence is coding for full-length TcAkt (52.1 kDa). The TcAkt (aa 1–458) and TcAkt-PH (aa 1–105) constructs were codon-optimized for E. coli, synthesized and inserted into the pET-28a(+) standard vector with a C-terminal 6xHis-tag (see SI Cloning).
Protein expression and purification
All experiments were performed with E. coli BL21(DE3) (Novagen®). Plasmids were transformed into BL21 cells by electroporation (1.70 kV, 600 Ohm, 10 µF). Single colonies were selected from agar plates, cultured overnight at 37 °C in 10 mL LB medium containing kanamycin (50 mg/mL) and stored in cryo stocks at − 80 °C.
For isotopic labeling, 1000 mL M9 minimal medium (Tables S7 and S8) containing 0.15% 15N-NH4Cl, 0.3% 13C-Glucose and 50 mg/mL kanamycin were inoculated with the overnight-culture (ONC) and incubated at 37 °C, 180 rpm until the OD600 reached 0.6–0.8. Induction of protein expression was performed by adding 0.5 mM isopropyl 1-thio-ß-D-galactopyranoside (IPTG) and cells were incubated overnight at 25 °C in a baffled shaking flask (180 rpm).
Cells containing TcAkt-PH protein were pelleted at 6000 xg, resuspended in 20 mL lysis buffer (50 mM KPi buffer pH 8.0, 10 mM imidazole, 300 mM NaCl) with protease inhibitor (Mix HP, Serva) and disrupted by sonication (45% amplitude, 2 s on, 2 s off) for 15 min on ice. Cell lysates were centrifuged at 4 °C for 1 h at 20,000 xg and the supernatant was loaded onto a pre-equilibrated Ni2+-NTA gravity column (Cube Biotech GmbH). The column was washed with 5 column volumes (CV) wash buffer W1 (50 mM KPi pH 8.0, 10 mM imidazole, 1 M NaCl), W2 (50 mM KPi pH 8.0, 20 mM imidazole, 300 mM NaCl) and W3 (50 mM KPi pH 8.0, 35 mM imidazole, 300 mM NaCl), respectively. The elution of His-bound protein was done with 10–12 mL elution buffer (50 mM KPi pH 8.0, 350 mM imidazole, 300 mM NaCl). The sample was loaded onto a pre-equilibrated size exclusion chromatography (SEC) column (HiLoad 26/600 Superdex 75, GE Healthcare) and the protein was isocratically eluted with SEC buffer (50 mM KPi pH 7.0, 150 mM NaCl, 0.02% NaN3) into 4 mL fractions. Fractions containing the pure protein were pooled, dialyzed against NMR buffer (50 mM KPi pH 6.5, 100 mM NaCl, 0.02% NaN3) in 3.5 kDa by dialysis tubes (Spectra/Por, Repligen) and concentrated with ultrafiltration devices (Amicon Ultra, MWCO 3000, Millipore).
Cells containing full-length TcAkt protein were pelleted at 6000 xg, resuspended in 20 mL lysis buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl) supplemented with protease inhibitor cocktail containing 100 mM PMSF (BioBasic), 100 mM Benzamidine, 0.5 mg/ml Leupeptin, and 70 mg/ml Pepstatin A (BioShop), and sonicated on ice for 4 min (18% amplitude, 60 s on, 60 s off). The mixture was centrifuged at 26,800 × g at 4 °C for 1 h, and the soluble protein sample was subjected to Ni2+ affinity chromatography (HisTrap, GE Healthcare Life Sciences). The column was washed with 5%, 7%, and 12% (v/v) imidazole of the elution buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 250 mM imidazole) 5 CV each. The protein was obtained with 100% elution buffer in 1.2 mL fractions. Then, the sample was diluted with 50 mM Tris–HCl pH 8.0 buffer to a final concentration of 125 mM NaCl, filtered using a 0.2 µm filter, and subjected to anion exchange chromatography on a Mono Q™ 10/100 GL column (Cytiva). The column was pre-equilibrated with buffer (50 mM Tris–HCl pH 8.0, 125 mM NaCl) and the protein was eluted with 500 mM NaCl using a stepwise gradient (125–500 mM) with a flow rate of 1.0 mL/min for 60 min. Pure protein fractions were collected, buffer exchanged (50 mM Tris–HCl pH 8.0, 125 mM NaCl), and concentrated using ultrafiltration devices (Amicon Ultra, MWCO 3000, Millipore). Finally, the concentrated protein fraction was loaded onto the SEC column, pre-equilibrated with buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl), and protein elution was performed utilizing the same buffer at a flow rate of 0.2 mL/min.
Protein concentration was determined by measuring the absorbance at 280 nm with the specific extinction coefficients for full-length TcAkt (ɛ = 55,700 M−1 cm−1) and TcAkt-PH (ɛ = 23,900 M−1 cm−1). The purity of the proteins was confirmed by SDS-PAGE, followed by Coomassie blue staining.
