
Abstract
Cation exchanger (CAX) genes play an important role in plant growth/development and response to biotic and abiotic stresses. Here, we tried to obtain important information on the functionalities and phenotypic effects of CAX gene family by systematic analyses of their expression patterns, genetic diversity (gene CDS haplotypes, structural variations, gene presence/absence variations) in 3010 rice genomes and nine parents of 496 Huanghuazhan introgression lines, the frequency shifts of the predominant gcHaps at these loci to artificial selection during modern breeding, and their association with tolerances to several abiotic stresses. Significant amounts of variation also exist in the cis-regulatory elements (CREs) of the OsCAX gene promoters in 50 high-quality rice genomes. The functional differentiation of OsCAX gene family were reflected primarily by their tissue and development specific expression patterns and in varied responses to different treatments, by unique sets of CREs in their promoters and their associations with specific agronomic traits/abiotic stress tolerances. Our results indicated that OsCAX1a and OsCAX2 as general signal transporters were in many processes of rice growth/development and responses to diverse environments, but they might be of less value in rice improvement. OsCAX1b, OsCAX1c, OsCAX3 and OsCAX4 was expected to be of potential value in rice improvement because of their associations with specific traits, responsiveness to specific abiotic stresses or phytohormones, and relatively high gcHap and CRE diversity. Our strategy was demonstrated to be highly efficient to obtain important genetic information on genes/alleles of specific gene family and can be used to systematically characterize the other rice gene families.
Similar content being viewed by others

Introduction
In a plant species with a wide geographic distribution, different members of a gene family may often become functionally differentiated. This functional differentiation among different members of a gene family, reflected by their sequence diversity among related species, as well as among different populations of a species, is of great interest to plant scientists because it is the consequences of the plant species or populations adapted to different ecological environments. Thus, dissecting the genetic diversity of different members of a gene family in a plant species is expected to provide important information on their functionalities and, thus, their potential uses in plant improvement.
Calcium (Ca2+) serves as a widespread secondary messenger in plant cells, facilitating a range of intricate responses and regulatory mechanisms in plants in response to biotic and abiotic stress through signal transduction1. Changes in Ca2+ concentration within the plant cytoplasm are crucial for signal transduction. Stimulation of plants by the external environment leads to rapid fluctuations in cytoplasmic Ca2+ levels, triggering a response of calcium signaling channels2,3,4,5,6. This process is achieved through interactions between numerous channel proteins and transport proteins on the cell membrane, which in turn regulate gene expression and various physiological reactions in plants7. Various signals are initiated by different Ca2+ sensors, including calmodulin (CaM), CaM-like proteins (CMLs), and calcineurin B-like proteins (CBLs), in order to elicit specific and tailored responses, thereby regulating gene expression and metabolism in plants8. Among these Ca2+ sensors, the cation exchanger (CAX) gene family has garnered significant attention. CAX represents a subset of the CaCA (Ca2+/Cation antiporter) superfamily, primarily situated in the vacuolar membrane9. Its function involves the of cellular cation homeostasis by transporting cytoplasmic cations, such as calcium, into vesicular storage using the pH gradient generated by the proton pump10,11. Strong evidence from Arabidopsis and other plants indicates that CAX genes play a crucial role in polycation tolerance, metal transport, element distribution, ion homeostasis, and abundance in response to abiotic stress10,12. In these processes, CAX genes function to restore Ca2+ in both intracellular and extracellular reservoirs of the cytoplasm, as well as distribute Ca2+ into plant vacuoles for continual signal perception, ensuring the progress of various biochemical reactions by interacting with Ca2+ channels9,11,13,14. For instance, in Arabidopsis, AtCAX1 expression is significantly induced by cold stress, while the CAX2 mutant exhibits flooding tolerance15. Additionally, in rice, OsCAX1a, OsCAX1c, and OsCAX4 may be involved in the absorption and transport of cadmium15,16, with OsCAX1a and OsCAX3 conferring tolerance to manganese17. The CAX gene families in different plant species vary considerably in number and sequence due to their diverse polyploid histories during evolution18,19,20. Therefore, understanding the functional differentiation of duplicated members of the CAX gene family and their genetic diversity within and among different populations has been a subject of widespread interest.
As one of the most important food crops, rice is grown in diverse ecologies worldwide and often faces a wide range of biotic and abiotic stress. Rice is also a model plant species for functional and population genomic research, with more than 4600 rice genes cloned and functionally characterized at the molecular level (https://funricegenes.github.io/)21,22. The past few decades have also witnessed tremendous progress in the genetic and molecular dissection of complex traits of rice using DNA marker-facilitated genetic mapping and various omics tools, such as transcriptomic, proteomic, and metabolomic analyses, generating huge amounts of data on the genetic and molecular mechanisms underlying complex traits and responses to biotic and abiotic stress in rice. To facilitate the integration of research advances in rice functional genomic research for rice improvement, rice population genomic research has proceeded more recently at an unprecedented rate. To date, more than 10,000 rice germplasm accessions worldwide have been re-sequenced23,24,25. Rice population genomic research has revealed tremendous amounts of genomic diversity in rice germplasm accessions and their organization within and among different rice populations, including almost unlimited numbers of single nucleotide polymorphisms (SNPs), huge numbers of structural variations (SVs) of > 30 bps, gene presence/absence variations (PAVs) for about 50% of rice genes, extremely rich gene-coding sequence (CDS) haplotype (gcHap) diversity in most rice genes, and gene copy number variation (CNVs) for a significant portion of rice genes23,26,27,28,29. One of the exciting discoveries from the 3010 rice genomes project (3KRGP) was the impact of modern breeding on the gcHap diversity of 45,963 rice genes, reflected primarily by changes in gcHap diversity and frequency shifts of the predominant (ancient) alleles in modern rice varieties compared to the landraces of two rice subspecies resulting from artificial selection during modern breeding27. In contrast to most SNPs in the inter-genic regions and introns of genes, gene PAVs, CNVs, and SVs that occur in genic regions and major gcHaps are expected to have large phenotypic effects and thus are powerful markers for genetic and molecular dissection of complex traits, as demonstrated more recently in the cases of Sub1a GL7 and qSW530,31,32. Using a rice super pan‐genome, natural allelic variation in TTL1 has been shown to control thermotolerance and grain size33. Several large and comprehensive databases have been established from rice functional and population genomic studies and have been made publicly available to all researchers34. However, it remains a great challenge to integrate the advances and huge amounts of information from rice functional and population genomic research into more efficient strategies for rice improvement. This is particularly true for > 90% rice genes with their functions unknown and the presence of many functional but uncharacterized alleles in each of the rice genes. Thus, one important question arises regarding how to obtain essential information on gene functionality and phenotypic effects more efficiently without going through the time-consuming efforts of the classical gene cloning process, given the presence of so much genetic and molecular information on rice genes in the public domain.
In this study, we reported an effort in functional characterization and allelic mining of the rice CAX gene family containing six members (OsCAX1a, OsCAX1b, OsCAX1c, OsCAX2, OsCAX3, and OsCAX4) by systematic analyses of their homology relationships among different plant species, cis-regulatory element (CRE) variation in promoter regions, expression patterns, genetic diversity in different rice populations, and associations with tolerance to different abiotic stress using publicly available data. We also analyzed their associations with salt tolerance using 3KRGP and eight introgression line (IL) populations. Our results demonstrated a powerful and highly efficient strategy for obtaining important genetic information on rice genes of unknown function essential for improving abiotic stress tolerance in rice using breeding by design in the future.
