
Abstract
Streptococcus gallolyticus is a non-motile, gram-positive bacterium that causes infective endocarditis. S. gallolyticus has developed resistance to existing antibiotics, and no vaccine is currently available. Therefore, it is essential to develop an effective S. gallolyticus vaccine. Core proteomics was used in this study together with subtractive proteomics and reverse vaccinology approach to find antigenic proteins that could be utilized for the design of the S. gallolyticus multi-epitope vaccine. The pipeline identified two antigenic proteins as potential vaccine targets: penicillin-binding protein and the ATP synthase subunit. T and B cell epitopes from the specific proteins were forecasted employing several immunoinformatics and bioinformatics resources. A vaccine (360 amino acids) was created using a combination of seven cytotoxic T cell lymphocyte (CTL), three helper T cell lymphocyte (HTL), and five linear B cell lymphocyte (LBL) epitopes. To increase immune responses, the vaccine was paired with a cholera enterotoxin subunit B (CTB) adjuvant. The developed vaccine was highly antigenic, non-allergenic, and stable for human use. The vaccine's binding affinity and molecular interactions with the human immunological receptor TLR4 were studied using molecular mechanics/generalized Born surface area (MMGBSA), molecular docking, and molecular dynamic (MD) simulation analyses. Escherichia coli (strain K12) plasmid vector pET-28a ( +) was used to examine the ability of the vaccine to be expressed. According to the outcomes of these computer experiments, the vaccine is quite promising in terms of developing a protective immunity against diseases. However, in vitro and animal research are required to validate our findings.
Similar content being viewed by others


Introduction
Streptococcus gallolyticus is a non-motile, gram-positive bacterium that was formerly known as Streptococcus bovis. It is a phenotypically diverse strain of bacteria from the Lancefield Group D Streptococci1. Although frequently found in microflora, 2.5-15% of it is found in a healthy person's gastrointestinal system and develops into an opportunistic pathogen that causes a variety of diseases, including colon cancer, infectious endocarditis, septicemia, and meningitis2.
Infective endocarditis incidence has increased significantly globally over the past 20 years3,4,5. Streptococcal infections contributed significantly to 2.6-7 occurrences of endocarditis per 100,000 people recorded each year, with incidences of 31% in European nations, 39% in South America, 17% in North America, and 32% in the rest of the world5. This condition primarily affects older patients, with a median age of 586. S. gallolyticus endocarditis is more likely to occur in those who consume fresh dairy products or raw meat, have a history of hepatic illnesses, have weakened immune systems, and have comorbid conditions such as rheumatic diseases and diabetes mellitus7.S. gallolyticus attempts to injure the endocardium when there is a primary infection, a metabolic disease, or an immune-compromised state. Following this injury, the loss of fibrin and platelets causes the formation of a thrombus. After thrombus development, the bacteria travel through the thrombus into the bloodstream. S. gallolyticus contains virulence traits that enable it to enter the bloodstream paracellularly and adhere to the endocardium, the collagen-rich surface of the damaged heart valve, without triggering a major immune response. This bacterium multiplies and forms a biofilm after adhering to the endocardium, inflaming the heart's lining and leading to endocarditis8,9.
Commonly prescribed antibiotics for infectious endocarditis include Gentamycin, Penicillin G, and streptomycin. Alternative options for individuals with penicillin allergies include vancomycin and gentamicin-related Ceftriaxone10. A costly surgical intervention may be required for patients who have a chronic fever and are resistant to medicinal therapy. S. gallolyticus is penicillin-resistant, and one strain of S. gallolyticus has also been proven to be tetracycline-resistant11. Hence, the development of an effective endocarditis treatment strategy, innovative therapeutic targets, and effective vaccines are urgently needed.
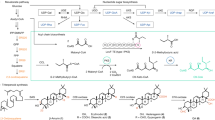
Vaccines offer protection against infectious diseases12. Vaccines stimulate the immune system, and their immunogenicity is critical in achieving protection against specific pathogens. It takes a long time for a vaccine candidate to successfully develop to the point of commercial release because of the numerous clinical trial phases that must be completed and the substantial financial commitment required.However, the vaccine's success rate can be bleak at times. In such a scenario, the immunoinformatics approach is beneficial in reducing the time required for vaccine development when compared to conventional approaches. There are examples of successful vaccine candidate development using in silico approaches13,14,15,16,17,18,19. Numerous computer methods have been developed for quick identification, such as subtractive genomic and core genomic approaches, which enable us to find the core essential genes that are not homologous to the human genome18,19,20,21. This study's goal is to employ in silico techniques to discover potential vaccine targets by connecting the proteome and genetic data of the S. gallolyticus species. In this study, the core proteome of seven strains of S. gallolyticus was analyzed to find potential vaccine candidates. S. gallolyticus proteins have been targeted to forecast B cell, IFN, and T cell epitopes. Subunit vaccines were developed by selecting conserved and high antigenic epitopes and then combining them with linkers and adjuvants (Figure 1). The vaccine's physicochemical properties, allergenicity, structural features, and antigenicity were investigated using online tools. Furthermore, docking among the TLR-4 and vaccine was performed. The advanced polyprotein formation was ultimately investigated via in-silico cloning. This study could open the path for the creation of dynamic and effective vaccines that comprise a special blend of several antigenic peptides produced from S. gallolyticus to suppress S. gallolyticus infection.