Akt activity assay
The kinase activity of purified recombinant TcAkt-6His was measured using a solid phase enzyme-linked immuno-absorbent assay kit (Abcam, ab139436), according to the manufacturer’s instructions. Briefly, 200 ng purified protein was incubated with ATP (1 µg/µl) for 60 min at 30 °C. The phosphorylation of the synthetic peptide was detected with a phospho-specific substrate antibody incubated 60 min at 21 °C. Subsequently, multiple washes were performed, and anti-rabbit IgG:HRP conjugate (1 µg/mL) and TMB substrate were added. The colorimetric detection was measured at 450 nm in a spectrophotometer (Varioskan Flash Multimode Reader, Thermo Scientific). The human HsAkt3 was used as a positive control and kinase assay dilution buffer was used as blank. Each reaction was performed in triplicate, and the results were expressed as relative kinase activity. Data were analyzed using GraphPad Prism 8.0.1.
DSF experiments
The Tm of TcAkt-6His was determined by monitoring the fluorescence intensity of SYPRO Orange dye (Thermo Fisher) bound to protein as a function of temperature. The protein was diluted to 1.6 µM in a buffer containing 50 mM Tris–HCl (pH 8.0) and 150 mM NaCl in the presence or absence of the indicated divalent cations (2 mM MgCl2 and 2 mM MnCl2 ∙ 4H2O) with Sypro 5 × at a final volume of 25 μl into the wells of a 96-well thin wall PCR plate. The nucleotides were evaluated at 16 μM. Thermal scanning (20–95 °C at 1.0 °C/min) was carried out in the real-time PCR equipment (CFX Connect, Biorad) measuring the intensity of fluorescence every 10 s with the SYBR channel. The melting temperature and the first derivative curve were calculated using the software of the equipment. Data are shown as mean from three independent experiments. Reactions without protein, in the presence of reaction buffer, and SYPRO, were included as controls.
NMR experiments
All spectra were recorded in 3 mm NMR tubes at 25 °C with a 700 MHz Bruker Avance III NMR spectrometer, equipped with a cryogenically cooled 5 mm TCI probe. 13C-NOESY99,100, 13C-HSQC101 and HCCH-TOCSY102,103,104 were recorded from a 500 µM 15N- and 13C-labeled sample in 100% D2O, all other experiments for backbone and side chain assignment were recorded with a 500 µM 15N- and 13C-labeled sample in 90% NMR buffer (50 mM KPi pH 6.5, 100 mM NaCl, 0.02% NaN3) and 10% (v/v) D2O. Spectra were processed with NMRPipe (NMRDraw v5.6 Rev)105 and analyzed with CcpNmr Analysis 2.4.2.106. Molecular images were created with PyMOL (v2.0 Schrödinger, LLC) and UCSF ChimeraX (RBVI)60.
Backbone and side chain assignment
Sequential backbone assignments were determined from the following 3D experiments: 15N-HSQC107, HNCO108,109, HN(CA)CO109,110, HNCA108,109,111, HN(CO)CA108,109, HNCACB112,113, HN(CO)CACB114, CC(CO)NH115,116. For side chain resonance assignments (13C and 1H) we used 13C-HSQC, H(CCO)NH115,116, HCCH-TOCSY102,103,104 and CC(CO)NH spectra.
Interproton distance restraints
Interproton distance restraints (NOEs) were obtained from a 3D 15N-NOESY99,100,117 (80 ms mixing time) and a 3D 13C-NOESY (130 ms mixing time) experiment. NOE assignment was achieved by a combination of CYANA-automated NOE assignment118,119 and manual assignment. Secondary structure predictions were done using backbone assignments and TALOS+58 (Fig. S24).
CS-Rosetta structure calculation
For the CS-Rosetta fragment generation we used the provided CS-Rosetta server as described by Shen et al.57 using the following input parameters: backbone chemical shifts (Cα, Cβ, C’, Hα and HN), TALOS+ restraints58 and NOEs between the following protons: HN-HN, Hα-HN and Hα-Hα (82 NOEs, including 26 long-range NOEs: i − i + (10–100)) (Table S1). Using TALOS+ predictions, the flexible termini were excluded from the subsequent calculations of TcAkt-PH (aa 2–105) (Fig. S24). For the structure generation we used an installed version of Rosetta 3.1357,58 and additionally included side chain NOEs (59 NOEs including 50 long-range NOEs: i − i + (10–100)) (Table S2). 10 000 structures were calculated. The output was validated by plotting the Cα-RMSD of each structure to the lowest-energy structure (S_07667), against the all-atom energy of each structure (Fig. 1a). The plot reveals a clear funnel towards the lowest-energy model, thereby indicating that the CS-Rosetta structure calculation is converged. Ten structures were selected according to the lowest Cα-RMSD to the lowest-energy structure resulting in an averaged Cα-RMSD value of 1.7 Å, additionally confirming successful structure calculation. Refinement statistics were determined via the PSVS server (PMID: 17186527) (Table S3). The best-scored model (S_07667) was used for further structural analysis.
Calculation of electrostatic surface
The coulombic electrostatic potential was calculated from atomic partial charges and coordinates according to Coulomb’s law:
φ… potential, q…atomic partial charges, d…distances from the atoms, ε…dielectric constant.