Materials and methods
Homology analysis, protein structure and domain prediction of CAX genes in different species
Six rice CAX genes (OsCAX1a, OsCAX1b, OsCAX1c, OsCAX2, OsCAX3, and OsCAX4) from the CAX gene family were examined in this study. Among these genes, only OsCAX1a was reported to be functionally related to rice panicle degeneration, while the functions of the other five rice CAX genes remain unknown35,36. To investigate the relationships between CAX genes in different species, the sequences of the six rice CAX genes were compared with those of Arabidopsis thaliana (At), Triticum aestivum (Ta), Zea mays (Zm), Glycine max (Glyma), Hordeum vulgare (Hv), Solanum tuberosum (St), Solanum lycopersicum (Sl), Saccharomyces cerevisiae (Sc), Synechococcus sp (Ssp), and Escherichia coli (Ec). Phylogenetic analysis of the CAX genes in these species was conducted using MEGA X software. The protein sequences of the CAX genes in the aforementioned species were compared using a logarithmic expected multi-sequence comparison method called CDS DNA sequence comparison. Names and the protein sequences of 62 CAX genes were listed in Table S1. Phylogenetic trees based on full-length protein sequence alignment were constructed using the neighbor-joining (NJ) method with 1000 bootstrap replicates. The CAXs protein structure and domain was predicted on https://swissmodel.expasy.org/ and https://www.genome.jp/37. Number of motifs were listed in Table S2.
Collinearity relationship and promoter cis-regulatory element analysis of the OsCAX gene family
Gene duplication patterns and collinearity relationships of the OsCAX gene family were identified and analyzed using MCScanX38. The ratio of synonymous mutations to non-synonymous mutations (Ka/Ks) between gene pairs was calculated using TBtools39 to determine the evolutionary selection pressure on each member of the OsCAX gene family. The 2000 bp sequences upstream of the translation start sites of each OsCAX gene were retrieved from the Os-Nipponbare-Reference-IRGSP-1.0 reference genome as the promoter sequence where the cis-regulatory elements were predicted using PlantCARE (Table S3) (http://bioinformatics.psb.ugent.be/webtools/plantcare)40. The results were visualized using TBtools. We also selected 25 Xian (indica) and 25 Geng (japonica) accessions from 33 and 111 high-quality rice genomes26,29. The 50 high-quality accessions information are available in the Table S4. We used “blastp” with setting the alignment threshold to 1e-10, to align and compare the CAX protein sequences of the Nipponbare reference genome with those of 50 samples. Subsequently, we used a Perl script to extract the promoter sequences 2000 bp upstream of each of the CAX genes and analyzed their variation in the predicted type and number/frequency of CREs using PlantCARE and a shell script.
Expression profile analysis of the rice CAX genes
We downloaded the expression profiles of the six rice CAX genes from three rice expression databases: one was built from the Geng variety Nipponbare (http://expression.ic4r.org/) (https://www.mbkbase.org/rice)41,42, and the other from two Xian varieties R498 and IR6429,43. The databases included rice gene expression data derived from RNA sequencing (RNA-Seq) analyses of different tissues, such as aleurone, anther, callus, leaf, panicle, pistil, root, seed, and shoot, at different growth stages, as well as under a variety of biotic and abiotic treatments (such as Cd, Pi, drought, cold and salt). The expression profiles of all rice CAX genes are available in the Table S5. The gene expression levels were calculated based on the log transformation of the FPKM values. Heatmaps of the expression patterns of all rice CAX genes were plotted using TBtools.
Construction of gene gcHap networks of rice CAX genes
When compared in IRGSP-1.0, all SNPs identified within the CDS regions of the six OsCAX genes were used to obtain the number of gene CDS haplotypes (gcHapN) for each of the OsCAX genes. Shannon’s equitability (EH) was used to evaluate the level of gcHap diversity at all rice CAX loci in specific rice populations or the whole species44. A haplotype network was built for each of the rice CAX genes based on pairwise differences between two adjacent gcHaps using the R package “pegas”45. To plot the network, we used only the major haplotypes, each carried by at least 50 accessions in the 3KRGP accessions. The network was produced using statistical parsimony, such that the most closely related haplotypes were connected first via the smallest number of mutations46.
To understand how modern breeding in past decades affected the gcHap diversity of rice CAX genes, we compared the differences in EH estimates and frequencies of the predominant gcHaps between 732 Xian (indica) landraces (LANs-Xian) and 328 modern Xian varieties (MVs-Xian) and between 358 Geng (japonica) landraces (LANs-Geng), and 139 modern Geng varieties (MVs-Geng) (https://www.rmbreeding.cn/Index)47 using an R script (https://github.com/isaac-Tsang/gcHap_diversity_LANs_MVs-) and Z tests27,48. We also compared the differences between LANs-Xian and MVs-Xian and between LANs-Geng and MVs-Geng for possible newly emergent gcHaps at each of the CAX genes in rice MV varieties. Finally, the data above were plotted using GraphPad Prism 9 software and the “ggplot2” package in R.
Genome-wide association study (GWAS) based on SNPs/gchaps
Based on the haplotype network analysis, the predominant gcHaps of the CAX genes in 3KRGP were determined and analyzed for their associations with several agronomic traits, including culm length (CL, cm); culm number (CN, count); grain length (GL, mm); 1000-grain weight (TGW, g); grain width (GW, mm); and days to heading (HD, day) from the IRRI (snp-seek.irri.org/)49 and RFGB databases (https://www.rmbreeding.cn/)47. The major haplotypes of all rice CAX genes, each carried by at least 50 accessions in the 3KRGP accessions, were obtained using an R script (https://github.com/isaac-Tsang/haplotype_network). Finally, the associations of major haplotypes with these agronomic traits across 3010 diverse rice accessions were achieved using an R script (https://github.com/isaac-Tsang/functional-importance-). The differences between the haplotypes were calculated by a two-way analysis of variance (ANOVA), and statistically significant differences were based on Duncan’s multiple range test at P < 0.05. GraphPad Prism 9 software was used to generate the phenotype and haplotype box graphs from the GWAS analyses.
GWAS was conducted to determine the associations of the major gcHaps for each of the CAX genes conferring salt tolerance, using historical phenotypic data for salt tolerance at the germination stage of the 478 rice accessions in 3KRG50. Moreover, validation of the associations of the CAX genes with salt tolerance was conducted using the agronomic trait data of 496 Huanghuazhan (HHZ) ILs under salt tolerance. The GWAS results are available in the Table S6. Manhattan plots of the GWAS results were analyzed using a mixed linear model (MLM) and plotted using the R package “rMVP”. We defined the LD decay distance to be chosen in such a way that the LD coefficient (r2) is reduced to half of its maximum value51. The significant SNP was defined as the SNP with the P value < 10–5. The candidate gene was identified within 200 kb upstream and downstream of the significant SNP.
Genome-wide variant detection and analysis of SNPs, SVs, and PAVs in the rice CAX family
For the 3KRG SNP dataset, we selected the 4.8mio SNPs subset (https://snp-seek.irri.org/) with 478 rice accessions49,50, and 3,355,717 SNPs were filtered. For HHZ ILs, we used “gatk” to call SNPs and “delly” to call the SVs with default parameters52. 1,345,828 SNPs were filtered based on the following criteria: (1) minor allele frequencies (MAFs) ≥ 0.05; (2) missing rate < 20%. For 111 high-quality rice genomes, we used “delly” to call the SVs of the CAX gene family with default parameters, and the genomes of nine HHZ IL parents were included in the 111 rice genomes.
Results
Phylogenetic relationship of CAX genes among different species
To investigate the evolutionary relationships of OsCAXs with CAXs from different species, an unrooted phylogenetic tree was constructed based on the aligned amino acid sequences of 62 CAX proteins (Fig. 1). The 62 CAX proteins were classified into three large clusters (IA, IB, and IC) containing 26 (IA), 33 (IB) and three (IC) proteins, respectively. Among the six OsCAX proteins, OsCAX2, OsCAX3, and OsCAX4 were grouped in three subgroups of cluster IB. OsCAX2 and OsCAX4 showed high homology with HORVU4Hr1G037210 and HORVU6Hr1G019470 of Hordeum vulgare in Cluster IB. OsCAX1a, OsCAX1b, and OsCAX1c were classified in the same subgroup of Cluster IA, though the former two were more closely related to each other. Clusters IA and IB contained two and five maize CAX genes related to OsCAX1a-1c, and OsCAX2-4, respectively. Moreover, OsCAX1a and OsCAX1b displayed high homology with HORVU3Hr1G049060 in Cluster IA. The soybean CAX family comprised 14 genes. And the homology analysis of CAX in bacteria and yeast was consistent with previous studies, indicating a separate third type. YNL321W in yeast may be more closely related to plants. The prediction of protein structures in rice, bacteria, and yeast revealed that OsCAX1a, OsCAX1b, and OsCAX1c, OsCAX2, and OsCAX3 have similar protein structures, each with 11 transmembrane (TM) helices, while OsCAX4 has only nine helices (Fig. S1). However, there were significant differences between the protein structures of CAX in rice and bacteria and yeast. CAXs in non-plant species were more tightly packed in membranes than those in rice, suggesting functional differentiation in the CAX family.

The phylogenetic relations of the CAX gene family in different species. The phylogenetic relations of CAX from Oryza sativa based on the Os-Nipponbare-Reference-IRGSP-1.0 reference genome and other species, including Arabidopsis thaliana (At), Triticum aestivum (Ta), Zea mays (Zm), Glycine max (Glyma), Hordeum vulgare (Hv), Solanum tuberosum (St), Solanum lycopersicum (Sl), Saccharomyces cerevisiae (Sc), Synechococcus sp (Ssp), and Escherichia coli (Ec). The 11 species are depicted by different colored shapes.
When the genomic locations of the six rice CAX genes were examined, we found that two pairs of homologous OsCAX genes, OsCAX1a/OsCAX1b and OsCAX2/OsCAX3, were located on segments of chromosomes 1/5 and 3/4 (Fig. S2), consistent with the duplicated chromosomal segments from the whole genome duplication (WGD) event in Poaceae inferred to have occurred about 65 million years ago (mya)53. The locations of OsCAX1a and OsCAX1b on rice chromosomes 1 and 5 suggested that additional duplication events occurred during the diploidization process after WGD. Moreover, the Ka/Ks ratios of all rice CAX pairs were < < 1.0, suggesting that all rice CAX genes had gone through strong purification selection during evolution (Table S7). These results suggest that the formation of the six rice CAX genes resulted from the well-known WGD event in Poaceae, while the close relationships of the rice CAX genes with maize CAX genes suggest that the rice CAX genes might share similar functionalities to those in maize. But whether there is a similar function between them still needs to be further verified by experiments.
Genetic diversity of rice CAX genes
To reveal the total genetic diversity of the six OsCAX genes in rice populations, we first examined their PAV diversity in 453 representative accessions of 3KRGP54. Of the six CAX genes, OsCAX2 and OsCAX3 were core genes in 453 accessions without PAV variation (Table S8). OsCAX1a was present in 453 accessions but absent in six Geng accessions of the subtropical group (GJsubtrop). OsCAX1b was present in all accessions except four accessions of population ADM. Similarly, OsCAX1c was present in all accessions except one of the Xian accessions from South Asia (XI2). OsCAX4 was present in all rice accessions except one Xian accession from China (XI1). We also examined their PAV diversity in the HHZ IL parental genomes, found that no PAVs were detected in all OsCAX genes (Table S9). This suggests that all six rice CAX genes are functionally important. For the correlation between geographic environmental conditions and CAXs, we analyzed the frequency of rice CAX genes existence in varieties at different geographical locations (Table S10). Remarkably, a significant geographic variation is observed in the distribution of the OsCAX1a and OsCAX1b genes. Five out of the six accessions lacking the OsCAX1a gene are from Southeast Asia, while the remaining one of the six accessions is from South Asia. All four accessions lacking the OsCAX1b gene are from Africa. The other four OsCAX genes showed almost no differences in geographical locations.
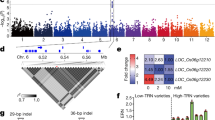
We then analyzed the gcHap diversity of the six OsCAX genes in 3KRG (Table S11). Of the six rice CAX genes, OsCAX1a and OsCAX2 were highly conserved (EH = 0.012 and 0.058, and gcHapN = 8 and 20), each with a single predominant allele plus three or two major alleles in rice populations (Fig. 2). The remaining four OsCAX genes were moderately diverse, with EH (gcHapN) ranging from 0.120 (41) for OsCAX1b to 0.197 (83) for OsCAX3 (Table S11). Interestingly, a significant portion of the gcHaps of the six OsCAX genes were unique for specific populations, including 19.5 Xian-unique gcHaps and 7.3 Geng-unique gcHaps. OsCAX1c was the only Xian–Geng divergent gene having two predominant alleles, with Hap1 present only in all Xian accessions and Hap2 in all Geng accessions (Fig. 2). We also identified a significant level of gcHap diversity among the nine HHZ IL parents (Table S12). Specifically, the gcHapN detected in the nine HHZ IL parents was four in OsCAX1a, six in OsCAX1b, nine in OsCAX1c and OsCAX4, and three in OsCAX2 and OsCAX3. Interestingly, all 34 alleles of the OsCAX genes identified in the HHZ IL parents were rare in the Xian accessions of 3KRG (Tables S12 and S13). To determine whether the OsCAX genes were of potential value in rice improvement, we compared the predominant gcHap frequencies between the landraces and modern varieties of subspecies Xian and Geng (Fig. 2). Surprisingly, the predominant gcHap (Hap1s) frequencies of all six OsCAX genes dropped significantly in the modern varieties when compared to the landraces in both subspecies, replaced by either rare ones or newly emerged ones that were absent in the landraces but present only in modern varieties. This clearly indicates that the ancient predominant alleles of all six OsCAX genes were not favored by artificial selection during the modern breeding of both subspecies.

Haplotype networks of OsCAX gene family and their associations with six traits in 3KRG. (a) OsCAX1a, (b) OsCAX1b, (c) OsCAX1c, (d) OsCAX2, (e) OsCAX3, (f) OsCAX4. Within each haplotype network, two adjacent gcHaps are separated by mutational changes with hatches indicating differences between the two most related haplotypes. The right side of each gene haplotype network corresponds to the phenotypic variation among the haplotypes. Boxplots are shown for the following traits: culm length (CL, cm); culm number (CN, count); grain length (GL, mm); 1000 grain weight (TGW, g); grain width (GW, mm); and days to heading (HD, day). The P values under trait names indicate differences between the haplotypes assessed by two-way ANOVA, with different letters on the boxplots indicating statistically significant differences at P < 0.05 based on Duncan’s multiple range test. The bar charts on the right show the differences in frequency of the predominant gcHaps between landraces (LANs) and modern varieties (MVs) in Xian and Geng. Chi-square tests were used to determine significant differences in the proportions of the same gcHap between the different populations with ****P < 0.0001, **P < 0.01, *P < 0.05, and n.s., not significant.