The resulting potential is in units of kcal/(mol·e) at 298 K. Standard amino acids are assigned atomic partial charges and types from the recommended force field versions in AmberTools 20 (for proteins: ff14SB)61,62.
NMR relaxation experiments
15N T1 spin–lattice relaxation times were determined from an inversion recovery experiment (Bruker pulse sequence: hsqct1etf3gpsi3d.2) with delay times of 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 1.0, 1.5, 2.0, 3.0 and 3.5 s. 15N T2 spin–spin relaxation times were determined from a spin echo CPMG experiment (Bruker pulse sequence: hsqct2etf3gpsi3d) with delay times of 0.017, 0.034, 0.051, 0.068, 0.085, 0.102, 0.119, 0.136, 0.153, 0.170, 0.187, 0.204, 0.237 and 0.330 s. Spectra were analyzed using CcpNmr Analysis 2.4.2.106. The rotational correlation times τC were calculated for each residue from Eq. (2) (Fig. S2b) and the mean value was calculated:
\({\uptau }_{{\text{C}}}\)…rotational correlation time [s], \({\upnu }_{{\text{N}}}\)…15N resonance frequency [Hz], \({{\text{T}}}_{1}\)…T1 relaxation time [s], \({{\text{T}}}_{2}\)…T2 relaxation time [s].
HetNOE values were measured with the pulse sequence hsqcnoef3gpsi. Spectra were analyzed using CcpNmr Analysis 2.4.2.106. HetNOE values were determined from the measured intensities of a saturated 15N-HSQC spectrum relative to a reference spectrum:
\({{\text{NOE}}}_{{\text{het}}}\)…{1H}-15N heteronuclear NOE, \({{\text{I}}}_{{\text{sat}}}\)…Intensities of saturated spectrum, \({{\text{I}}}_{{\text{ref}}}\)…Intensities of reference spectrum.
NMR CSP experiments
For CSP experiments, a 170 µM 15N-labeled sample and a 30 mM stock of Ins(1,3,4,5)P4 in NMR buffer were prepared. Small volumes of Ins(1,3,4,5)P4 were added stepwise to the protein sample up to a ratio of 1:35 (protein:ligand). 15N-HSQC spectra were recorded after each step to follow the shift changes. Spectra were analyzed using CcpNmr Analysis 2.4.2.106.
Euclidean distances, also called d-values, were calculated as described by Williamson53:
\(d\)…Euclidean distance, \({\delta }_{N}\)…15N chemical shift changes, \({\delta }_{H}\)…1H chemical shift changes, \(\alpha\)…scaling factor (glycines \(\alpha\)=0.2, all other amino acids \(\alpha\)=0.14).
A threshold value was determined according to the procedure described by Schumann et al.120 to exclude residues with very small shift changes.
The 15N chemical shifts were weighted with a scaling factor α = 0.14. The dissociation constant (Kd) was then fitted for each amino acid individually with the following equation:
\(\Delta {\updelta }_{{\text{obs}}}\)…change in observed shift, \(\Delta {\updelta }_{{\text{max}}}\)…maximum shift change on saturation, \({[{\text{P}}]}_{{\text{t}}}\)…total protein concentration, \({[{\text{L}}]}_{{\text{t}}}\)…total ligand concentration, \({{\text{K}}}_{{\text{d}}}\)…dissociation constant.
Kd values with d-values below the calculated threshold (d-value < 0.008) were excluded from the dataset. Outliers were identified via boxplot analysis and excluded from the dataset (Kd > 90 µM). Mean and standard deviation were calculated from the resulting 64 values (Table S4).
MD simulations
To model the TcAkt-PH structure with ligands (Ins(1,3,4,5)P4, Ins(1,4,5)P3, Ins(1,3,4)P3, Ins(1,3,5)P3), we have used the experimental structure of the PH domain of HsAkt (PDB: 1UNQ) with bound Ins(1,3,4,5)P4 as a template. The coordinates of the TcAkt-PH structure were aligned with the HsAkt-PH structure. Subsequently, using the builder tool in Maestro (Schrödinger, LLC)121, we have utilized the coordinates of the bound ligand to manipulate the structure of the original ligand and model other ligands with the aligned structure of TcAkt-PH. Subsequently, hydrogen atoms were added using the protein preparation wizard122 in Maestro. Each modeled TcAkt-PH structure without ligands and with ligands then undergoes an energy minimization step only for the H atom, followed by solvation and neutralization.
For equilibration, the system was subjected to 100 ps of Brownian Dynamics NVT at 10 K with restraints on solute-heavy atoms, followed by short 12 ps NVT and 12 ps NPT at 10 K with restraints on solute-heavy atoms. Later, the temperature was increased to 300 K for another 12 ps NPT run with restraints on solute-heavy atoms. Finally, all restraints were removed, and a short 24 ps NPT run was performed, followed by another 1 µs long NPT at 300 K.
The equilibrated system was then further simulated for 1 µs long production runs at 300 K. In total, each system was simulated for 2 µs. For these simulations, the program Desmond123 was used with the OPLS4124 all-atom forcefield. A time step of 2 fs was used throughout the simulations, employing a Nose–Hoover125,126 thermostat and a Martyna-Tobias-Klein127 barostat, with relaxation times of 1.0 and 2.0 ps, respectively. The particle mesh Ewald128 method was used to treat long-range interactions, and a nonbonded cutoff of 9.0 Å was used for short-range interactions. For analysis, the last 1 µs production run has been used.
AF calculations
Ab-initio models for TcAkt were calculated using an AlphaFold 2.3 installation in standard configuration for monomers with full databases and monomer model weights69. With the full-length sequence (aa 1–458) as input, 25 models were generated and ranked by the highest pTM score. The top-ranked model (see Fig. 6) reached a pTM score of 80.6.
RoseTTAFold All-Atom assembly calculation
The models of the TcAkt kinase domain bound to ATP were calculated on a RoseTTAFold All-Atom72 installation in standard configuration. A set of 5 models was generated with the domain sequence (331 aa) and the chemical structure of ATP as input. All 5 predicted models show ATP at the same binding site. The models were ranked by the lowest pae_inter score, the top-ranked model reached a score of 7.6. The models were relaxed using amber99sb129 and GAFF130 force fields with parameters oriented to the relaxation algorithm of AlphaFold-Multimer69,131. The highest ranked model is shown in Fig. 7.
Molecular docking
The structure of HsAkt was retrieved from the RCSB PDB under accession code 4GV155. Non-protein atoms were then removed from the structure, leaving only the bound inhibitor, Capivasertib. Subsequently, protein preparation was conducted using Maestro121 which involved the addition of hydrogen atoms following an energy minimization process to refine the positions of the added hydrogen atoms. The coordinates of a modeled TcAkt structure were aligned with those of the 4GV1 structure, utilizing the bound inhibitor’s coordinates for grid generation. A receptor grid was generated around the bound inhibitor to facilitate docking. The inhibitor molecule underwent a separate ligand preparation step to explore its possible conformations and stereoisomers. Generated ligand conformations were then subjected to docking into the receptor using the extra precision protocol of Glide software132. Furthermore, a similar procedure was repeated for docking the PIT-1 (CAS 53501-41-0) inhibitor with the PH-domain of HsAkt, employing both the PDB:1UNQ structure and the PH domain of the AF structure of TcAkt.
Data availability
The NMR solution structure of TcAkt-PH has been deposited in the PDB with ID 8OZZ. NMR chemical shift assignments have been deposited in the BMRB data bank with accession number 52088. Most data generated and/or analyzed during the current study are included in this article and its supplementary information files. Corresponding raw data tables are available upon request to the corresponding authors.
References
World Health Organization. Chagas disease (also known as American trypanosomiasis). https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (2023).
Martín-Escolano, J. et al. An updated view of the Trypanosoma cruzi life cycle: Intervention points for an effective treatment. ACS Infect. Dis. 8, 1107–1115 (2022).
Abras, A. et al. Worldwide control and management of Chagas disease in a new era of globalization: A close look at congenital Trypanosoma cruzi infection. Clin. Microbiol. Rev. 35, 1–33 (2022).
Alonso-Padilla, J., Pinazo, M. J. & Gascón, J. Chagas disease in Europe. In Chagas Disease (eds Altcheh, J. & Freilij, H.) 111–123 (Springer, 2019).
Eberhard, F. E., Cunze, S., Kochmann, J. & Klimpel, S. Modelling the climatic suitability of Chagas disease vectors on a global scale. Elife 9, 1–16 (2020).
Garza, M. et al. Projected future distributions of vectors of Trypanosoma cruzi in North America under climate change scenarios. PLoS Negl. Trop. Dis. 8, e2818 (2014).
Coura, J. R. The main sceneries of Chagas disease transmission. The vectors, blood and oral transmissions: A comprehensive review. Mem. Inst. Oswaldo Cruz 110, 277–282 (2015).
Beatty, N. L., Forsyth, C. J., Burkett-Cadena, N. & Wisely, S. M. Our current understanding of Chagas disease and Trypanosoma cruzi infection in the state of Florida: An update on research in this region of the USA. Curr. Trop. Med. Rep. 9, 150–159 (2022).
Lascano, F., García Bournissen, F. & Altcheh, J. Review of pharmacological options for the treatment of Chagas disease. Br. J. Clin. Pharmacol. 88, 383–402 (2022).
Pérez-Molina, J. A. & Molina, I. Chagas disease. Lancet 391, 82–94 (2018).
Apt, W. Treatment of Chagas Disease. American Trypanosomiasis Chagas Disease: One Hundred Years of Research 2nd edn. (Elsevier Inc., 2017). https://doi.org/10.1016/B978-0-12-801029-7.00032-0.
Linciano, P. et al. Aryl thiosemicarbazones for the treatment of trypanosomatidic infections. Eur. J. Med. Chem. 146, 423–434 (2018).
de Barros Dias, M. C. H. et al. 1,3-Thiazole derivatives as privileged structures for anti-Trypanosoma cruzi activity: Rational design, synthesis, in silico and in vitro studies. Eur. J. Med. Chem. 257, 115508 (2023).