We then analyzed the SVs in the six OsCAX genes in 453 representative accessions of 3KRGP with a sequencing depth of 20X and detected an around 4-kb deletion in OsCAX1c and a 300 bp deletion in OsCAX3. Because of the low power in detecting SVs from the NGS-generated short sequences, we analyzed 111 high-quality genomes from third-generation sequencing (TGS) technology29 and detected 636 SVs of various sizes in the OsCAX genes. These SVs included 48.8% deletions, 48.9% insertions, 2.1% duplications, and one copy number variation ranging from 35 to 35,370 bp. Among these SVs, 58% occurred in intergenic regions, 33% in upstream promoter regions, and 9% in genic regions. Specifically, the number of SVs detected was 461 in OsCAX1a, 90 in OsCAX4, 64 in OsCAX3, 15 in OsCAX1c, four in OsCAX2, and two in OsCAX1b (Table S14). In the HHZ IL parental genomes, SVs were detected in all OsCAX genes except OsCAX1c (Fig. S3 and Table S15). Multiple small insertions and deletions in the intergenic region of OsCAX1a were detected among all parents except for IR50 and IR64, ranging from two SVs in HHZ, OM1723, and Phalguna to ten SVs in IR64. In OsCAX1b, we detected a single deletion of about 810 bp in the promoter region of PsBRC28, IR64, OM1723, and Phalguna but not in the other five parents. A single deletion of about 266 bp was detected in the promoter region of OsCAX2 in all nine parents. In OsCAX3, a 30 bp deletion and a 385 bp insertion were detected in the promoter regions of all parents except IR64, in addition to a 51 bp deletion in the intergenic regions of HHZ and Teqing. A single insertion of sizes ranging from 208 to 251 bp in nine parents was identified in the promoter region of OsCAX4. There were one or more SVs in all six OsCAX genes among the different rice accessions, particularly in OsCAX1a, and all of these SVs occurred in the intergenic or intronic regions.
Another aspect of the genetic diversity and functional differentiation of the six OsCAX genes was reflected by the CRE variation in the 2-kb promoter regions because the number and type of CREs in the promoter region of a gene are known to determine its expression profile, its phenotypic effects, and how it is regulated. Table 1 shows the 25 CREs predicted in the 2-kb regions of the six OsCAX genes in the 50 representative high-quality rice genomes (25 accessions for Xian or Geng). These CREs were roughly classified into five large functional categories (Fig. S4) according to their binding transcriptional factors (TFs), including six light-responsive elements, four stress-responsive elements, eight hormone-responsive elements, five developmental-related elements, and two MYB-related elements. However, considerable variation was present in the type and copy number of specific CREs in the promoters of the six OsCAX genes and their frequencies in populations Xian and Geng. Of the 25 CREs, eight CREs (Box-4, MYB, MYC, ARE, STRE, as-1, CGTCA-motif, and TGACG-motif) were common ones detected in the promoters of all six OsCAX genes, each in a significant portion of the examined rice accessions. ARBE and ERE were also common and present in six of the OsCAX genes, except OsCAX1c and OsCAX1b. Most of the common CREs belonged to the abiotic stress and hormone-responsive groups, consistent with their expected primary functions. The remaining CREs were highly specific, each present in promoters of 1–4 of the OsCAX genes in all rice accessions or a portion of a rice population, indicating that they contribute to the specific functions of different OsCAX genes. For example, a light-responsive CRE, GA-motif, was detected in OsCAX2 in 90% of Geng and 50% of Xian accessions, while ARE was detected in OsCAX1a and OsCAX1b in approximately 50% of the rice accessions and in OsCAX4 in 40% of Geng accessions. LTR was detected in OsCAX1a, OsCAX2, and OsCAX4 in > 50% of rice accessions. We observed minor and quantitative differences between populations Xian and Geng regarding the average number and type of common CREs in the OsCAX gene promoters, indicating their evolutionary conservativeness. However, qualitative differences between the Xian and Geng accessions were observed in many specific CREs, particularly in the OsCAX2 and OsCAX4 promoters. In the OsCAX4 promoter, four light-responsive CREs (AE-box, G-box, I-box, and MRE), a TGA-element, and an RY-element were present in all or most accessions of one subspecies but not in the other. Highly significant quantitative differences were also detected between the Xian and Geng accessions for the frequencies of the three hormone-responsive elements in the OsCAX4 promoter. In the OsCAX2 promoter, four CREs (I-box, ARE, DRE-core, and TGA-element) were present in most Geng accessions but absent in the Xian accessions. These results suggest that the emergence of OsCAX2 and OsCAX4 might be accompanied by the well-known Xian–Geng subspecific differentiation and might have contributed to the adaptations of Geng rice to long-day and colder environments. In order to explore the difference of monocots and dicots, we also analyzed the CREs between them and observed a higher average number of CRE species in monocots, particularly in development-related CREs (Fig. S4). This could indicate that these CREs were more effectively regulated by hormones in monocotyledons to facilitate their growth. Furthermore, stress-responsive CREs were less prevalent in monocots compared to dicots, indicating that despite their faster growth, monocots are more vulnerable to environmental stress.
In addition, we analyzed of the domains of rice, Arabidopsis, bacteria, and yeast CAX genes, which revealed that Na_Ca_ex is present in all genes, indicating a relation to family characteristics and response to stress through the regulation of calcium ion transport. Furthermore, we observed that FA_desaturase and Glyco_hydro_59 were exclusive to plants, while YccF, Na_K-ATPase, and SieB were exclusive to non-plant species. This observation suggests functional differentiation of CAX genes (Fig. S5).
Functional differentiation of rice CAX genes
The promoter CRE contents and gene coding sequence divergence of the six OsCAX genes from the above analyses suggest their obvious functional differentiation. To obtain additional evidence for this, we examined the expression profiles of the OsCAX genes (Figs. 3 and 4) using the following observations. In Nipponbare (Geng), different OsCAX genes showed strikingly different expression patterns at different developmental stages and in response to different abiotic stress or to ABA and JA treatments. OsCAX1a was expressed strongly and constitutively in all plant tissues at all developmental stages and under all abiotic stress or ABA/JA treatments. OsCAX2 and OsCAX3 showed similar expression patterns to OsCAX1a, although their expression levels were slightly weaker (Figs. 3 and 4), suggesting their important roles in the tolerance of rice to these abiotic stresses. In contrast, the expression of the remaining three OsCAX genes was more tissue or developmentally specific. The expression of OsCAX1b was strong at the seedling stage but became weaker at the reproductive stage (Fig. 3), although its expression in the shoot was strongly upregulated under all abiotic stress and ABA/JA treatments (Fig. 4). The expression of OsCAX1c was weak in the roots under all abiotic stress and ABA/JA treatments. However, its expression in shoots was strongly upregulated under all abiotic stress but downregulated under submergence and ABA/JA treatments (Figs. 3 and 4). The expression of OsCAX4 was weak at the seedling stage and strong in the panicles. Its expression in shoots was also weak under all abiotic stress and hormone treatments. However, its expression in roots was strongly upregulated under salt and JA treatments, suggesting its possible role in rice salt tolerance.

Expression profiles of the OsCAX genes in different tissues and developmental stages. (a) Different tissues and developmental stages expression patterns of the OsCAX gene family in Nipponbare (Geng) and R498 (Xian). (b) Different tissues and developmental stages expression patterns of the OsCAX gene family in IR64 (Xian).

Expression profiles of the OsCAX genes under different abiotic stresses. (a) Expression profiles of OsCAX genes in Nipponbare (Geng) under salt, drought, Pi, Cd, Submergence and cold treatment in shoot and root. (b) Expression profiles of OsCAX genes in Nipponbare (Geng) under ABA and JA treatment in shoot and root. (c) Expression profiles of OsCAX genes in IR64 (Xian) under salt, drought, nitrogen, Submergence, cold and GA treatment in Shoot. (d) Expression profiles of OsCAX genes in IR64 (Xian) under drought, Al and nitrogen treatment in root.