Castro Fernandes Melo Reis, R. et al. Design and synthesis of new 1,2,3-triazoles derived from eugenol and analogues with in vitro and in vivo activity against Trypanosoma cruzi. Eur. J. Med. Chem. 258, 115622 (2023).
García-Huertas, P. & Cardona-Castro, N. Advances in the treatment of Chagas disease: Promising new drugs, plants and targets. Biomed. Pharmacother. 142, 112020 (2021).
Hargrove, T. Y. et al. Complexes of Trypanosoma cruzi sterol 14α-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: Structural basis for pathogen selectivity. J. Biol. Chem. 288, 31602–31615 (2013).
Doyle, P. S. et al. A nonazole CYP51 inhibitor cures Chagas’ disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 54, 2480–2488 (2010).
Beltran-Hortelano, I., Perez-Silanes, S. & Galiano, S. Trypanothione reductase and superoxide dismutase as current drug targets for Trypanosoma cruzi: An overview of compounds with activity against Chagas disease. Curr. Med. Chem. 24, 1066–1138 (2017).
de Oliveira, R. G., Cruz, L. R., Mollo, M. C., Dias, L. C. & Kratz, J. M. Chagas disease drug discovery in Latin America: A mini review of antiparasitic agents explored between 2010 and 2021. Front. Chem. 9, 1–7 (2021).
Padilla, A. M. et al. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat. Microbiol. 7, 1536–1546 (2022).
Tarleton, R. L. Effective drug discovery in Chagas disease. Trends Parasitol. 39, 423–431 (2023).
De Rycker, M., Wyllie, S., Horn, D., Read, K. D. & Gilbert, I. H. Anti-trypanosomatid drug discovery: progress and challenges. Nat. Rev. Microbiol. 21, 35–50 (2023).
Bustamante, C. et al. In silico, in vitro, and pharmacokinetic studies of UBMC-4, a potential novel compound for treating against Trypanosoma cruzi. Pathogens 11, 616 (2022).
Ochoa, R., Rocha-Roa, C., Marín-Villa, M., Robledo, S. M. & Varela-M, R. E. Search of allosteric inhibitors and associated proteins of an AKT-like kinase from trypanosoma cruzi. Int. J. Mol. Sci. 19, 3951 (2018).
Bae, H. et al. PH domain-mediated autoinhibition and oncogenic activation of Akt. Elife 11, 1–27 (2022).
Ebner, M., Lučić, I., Leonard, T. A. & Yudushkin, I. PI(3,4,5)P3 engagement restricts Akt activity to cellular membranes. Mol. Cell 65, 416-431.e6 (2017).
Varela, R. E. M., Ochoa, R., Muskus, C. E., Muro, A. & Mollinedo, F. Identification of a RAC/AKT-like gene in Leishmania parasites as a putative therapeutic target in leishmaniasis. Parasites and Vectors 10, 1–10 (2017).
Chu, N. et al. The structural determinants of Ph domain-mediated regulation of Akt revealed by segmental labeling. Elife 9, 1–23 (2020).
Truebestein, L. et al. Structure of autoinhibited Akt1 reveals mechanism of PIP3-mediated activation. Proc. Natl. Acad. Sci. U. S. A. 118, 1–11 (2021).
Shaw, A. L. et al. ATP-competitive and allosteric inhibitors induce differential conformational changes at the autoinhibitory interface of Akt1. Structure 31, 343-354.e3 (2023).
Lučic, I. et al. Conformational sampling of membranes by Akt controls its activation and inactivation. Proc. Natl. Acad. Sci. U. S. A. 115, E3940–E3949 (2018).
Meuillet, E. J. Novel inhibitors of AKT: Assessment of a different approach targeting the pleckstrin homology domain. Curr. Med. Chem. 18, 2727–2742 (2011).
Lu, S. et al. The mechanism of ATP-dependent allosteric protection of Akt kinase phosphorylation. Structure 23, 1725–1734 (2015).
Modi, V. & Dunbrack, R. L. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 116, 6818–6827 (2019).
Li, X. et al. Autophosphorylation of Akt at threonine 72 and serine 246: A potential mechanism of regulation of Akt kinase activity. J. Biol. Chem. 281, 13837–13843 (2006).
Manning, B. D. & Toker, A. AKT/PKB signaling: Navigating the network. Cell 169, 381–405 (2017).
Nakayasu, E. S., Gaynor, M. R., Sobreira, T. J. P., Ross, J. A. & Almeida, I. C. Phosphoproteomic analysis of the human pathogen Trypanosoma cruzi at the epimastigote stage. Proteomics 9, 3489–3506 (2009).
Zhang, H. et al. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 277, 39379–39387 (2002).
Huang, Q. et al. Akt2 kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis in ovarian cancer cells via phosphorylating gapdh at threonine 237 and decreasing its nuclear translocation. J. Biol. Chem. 286, 42211–42220 (2011).
Hermida, M. A., Dinesh Kumar, J. & Leslie, N. R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 65, 5–15 (2017).
Souza, D. H. F. et al. Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase: Structure, catalytic mechanism and targeted inhibitor design. FEBS Lett. 424, 131–135 (1998).
Ferguson, K. M. et al. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell 6, 373–384 (2000).
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341 (2010).
Booth, L. A. & Smith, T. K. Lipid metabolism in Trypanosoma cruzi: A review. Mol. Biochem. Parasitol. 240, 111324 (2020).
Yamamoto, E., Kalli, A. C., Yasuoka, K. & Sansom, M. S. P. Interactions of pleckstrin homology domains with membranes: Adding back the bilayer via high-throughput molecular dynamics. Structure 24, 1421–1431 (2016).
Le Huray, K. I. P., Wang, H., Sobott, F. & Kalli, A. C. Systematic simulation of the interactions of pleckstrin homology domains with membranes. Sci. Adv. 8, 1–15 (2022).
Schoijet, A. C. et al. A Trypanosoma cruzi phosphatidylinositol 3-kinase (TcVps34) is involved in osmoregulation and receptor-mediated endocytosis. J. Biol. Chem. 283, 31541–31550 (2008).
Gimenez, A. M. et al. Phosphatidylinositol kinase activities in Trypanosoma cruzi epimastigotes. Mol. Biochem. Parasitol. 203, 14–24 (2015).
Bahia, D. et al. The TryPIKinome of five human pathogenic trypanosomatids: Trypanosoma brucei, Trypanosoma cruzi, Leishmania major, Leishmania braziliensis and Leishmania infantum—New tools for designing specific inhibitors. Biochem. Biophys. Res. Commun. 390, 963–970 (2009).
Tirado-Duarte, D. et al. The Akt-like kinase of Leishmania panamensis: As a new molecular target for drug discovery. Acta Trop. 177, 171–178 (2018).
Queiroz, R. M. L. et al. Quantitative proteomic and phosphoproteomic analysis of Trypanosoma cruzi amastigogenesis. Mol. Cell. Proteom. 13, 3457–3472 (2014).
Becker, W., Bhattiprolu, K. C., Gubensäk, N. & Zangger, K. Investigating protein-ligand interactions by solution nuclear magnetic resonance spectroscopy. ChemPhysChem 19, 895–906 (2018).
Williamson, M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 (2013).
Miao, R. et al. Akt: A potential drug target for metabolic syndrome. Front. Physiol. 13, 822333 (2022).
Addie, M. et al. Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J. Med. Chem. 56, 2059–2073 (2013).
Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol. Res. 200, 107059 (2024).
Shen, Y. et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. U. S. A. 105, 4685–4690 (2008).
Shen, Y., Vernon, R., Baker, D. & Bax, A. De novo protein structure generation from incomplete chemical shift assignments. J. Biomol. NMR 43, 63–78 (2009).
Lemmon, M. A. & Ferguson, K. M. Pleckstrin homology domains. Curr. Top. Microbiol. Immunol. 228, 39–74 (1997).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Maier, J. A. et al. ff14SB: Improving the Accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Case, D. A. et al. AMBER 2020 (2020).
Fusani, L., Palmer, D. S., Somers, D. O. & Wall, I. D. Exploring ligand stability in protein crystal structures using binding pose metadynamics. J. Chem. Inf. Model. 60, 1528–1539 (2020).
Jean, S. & Kiger, A. A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 127, 923–928 (2014).
Frech, M. et al. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J. Biol. Chem. 272, 8474–8481 (1997).
Franke, T. F., Kaplan, D. R., Cantley, L. C. & Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinosi-3,4-bisphosphate. Science 275, 665–668 (1997).
Wang, D. D., Ou-Yang, L., Xie, H., Zhu, M. & Yan, H. Predicting the impacts of mutations on protein-ligand binding affinity based on molecular dynamics simulations and machine learning methods. Comput. Struct. Biotechnol. J. 18, 439–454 (2020).
Khazanov, N. A. & Carlson, H. A. Exploring the composition of protein-ligand binding sites on a large scale. PLoS Comput. Biol. 9, e1003321 (2013).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Biondi, R. M. et al. High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J. 21, 4219–4228 (2002).
Pascuccelli, V., Labriola, C., Téllez-Iñón, M. T. & Parodi, A. J. Molecular and biochemical characterization of a protein kinase B from Trypanosoma cruzi. Mol. Biochem. Parasitol. 102, 21–33 (1999).
Krishna, R. et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science. 2528, 1–17 (2024).
Yang, J. et al. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 9, 940–944 (2002).
Yudushkin, I. Control of Akt activity and substrate phosphorylation in cells. IUBMB Life 72, 1115–1125 (2020).
Akhoon, B. A., Gandhi, N. S. & Pandey, R. Computational insights into the active structure of SGK1 and its implication for ligand design. Biochimie 165, 57–66 (2019).
Kornev, A. P., Taylor, S. S. & Ten Eyck, L. F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U. S. A. 105, 14377–14382 (2008).
Meharena, H. S. et al. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 11, e1001680 (2013).
Stehle, J., Weisner, J., Eichhorn, L., Rauh, D. & Drescher, M. Insights into the conformational plasticity of the protein kinase Akt1 by multi-lateral dipolar spectroscopy. Chem. A Eur. J. https://doi.org/10.1002/chem.202203959 (2023).