The OsCAX genes in Xian variety R498 showed similar expression patterns as in Nipponbare except for OsCAX1a, whose expression was weaker at the booting and flowering stage. In IR64 (Xian), the six OsCAX genes showed similar expression patterns in terms of tissue and developmental specificities, except that the expression of OsCAX1a and OsCAX2 were strongly upregulated under nitrogen and aluminum stress. Additionally, in IR64, the expression of all six OsCAX genes was strongly upregulated in shoots but strongly downregulated in roots under drought, except for OsCAX2 and OsCAX3, whose expression in roots was also strongly upregulated under drought and GA treatments. OsCAX4 showed minimal response to nitrogen and GA treatments (Figs. 3 and 4). These results indicated that the functional differentiation of the OsCAX genes was at least partially reflected by their different expression patterns. We analyzed the expression differences between Nipponbare and R498 at various growth stages, as well as between Nipponbare and IR64 under different stress conditions (Figs. S6 and S7). No significant expression difference was found between Nipponbare and R498 at different growth stages. However, OsCAX1a showed significant upregulation in the panicle of Nipponbare from the booting to filling stage. This suggests that OsCAX1a may be specifically expressed in the panicle, consistent with Gan et al.'s study indicating its involvement in rice panicle development36. Under stress treatments, the expression levels of six rice CAX genes in Nipponbare were higher than those in IR64, possibly due to the fact that IR64 is a stress-sensitive variety. Therefore, there is a connection between gene expression and the characteristics of the variety.
Associations of OsCAX genes with agronomic traits and abiotic stress tolerance
To gain evidence of the functional importance of the OsCAX genes, we performed GWAS analyses to detect possible associations between the major alleles of the six OsCAX genes and several agronomic traits. Major alleles at OsCAX1a were significantly associated with CL (P = 4.1 × 10−8) (Fig. 2a). Very strong associations were detected for major alleles in OsCAX1b with CL (P = 3.0 × 10−31), TGW (P = 5.9 × 10−22), GW (P = 5.2 × 10−43), and HD (P = 1.1 × 10−37) (Fig. 2b). Major alleles in OsCAX1c were strongly associated with GW (P = 8.0 × 10−70) and CN (P = 4.2 × 10−28) but weakly associated with CL (P = 2.2 × 10−10) (Fig. 2c). For OsCAX2, strong associations were detected with CN (P = 3.8 × 10−18) and HD (P = 3.9 × 10−62) (Fig. 2d). OsCAX3 was also associated with CN and HD, but the associations were weaker than OsCAX2 (Fig. 2e). The major alleles in OsCAX4 were significantly associated with all six traits, although the association was strongest for GW (Fig. 2f).
Using the previously reported phenotypic data of salt treatment during the germination stage in 3KRG50 and agronomic traits under salt treatment in HHZ ILs, we performed GWAS to determine whether the six OsCAX genes were associated with salt tolerance (Fig. S8). OsCAX1a was associated with agronomic traits (HD, days to heading) under salt treatment (Fig. S8c). OsCAX1c was strongly associated with the 48 h imbibition rate at the germination stage (Fig. S8a) and panicle length under salt stress (Fig. S8b).
Discussion
Despite the great achievements in the global efforts of rice functional genomics over the past decades, applications of this theoretical research progress to rice improvement remain very limited for two primary reasons. First, cloned rice genes still account for a small portion of the total genes in the species. Second, the current information on most cloned genes is incomplete or even lacking regarding the number of traits affected, the phenotypic differences among major alleles of each rice gene, and in particular, the functional/genetic relationships between and among different genes (epistasis) /alleles (dominance) and their interactions with the environment. Thus, the greatest challenge or question is how to obtain the most genetic information for genes of unknown function required for improving complex traits through breeding by design (BBD) without going through the tedious process of gene cloning. In this study, we demonstrated a highly efficient strategy for obtaining important information on the functionalities and phenotypic effects of six OsCAX genes. Differing from the classical gene cloning approach, which typically follows single genetic variations of large phenotypic effects on specific traits of interest, this strategy started by selecting a gene family with potentially important but largely unknown functions and then revealed their possible functionalities by systematic analyses of their genetic diversity, expression patterns, associations with traits of agronomic importance from random and experimental populations using publicly available data, and responsiveness to artificial selection during modern breeding. This strategy took several steps to reveal the functionalities of the OsCAX genes that merit further discussion.
Of the six OsCAX genes, OsCAX1a (cluster IA) and OsCAX2 (cluster IB) were inferred to be the original ones presumably from the two ancestor plant species before the WGD event of about 65 mya55, suggested by their extreme genetic conservativeness (as core genes with low genetic diversity) and greater functional versatility (constitutive expression across different plant tissues and upregulated by many abiotic stresses/plant hormones). The specificity of CAX depends on the diversity of amino acid sequences between different CAX, resulting in functional differences between CAX proteins. This was consistent with their known functions as cation transporter proteins10,12. Thus, the reported involvement of OsCAX1a in rice panicle degeneration may not be its primary function36. OsCAX3 and OsCAX4 were apparently derived from OsCAX2 at early time during the long diploidization process after the WGD event while OsCAX1b and OsCAX1c were derived from OsCAX1a through unequal crossing events more recently. The tissue- and development-specific expression patterns of these four OsCAX genes strongly suggested their functions contributing the adaptation of rice plants to specific environmental factors, as reported involvement of OsCAX1c, and OsCAX4 in rice cadmium (Cd2+) uptake and translocation35 and suggested by their associations with tolerance to specific abiotic stresses detected in our GWAS. Clearly, these later derived four OsCAX genes were differentiated from their ancestor ones and acquired new functions during evolution.
CAX represent a significant class of transmembrane transporters. They play crucial roles in maintaining Ca2+ homeostasis, enhancing abiotic stress resistance, and facilitating the transport of heavy metal ions in plants19,56,57. The primary substrate of CAXs is Ca2+, regulating cytoplasmic Ca2+ levels by modulating its flux across the membrane19,58. This involves the transportation of Ca2+. Reports suggest that CAXs in the tonoplast exhibit high capacity and low affinity for Ca2+ 59, indicating a potential role in scenarios with elevated cytoplasmic Ca2+ concentrations. AtCAX1 is localized in the tonoplast and primarily regulates cytoplasmic Ca2+ concentration, exhibiting high transport activity with low affinity for Ca2+ 60,61. This indicates that AtCAX1 may possess Ca2+ tolerance and facilitate Ca2+ uptake and transport under conditions of high Ca2+. Although AtCAX2 exhibits lower affinity for Ca2+ compared to that of AtCAX1, it has a broader range of cation selectivity60,62. This disparity in affinity is likely attributed to variations in key amino acids involved in Ca2+ transport. Notably, a reported mutation from valine to leucine at position of 203 in MdCAX3L-1 significantly enhances Ca2+ transport16,63, indicating that this position plays a pivotal role in determining its Ca2+ transport capacity. The variations in Ca2+ transport capacity correlate with the differentiation of CAXs functions, observed in both Arabidopsis thaliana and Malus domestica60,61,62,63. This suggests a genetic basis for this phenotypic trait. Recent studies have elucidated the molecular mechanism of CAXs in Ca2+ efflux and the regulation of Ca2+ homeostasis, which is vital for plant growth and immune responses. CAXs mediate the scavenging of excess cytoplasmic Ca2+ across various physiological conditions in plants64. Under normal soil growth conditions, plants utilize the Ca2+-CBL-CIPK-CAX1/3 pathway to regionalize Ca2+ into the tonoplast, thereby maintaining [Ca2+]cyt homeostasis. During immune responses, the FLS2-BAK1-BIK1/PBL1 pathway analogously activates CAX1/3, thereby modulating calcium signaling and immune responses64. This study focused on analyzing the physicochemical properties, structural features, phylogenetic relationships, cis-acting elements, and expression patterns of CAXs family members. Future research is necessary to further elucidate the specific functions of CAXs in ionic response, signal transduction, and abiotic stress tolerance, among others.