Salvador, R. R. S. et al. New carbohydrazide derivatives of 1H-pyrazolo[3,4-b]pyridine and trypanocidal activity. An. Acad. Bras. Cienc. 88, 2341–2348 (2016).
Wu, Y. T. et al. mTOR complex 2 targets Akt for proteasomal degradation via phosphorylation at the hydrophobic motif. J. Biol. Chem. 286, 14190–14198 (2011).
Thomas, C. C., Deak, M., Alessi, D. R. & Van Aalten, D. M. F. High-resolution structure of the pleckstrin homology domain of protein kinase B/Akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 12, 1256–1262 (2002).
James, S. R. et al. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 315, 709–713 (1996).
Milburn, C. C. et al. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem. J. 375, 531–538 (2003).
Craveur, P. et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2, 1–20 (2015).
Hua, H. et al. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 14, 1–25 (2021).
Abeyrathna, P. & Su, Y. The critical role of Akt in cardiovascular function. Vasc. Pharmacol. 74, 38–48 (2015).
Goyal, A., Agrawal, A., Verma, A. & Dubey, N. The PI3K-AKT pathway: A plausible therapeutic target in Parkinson’s disease. Exp. Mol. Pathol. 129, 104846 (2022).
Morel, M. et al. Compound library screening identified Akt/PKB kinase pathway inhibitors as potential key molecules for the development of new chemotherapeutics against schistosomiasis. Int. J. Parasitol. Drugs Drug Resist. 4, 256–266 (2014).
Zimmerman Savill, K. M. et al. Distinct resistance mechanisms arise to allosteric vs. ATP-competitive AKT inhibitors. Nat. Commun. 13, 1–17 (2022).
Oliveira, M. et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann. Oncol. 30, 1289–1297 (2019).
Turner, N. C. et al. BEECH: A dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA. Ann. Oncol. 30, 774–780 (2019).
Landel, I., Quambusch, L., Depta, L. & Rauh, D. Spotlight on AKT: Current therapeutic challenges. ACS Med. Chem. Lett. 11, 225–227 (2020).
Chiurillo, M. A., Jensen, B. C. & Docampo, R. Drug target validation of the protein kinase AEK1, essential for proliferation, host cell invasion, and intracellular replication of the human pathogen Trypanosoma cruzi. Microbiol. Spectr. https://doi.org/10.1128/Spectrum.00738-21 (2021).
Bachmaier, S. & Boshart, M. Kinetoplastid AGC kinases. In Protein Phosphorylation in Parasites: Novel Targets for Antiparasitic Intervention (eds Doerig, C. et al.) 99–122 (Wiley-VCH Verlag GmbH & Co., 2014). https://doi.org/10.1002/9783527675401.ch05.
Page, N. et al. Identification and development of a subtype-selective allosteric AKT inhibitor suitable for clinical development. Sci. Rep. 12, 15715 (2022).
Uhlenbrock, N. et al. Structural and chemical insights into the covalent-allosteric inhibition of the protein kinase Akt. Chem. Sci. 10, 3573–3585 (2019).
Quambusch, L. et al. Covalent-allosteric inhibitors to achieve Akt isoform-selectivity. Angew. Chem. 131, 18999–19005 (2019).
Weisner, J. et al. Kovalent-allosterische kinase-inhibitoren. Angew. Chem. 127, 10452–10456 (2015).
Marion, D. et al. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H–15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: Application to interleukin 1. beta. Biochemistry 28, 6150–6156 (1989).
Zuiderweg, E. R. P. & Fesik, S. W. Heteronuclear three-dimensional NMR spectroscopy of the inflammatory protein C5a. Biochemistry 28, 2387–2391 (1989).
Vuister, G. W. & Bax, A. Resolution enhancement and spectral editing of uniformly 13C-enriched proteins by homonuclear broadband 13C decoupling. J. Magn. Reson. 435, 69 (1991).
Olejniczak, E. T., Xu, R. X. & Fesik, S. W. A 4D HCCH-TOCSY experiment for assigning the side chain1H and13C resonances of proteins. J. Biomol. NMR 2, 655–659 (1992).
Bax, A., Clore, G. M. & Gronenborn, A. M. 1H1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J. Magn. Reson. 88, 425–431 (1990).
Kay, L. E., Xu, G.-Y., Singer, A. U., Muhandiram, D. R. & Forman-Kay, J. D. A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. Ser. B 101, 333–337 (1993).
Delaglio, F. et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Skinner, S. P. et al. CcpNmr AnalysisAssign: A flexible platform for integrated NMR analysis. J. Biomol. NMR 66, 111–124 (2016).
Davis, A. L., Keeler, J., Laue, E. D. & Moskau, D. Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. J. Magn. Reson. 98, 207–216 (1992).
Grzesiek, S. & Bax, A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 96, 432–440 (1992).
Kay, L. E., Xu, G. Y. & Yamazaki, T. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. Ser. A 109, 129–133. https://doi.org/10.1006/jmra.1994.1145 (1994).
Clubb, R. T., Thanabal, V. & Wagner, G. A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, and 13C’ chemical shifts in 15N–13C-labeled proteins. J. Magn. Reson. https://doi.org/10.1093/oseo/instance.00076703 (2017).
Schleucher, J., Sattler, M. & Griesinger, C. Coherence selection by gradients without signal attenuation: Application to the three-dimensional HNCO experiment. Angew. Chem. 32, 1489–1491 (1993).
Wittekind, M. & Mueller, L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J. Magn. Reson. Ser. B 101, 201–205. https://doi.org/10.1006/jmrb.1993.1033 (1993).
Muhandiram, D. R. & Kay, L. E. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. Ser. B 103, 203–216. https://doi.org/10.1006/jmrb.1994.1032 (1994).
Grzesiekt, S. & Bax, A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293 (1992).
Löhr, F. & Rüterjans, H. Correlation of backbone amide and side-chain 13C resonances in perdeuterated proteins. J. Magn. Reson. 156, 10–18 (2002).
Montelione, G. T., Lyons, B. A., Emerson, S. D. & Tashiro, M. An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J. Am. Chem. Soc. 114, 10974–10975 (1992).
Marion, D., Kay, L. E., Sparks, S. W., Torchia, D. A. & Bax, A. Three-dimensional heteronuclear NMR od 15N-labeled proteins. J. Am. Chem. Soc. 111, 1515–1517 (1989).
Güntert, P., Mumenthaler, C. & Wüthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 (1997).
Herrmann, T., Güntert, P. & Wüthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 (2002).
Schumann, F. H. et al. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR 39, 275–289 (2007).
Schrödinger. Maestro (2021).
Sastry, G. M., Adzhigirey, M. & Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. https://doi.org/10.1007/s10822-013-9644-8 (2013).
Bowers, K. J. et al. Molecular dynamics: Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing—SC ’06 (ACM Press, 2006). https://doi.org/10.1145/1188455.1188544.
Lu, C. et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. https://doi.org/10.1021/acs.jctc.1c00302 (2021).
Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Nosé, S. Molecular physics: An international journal at the interface between chemistry and physics a molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 (1984).
Martyna, G. J. et al. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 (1994).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N.log (N) method for Ewald sums in large systems. J. Chem. Phys. 10089, 1–5 (2020).
Hornak, V. et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. PROTEINS Struct. Funct. Bioinform. 65, 712–725 (2006).
He, X., Man, V. H., Yang, W., Lee, T. S. & Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 6–12 (2020).
Evans, R. et al. Protein complex prediction with RNSC. DeepMind https://doi.org/10.1007/978-1-61779-361-5_16 (2021).
Friesner, R. A. et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Acknowledgements
K.Z., T.P.K. and N.G. acknowledge the support of the field of excellence BioHealth at the University of Graz. L.O.J. acknowledges the support of CODI-Universidad de Antioquia, Coimbra Group Scholarship (University of Graz), as well as the Emerging Leaders in the Americas Program Canada (McGill University). We thank Yang Shen (NIDDK) for CS-Rosetta technical support. We thank Roberto Tejero (University of Valencia) for support with PSVS analysis. We thank Kyle Le Huray (University of Leeds) for providing the coordinate file of membrane-bound PH domain of HsAkt1, which we used for our illustrations. We also thank Lucy Maremmano for insightful discussions and motivation.
Funding
The authors acknowledge the financial support by the University of Graz. This research was funded by the Austrian Science Fund (FWF) [https://doi.org/10.55776/T1239 to N.G.; https://doi.org/10.55776/DOC50 to K.Z]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. This research received funding from the Land Steiermark and the City of Graz (to K.Z.), the MINCIENCIAS-Colombia (Grant Number 111577757016, to M.M. and L.O.J.), and a Project Grant from the Canadian Institutes of Health Research (Grant Number PJT-155941, to A.G). The authors also acknowledge the financial support by the University of Graz, the interuniversity programs NAWI Graz and BioTechMed for financial support. Financial contributions by the Land Steiermark infrastructure grant “Frontier NMR” (Project Number 1109) are also gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
+K.S. and L.O.J. contributed equally to this work. A.S.S. performed and analyzed the MD simulations and docking studies; C.B. performed the AF and RoseTTaFold calculations; T.B.O, A.G. and L.O.J. performed and analyzed the DSF experiments; K.S., L.O.J. and N.G. performed the NMR experiments; K.S. and N.G. analyzed the NMR experiments and determined the structure (PDB: 8OZZ); L.O.J. performed the activity assays; K.S. and N.G. analyzed the NMR and AF structure; K.G., T.P.K., A.G., S.P., M.M., K.Z. and N.G. supervised the work; K.S, L.O.J. and N.G. conceptualized and wrote the manuscript. All authors contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stadler, K.A., Ortiz-Joya, L.J., Singh Sahrawat, A. et al. Structural investigation of Trypanosoma cruzi Akt-like kinase as drug target against Chagas disease. Sci Rep 14, 10039 (2024). https://doi.org/10.1038/s41598-024-59654-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-59654-8
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