A second aspect of the OsCAX genes revealed in this study was the rich genetic diversity, particularly the rich gcHap diversity, at the four later derived ones among different rice accessions, even though the observed genetic diversity at the four OsCAX genes was lower than the average genetic diversity of all rice genes27. This was consistent with their expected function in the upstream regulatory role in signal transduction, and with their low gene PAVs observed in rice populations. Interestingly, we observed the significant frequency shifts of the predominant gcHaps at all six OsCAX gene loci in response to the artificial selection during modern rice breeding, suggesting that the OsCAX gene loci may also have functions involved in rice productivity, as suggested by their associations with agronomic traits detected in the GWAS. However, it remains unclear regarding which of the allele(s) were replacing the predominant one at each of the OsCAX gene loci and what specific trait(s) are associated with them. Our observations indicate that the CAX genes across various species exhibit N-terminal self-repression regulation, as extensively documented in previous reports61,65,66,67,68. Based on the 3D structure and Ca2+ transport mechanism of ScVCX169,70, we hypothesize that the inhibitory effect exerted by the N-terminal of the CAX protein arises from its interactions with amino acids situated at specific positions within the transmembrane region. Upon alteration of key amino acids within the transmembrane region, the interaction is significantly impacted or disrupted, subsequently abolishing the inhibitory effect of the self-inhibitory region17,61,68,71. This hypothesis aligns with findings from other species. Deletion or disruption of the N-terminal autoinhibitory region restores the Ca2+ transport capacity of plant CAX proteins. SOS2 activates the Ca2+ transport function of CAX proteins through interactions with the N-terminal autoinhibitory region72,73. Similarly, the preservation of Ca2+ transport activity and protein structure in ScVCX1 relies on specific amino acid interactions within transmembrane regions69. Recent research has demonstrated that in Arabidopsis, AtCAX1 interacts with and is activated by AtSOS2, a crucial component in salt stress resistance in Arabidopsis72. The significance of this observation lies in the N-terminal residue of the CAX protein, as numerous proteins engage with CAX through binding to its N-terminal74,75.
The current knowledge indicates that the number and types of CREs in gene promoter regions represent an important aspect of gene expression regulation and thus gene functionality. Indeed, we detected a total of 25 CREs of five major functional groups, most of which belonged to the abiotic stress and hormone-responsive groups, consistent with their expected primary functions. The presence of multiple CREs in each of the OsCAX gene promoters (mean = 14.5) was a sign of their functional versatility, consistent with their key roles in the upstream of plant signal transduction. In this respect, the significant level of genetic variation in both number and type of CREs in the OsCAX gene promoters observed among the 50 high quality representative rice genomes represent another dimension of genomic diversity within and among populations which has not been well characterized for almost all rice genes. This type of genetic variation in gene promoter regions may have important phenotypic consequences, as reported in the cases of ARE1, GWT2 and GSE5 where a single deletion/insertion or a SNP mutation in the gene promoter regions caused reduced gene expression associated with large phenotypic effects on nitrogen use efficiency/grain yield (ARE1), grain length/yield (GSE5) or grain width and weight (GWT2)76,77,78. In this respect, three interesting observations were noted regarding the CRE number and type variation in the OsCAX gene promoter regions. First, we found that the frequencies of specific CREs present in the OsCAX gene promoters varied considerably, with eight common CREs (Box-4, G-box, MYB, MYC, ABRE, as-1, CGTCA-motif and TGACG-motif) that were present in high frequency in all six OsCAX gene promoters. For example, of the 25 CREs detected, the average copy numbers in the six OsCAX gene promoters were the highest for MYC (4.41), MYB (3.95) and STRE (3.90) across the six OsCAX genes, followed by for CGTCA-motif (3.0), as-1 (3.0) and TGACG-motif (3.0), indicating the primary functions of most OsCAX genes are involved in abiotic stresses and responsive to plant hormones. Clearly, these common CREs provide key information on which TEs involved in the regulation of these common OsCAX genes and their primary functions. Second, each OsCAX gene promoter contains a unique set of CREs in most rice accessions, which were expected to be a major contributor to its functional differentiation. For example, the OsCAX1b promoter contains three strong CREs (MYB, MYC, STRE), six moderately strong CREs (G-box, ABRE, as-1, CGTCA-motif, TGACG-motif, TGA-element and CAT-box). The OsCAX1c promoter contained five strong CREs (as-1, CGTCA-motif, TGACG-motif, ARE and MYC) and three moderately strong CREs (box-4, MYB and TCA-element). OsCAX3 promoter contains two strong CREs (MYB and STRE), nine moderately strong CREs (Box-4, G-box, MYC, ARE, ABRE, as-1, CGTCA-motif, ERE, TGACG-motif and TGA-element). The OsCAX4 promoter contains three strong CREs (MYB, MYC and STRE), six moderately strong CREs (Box-4, ARE, as-1, LTR, CGTCA-motif and TGACG-motif). Thirdly, the remaining CREs each had a single copy in the OsCAX gene promoters in all rice accessions or most of a specific rice population, indicating they contributing to the specific functions of different OsCAX genes. For example, a light responsive motif, AE-box is present only in the promoters of OsCAX1a and OsCAX1b of most rice accessions, but not in other OsCAX genes. A light responsive element, MRE, and the circadian element are present only in the promoter of OsCAX2 of all rice accessions but not in other five OsCAX genes. Fourth, only minor and quantitative differences existed between populations Xian and Geng regarding the average number and type of strong and moderately CREs in the OsCAX gene promoters, indicating their evolutionary conserveness. However, qualitative differences between Xian and Geng were observed in many low copy CREs, particularly in the OsCAX2 and OsCAX4 promoters. In these cases, the CREs were present only in accessions of one subspecies but not in the other. In the OsCAX4 promoter, these CREs included AE-box, G-box, MRE and TGA-element which were present in all or most Geng accessions not but in all or most Xian accessions, and the opposite was true for Box-4, I-box, ERE, and RY-element in all or most Xian accessions but not in all or most Geng accessions. Other CREs included box-4 in OsCAX1b, I-box, ARE, DRE-core and TGA-element of OsCAX2 which were present in most Geng accessions but absent in the Xian accessions. These results suggested that the presence of these Geng-unique CREs in the OsCAX2 and OsCAX4 promoters might have contributed the adaptations of Geng rice to the long-day and colder environments. All these led us to a conclusion that the functional differentiation of different members of a gene family was at least partially achieved by recruiting new CREs in their promoter regions. This also implies that an improved efficiency of knock-in mutation by the CRISPR-case 9 technology can be achieved by creating new CREs in gene promoter regions, particularly for those conserved regulatory genes such as CAX genes. This later point should serve as a new direction in gene-editing efforts to be actively tested experimentally. Thus, future population genomic efforts should be taken to assess the types and average number of CREs in promoters of all rice genes across representative rice genomes, even though accurate assessment of gene promoter diversity within and among different rice populations requires high quality genomic sequence data.
Taking together, combined evidence from our analyses indicated that OsCAX1a and OsCAX2 appear to function as general signal transporters in many processes of rice growth and development and in rice responses to diverse environments, but they might be of less value in rice improvement because of their low genetic diversity. In contrast, OsCAX1b, OsCAX1c, OsCAX3 and OsCAX4 each was expected to be of potential value in rice improvement because of their associations with specific rice traits, their responsiveness to specific abiotic stresses or phytohormones, and relatively high gcHap and CRE diversity. However, additional efforts have to be taken to characterize and verify the ‘desirable’ allele(s) and their phenotypic effects at each of these OsCAX genes, which can be easily achieved by the CRISPR-9 mediated knockout or knock-in mutagenesis experiments48.
Finally, we have demonstrated a highly efficient strategy to obtain important genetic information on genes/alleles of specific gene family through comprehensive analyses using the public genomic and phenotypic data. Obviously, the obtained information on the function and phenotypic effects of different members of the gene family is incomplete. This is virtually true for all rice genes. Fortunately, as more and more high-quality population genomic and phenotypic data of rice are accumulating in the public domain and more powerful analytic tools become available, this strategy can be used to systematically characterize all the 12,000+ rice gene families, generating tremendous amounts of genetic information more applicable to rice improvement. Also, all rice traits of agronomic importance are not controlled by single genes, but by multiple genes acting in complex signaling pathways. Thus, this strategy can be easily extended to pathway-based multi-loci analyses, focusing on all regulatory genes involved in each of important signaling pathways. One important result expected from this type of pathway-based analyses would be the more accurate prediction and quantification of the epistatic relationships between and among different genes acting in each of the signaling pathways, which will eventually allow more accurate prediction of complex trait phenotypes of breeding progenies based on genetic variation of segregating loci, an essential step to realize BBD in future.
Conclusions
Tremendous functional differentiation of six rice CAX genes were revealed through comprehensive transcriptomic and genetic diversity analyses using population genomic and phenotypic variation data. OsCAX2 and OsCAX1a were inferred to be ancient ones and function as general signal transporters in many processes of rice growth and development and in rice responses to diverse environments, while OsCAX1b, OsCAX1c, OsCAX3 and OsCAX4 were derived ones and each acquired new functions for specific rice traits and/or adaptation to specific abiotic stresses during evolution.
Data availability
Data generated and analyzed during the current study are included in this article and its supplementary information files.
References
Weinl, S. & Kudla, J. The CBL–CIPK Ca2+-decoding signaling network: function and perspectives. New Phytol. 184, 517–528 (2009).
Allen, G. J. & Sanders, D. Two voltage-gated, calcium release channels coreside in the vacuolar membrane of broad bean guard cells. The Plant Cell 6, 685–694 (1994).
Snedden, W. A. & Fromm, H. Calmodulin, calmodulin-related proteins and plant responses to the environment. Trends Plant Sci. 3, 299–304 (1998).
Luan, S., Kudla, J., Rodriguez-Concepcion, M., Yalovsky, S. & Gruissem, W. Calmodulins and Calcineurin B–like Proteins. Plant Cell 14, S389–S400 (2002).
Harper, J. F., Breton, G. & Harmon, A. Decoding Ca2+ signals through plant protein kinases. Annu. Rev. Plant Biol. 55, 263–288 (2004).
Kudla, J. et al. Advances and current challenges in calcium signaling. New Phytol. 218, 414–431 (2018).
Ma, X. et al. The CBL–CIPK pathway in plant response to stress signals. Int. J. Mol. Sci. 21, 5668 (2020).
Resentini, F., Ruberti, C., Grenzi, M., Bonza, M. C. & Costa, A. The signatures of organellar calcium. Plant Physiol. 187, 1985–2004 (2021).
Sanders, D., Pelloux, J., Brownlee, C. & Harper, J. F. Calcium at the crossroads of signaling. The Plant Cell 14, S401–S417 (2002).
Hirschi, K. D. Expression of arabidopsis CAX1 in tobacco: Altered calcium homeostasis and increased stress sensitivity. The Plant Cell 11, 2113–2122 (1999).
Dodd, A. N., Kudla, J. & Sanders, D. The language of calcium signaling. Annual Rev. Plant Biol. 61, 593–620 (2010).
Choe, M., Choe, W., Cha, S. & Lee, I. Changes of cationic transport in AtCAX5 transformant yeast by electromagnetic field environments. J. Biol. Phys. 44, 433–448 (2018).
White, P. J. Calcium in plants. Annals Bot. 92, 487–511 (2003).
Demidchik, V., Shabala, S., Isayenkov, S., Cuin, T. A. & Pottosin, I. Calcium transport across plant membranes: Mechanisms and functions. New Phytologist 220, 49–69 (2018).
Bakshi, A., Choi, W. G., Kim, S. H. & Gilroy, S. The vacuolar Ca2+ transporter CATION EXCHANGER 2 regulates cytosolic calcium homeostasis, hypoxic signaling, and response to flooding in Arabidopsis thaliana. New Phytologist 240, 1830–1847 (2023).
Yang, J. et al. MdbHLH4 negatively regulates apple cold tolerance by inhibiting MdCBF1/3 expression and promoting MdCAX3L-2 expression. Plant Physiol. 191, 789–806 (2023).
Kamiya, T., Akahori, T. & Maeshima, M. Expression profile of the genes for rice cation/H+ exchanger family and functional analysis in yeast. Plant Cell Physiol. 46, 1735–1740 (2005).
Emery, L., Whelan, S., Hirschi, K. D. & Pittman, J. K. Protein phylogenetic analysis of Ca2+/cation antiporters and insights into their evolution in plants. Front. Plant Sci. 3, 16842 (2012).
Pittman, J. K., Hirschi, K. D. & Weber, A. CAX-ing a wide net: Cation/H+ transporters in metal remediation and abiotic stress signalling. Plant Biol. 18, 741–749 (2016).
Yu, H. et al. A route to de novo domestication of wild allotetraploid rice. Cell 184, 1156-1170.e1114 (2021).
Yao, W., Li, G., Yu, Y. & Ouyang, Y. funRiceGenes dataset for comprehensive understanding and application of rice functional genes. GigaScience 7, gix119 (2018).
Huang, F. et al. New data and new features of the funricegenes (functionally characterized rice genes) database: 2021 update. Rice 15, 23 (2022).
Wang, W. et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557, 43–49 (2018).
Wang, T. et al. A rice variation map derived from 10 548 rice accessions reveals the importance of rare variants. Nucleic Acids Res. 51, 10924–10933 (2023).
Zhou, Y. et al. Pan-genome inversion index reveals evolutionary insights into the subpopulation structure of Asian rice. Nature Commun. 14, 1567 (2023).
Qin, P. et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184, 3542-3558.e3516 (2021).
Zhang, F. et al. The landscape of gene–CDS–haplotype diversity in rice: Properties, population organization, footprints of domestication and breeding, and implications for genetic improvement. Mol. Plant 14, 787–804 (2021).
Shang, L. et al. A super pan-genomic landscape of rice. Cell Res. 32, 878–896 (2022).
Zhang, F. et al. Long-read sequencing of 111 rice genomes reveals significantly larger pan-genomes. Genome Res 32, 853 (2022).
Ecker, J. R. et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (pav) in genome content. PLoS Genet. 5, e1000734 (2009).
Hirsch, C. D., Evans, J., Buell, C. R. & Hirsch, C. N. Reduced representation approaches to interrogate genome diversity in large repetitive plant genomes. Brief. Funct. Genom. 13, 257–267 (2014).
Lu, F. et al. High-resolution genetic mapping of maize pan-genome sequence anchors. Nature Commun. 6, 1 (2015).
Lin, Y. et al. Identification of natural allelic variation in TTL1 controlling thermotolerance and grain size by a rice super pan-genome. J. Integr. Plant Biol. 65, 2541–2551 (2023).
Yu, Z. et al. Rice gene index: A comprehensive pan-genome database for comparative and functional genomics of Asian rice. Mol. Plant 16, 798–801 (2023).
Zou, W. et al. The rice cation/H+ exchanger family involved in Cd tolerance and transport. Int. J. Mol. Sci. 22, 8186 (2021).
Gan, Q. et al. Ca2+ deficiency triggers panicle degeneration in rice mediated by Ca2+/H+ exchanger OsCAX1a. Plant Cell Environ. 46, 1610–1628 (2023).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49 (2012).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742 (2023).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327 (2002).
Xia, L. et al. Rice expression database (RED): An integrated RNA-Seq-derived gene expression database for rice. J. Genet. Genom. 44, 235–241 (2017).
Peng, H. et al. MBKbase for rice: an integrated omics knowledgebase for molecular breeding in rice. Nucleic Acids Res. 48, D1085 (2019).
Du, H. et al. Sequencing and de novo assembly of a near complete indica rice genome. Nature Commun. 8, 15324 (2017).
Sheldon, A. L. Equitability indices: Dependence on the species count. Ecology 50, 466–467 (1969).
Paradis, E. pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420 (2010).
Templeton, A. R., Crandall, K. A. & Sing, C. F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III Cladogram estimation. Genetics 132, 619–633 (1992).
Wang, C. C. et al. Towards a deeper haplotype mining of complex traits in rice with RFGB v. 2.0. Plant Biotechnol. J. 18, 14–16 (2019).
Zeng, W. et al. Functional characterization and allelic mining of OsGLR genes for potential uses in rice improvement. Front. Plant Sci. 14, 1236251 (2023).
Mansueto, L. et al. Rice SNP-seek database update: new SNPs, indels, and queries. Nucleic Acids Res. 45, D1075–D1081 (2017).
Y. Cui, F. Zhang, Y. Zhou, The application of multi-locus GWAS for the detection of salt-tolerance loci in rice, Front. Plant Sci., 9 (2018).
Huang, X. et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genet. 42, 961–967 (2010).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Wang, X. et al. Genome alignment spanning major poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Molecular Plant 8, 885–898 (2015).
Sun, C. et al. RPAN: rice pan-genome browser for ∼3000 rice genomes. Nucleic Acids Res. 45, 597–605 (2017).
Salman-Minkov, A., Sabath, N. & Mayrose, I. Whole-genome duplication as a key factor in crop domestication. Nature Plants 2, 1 (2016).
Cheng, N.-H., Pittman, J. K., Shigaki, T. & Hirschi, K. D. Characterization of CAX4, an arabidopsis H+/cation antiporter. Plant Physiol. 128, 1245–1254 (2002).
Korenkov, V. et al. Enhanced Cd2+-selective root-tonoplast-transport in tobaccos expressing Arabidopsis cation exchangers. Planta 225, 403–411 (2006).
Ma, Y. & Berkowitz, G. A. Multimeric CAX complexes and Ca2+ signaling—beyond humdrum housekeeping. J. Exp. Bot. 68, 3997–3999 (2017).
Hirschi, K. D., Korenkov, V. D., Wilganowski, N. L. & Wagner, G. J. Expression of arabidopsis CAX2 in tobacco altered metal accumulation and increased manganese tolerance. Plant Physiol. 124, 125–134 (2000).
Cheng, N.-H. et al. Functional association of arabidopsis CAX1 and CAX3 Is required for normal growth and ion homeostasis. Plant Physiol. 138, 2048–2060 (2005).
Pittman, J. K. & Hirschi, K. D. Regulation of CAX1, an arabidopsis Ca2+/H+ antiporter identification of an N-terminal autoinhibitory domain. Plant Physiol. 127, 1020–1029 (2001).
Pittman, J. K. & Hirschi, K. D. Don’t shoot the (second) messenger: endomembrane transporters and binding proteins modulate cytosolic Ca2+ levels. Current Opin. Plant Biol. 6, 257–262 (2003).
Mao, K. et al. Genome-wide analysis of the apple CaCA superfamily reveals that MdCAX proteins are involved in the abiotic stress response as calcium transporters. BMC Plant Biology 21, 1 (2021).
Wang, C. et al. Mechanisms of calcium homeostasis orchestrate plant growth and immunity. Nature 627, 382–388 (2024).
Shigaki, T. et al. The expression of the open reading frame of arabidopsis CAX1, but not its cDNA, confers metal tolerance in yeast. Plant Biol. 12, 935–939 (2010).
Chung, M. Y. et al. Modest calcium increase in tomatoes expressing a variant of Arabidopsis cation/H+ antiporter. Plant Biotechnol. Reports 4, 15–21 (2009).
Edmond, C. et al. Comparative analysis of CAX2-like cation transporters indicates functional and regulatory diversity. Biochem. J. 418, 145–154 (2009).
Zhang, J. et al. GhCAX3 Gene, a novel Ca2+/H+ exchanger from cotton, confers regulation of cold response and ABA induced signal transduction. PLoS ONE 8, e66303 (2013).
Waight, A. B. et al. Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature 499, 107–110 (2013).
Pittman, J. K. & Hirschi, K. D. Phylogenetic analysis and protein structure modelling identifies distinct Ca2+/Cation antiporters and conservation of gene family structure within Arabidopsis and rice species. Rice 9, 1 (2016).
Park, S. et al. Increased calcium levels and prolonged shelf life in tomatoes expressing Arabidopsis H+/Ca2+ transporters. Plant Physiol. 139, 1194–1206 (2005).
Cheng, N.-H., Pittman, J. K., Zhu, J.-K. & Hirschi, K. D. The protein kinase SOS2 activates the arabidopsis H+/Ca2+ antiporter CAX1 to integrate calcium transport and salt tolerance. J. Biol. Chem. 279, 2922–2926 (2004).
Cheng, N.-H., Pittman, J. K., Barkla, B. J., Shigaki, T. & Hirschi, K. D. The Arabidopsis cax1 mutant exhibits impaired ion homeostasis, development, and hormonal responses and reveals interplay among vacuolar transporters. The Plant Cell 15, 347–364 (2003).
Shigaki, T. & Hirschi, K. D. Diverse functions and molecular properties emerging for CAX Cation/H+ exchangers in plants. Plant Biol. 8, 419–429 (2008).
Manohar, M., Shigaki, T. & Hirschi, K. D. Plant cation/H+ exchangers (CAXs): biological functions and genetic manipulations. Plant Biol. 13, 561–569 (2011).
Duan, P. et al. Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol. Plant 10, 685–694 (2017).
Wang, Q. et al. Genetic variations in ARE1 mediate grain yield by modulating nitrogen utilization in rice. Nature Commun. 9, 735 (2018).
Ruan, B. et al. Natural variation in the promoter of TGW2 determines grain width and weight in rice. New Phytologist 227, 629–640 (2020).
Funding
This research was financially supported by National Natural Science Foundation of China (U21A20214, 31971927, 32101768 and 32261143470); National Key Research and Development Program of China “Creation and Application of Novel Japonica Rice Germplasm with High Quality and Yeild in the Middle and Lower Reaches of Yangtze River” (2023YFD1200900); Key Research and Development Program of Anhui Province (202004a06020024) and National Key R&D Program of China (2022YFE0139400).
Author information
Authors and Affiliations
Contributions
L.M and M.L. formulated the research concept. S.L., Y.C., Y.Z., T.F., J.C., L.L., Y.Q., T.H., C.Z., F.W. and W.Z. performed the information collection and data analysis. S.L., Y.C., Y.Z. M.L. and L.M drafted the manuscript. M.L., L.M. and Z.L. revised the manuscript. M.L., L.M. generously sponsored the research. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lian, S., Chen, Y., Zhou, Y. et al. Functional differentiation and genetic diversity of rice cation exchanger (CAX) genes and their potential use in rice improvement. Sci Rep 14, 8642 (2024). https://doi.org/10.1038/s41598-024-58224-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58224-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