Graphical abstract.
Material and methods
Core proteomes identification
In this study, seven S. gallolyticus strains with complete assembly levels were considered for pan-genome analysis and their proteomes were downloaded from NCBI.. Humans were considered hosts. Their information is presented in Additional File 1: Table S1. The core proteome analysis was then performed on the proteomes using the Bacterial Pan Genome Analysis Tool (BPGA)22to identify conserved proteins shared by all strains to select potential vaccine candidates.
Vaccine candidate prioritization
It is believed that essential proteins are crucial for cell viability. The core proteome of S. gallolyticus strains was uploaded to the Geptop 2.0 server to find essential proteins. The cutoff value for essentiality was set at 0.24. Geptop can be used for any bacterial species with a sequenced genome23,24. Essential proteins were uploaded to BlastP using an e-value of 10-4, and query coverage and identity of more than 70 % and 30% respectively against the human proteome to find non-homologous protein sequences of S. gallolyticus25. Proteins having no similarity with the human proteome were considered non-homologous proteins. For genome analysis and genome annotation in bacterial infections, it is crucial to predict protein subcellular localization because these proteins may serve as the main targets for drugs or vaccines26. The BUSCA server was employed to forecast non-homologous protein subcellular localization27. Furthermore, their antigenicity and allergenicity were evaluated using the Vaxijen 2.0 and AllerTop tools, respectively28,29. The molecular weight (MW) and stability were calculated using the ProtParam program30. Further, the TMHMM server was utilized to forecast transmembrane helices31. Proteins having 0 transmembrane helices that are non-allergenic, antigenic, and stable were identified as prospective vaccine candidates.
Prediction of epitopes and selection phase
T-cell (HTL and CTL) and B-cell epitopes were predicted from the potential vaccine candidates. The MHC-I and MHC-II binding tools of the Immune Epitope Database (IEDB) were used to predict CTL and HTL epitopes, respectively32. The consensus approach was applied to a provided protein sequence in FASTA format. The human species was picked as the origin of all alleles. The epitopes with a consensus score of < 2were chosen based on their capacity to bind. ABCPred server was used to forecast linear B cell epitopes. To further evaluate the anticipated epitopes, ToxinPred, VaxiJen (v2.0), the MHC class I immunogenicity, and Allergen FP1.0 servers were employed33,34,35,36. Finally, the IFNepitope web server was utilized to screen HTL epitopes that induce IFN-gamma37
Population coverage analysis
IEDB's web-based population coverage analysis tool was utilized to determine the population distribution of predicted epitopes. This tool provides the frequency of HLA alleles in 78 various population subgroups across 11 various geographic locations38,39.
Designing and structural analysis of vaccine
Due to their small size, epitopes typically aren't able to elicit an immune response when used alone in a vaccine. In order to activate the innate and adaptive immune systems, they require a carrier that contains strong immunostimulatory adjuvants40. Thus, an EAAAK linker was used to join the vaccine's N terminal to the cholera enterotoxin subunit B (CTB) adjuvant. In order to replicate the immunogen's ability to function as a separate immunogen and produce higher antibody concentrations than a single immunogen, linkers are necessary in the vaccine's design41. The AAY, GPGPG, and KK linkers were used to join CTL, HTL, and LBL epitopes, respectively.The 3Dpro program from the SCRATCH suite was utilized to forecast the 3D structure of the vaccine42. The structure was refined further using an online galaxy refinement server43. The refined structure was further validated through the RAMPAGE server, Verify 3D, PROSAweb server, and ERRAT server44,45,46,47.
Post analysis of vaccine
Allergenicity testing can predict the vaccine's capacity to produce allergic responses. Hence, the AllerTop server was used30. VaxiJen v2.0 was utilized to analyze structural antigenicity28. The SCRATCH suite's SOLpro server was used to determine the vaccine's solubility48. To determine the nature and stability of the vaccine, stereochemical characteristics were predicted utilizing the ProtParam server30. Finding the secondary structure required the use of the SOPMA server49. Conformational and linear B cell epitopes of the vaccine were forecasted through the Ellipro server50.
Disulfide engineering
Disulfide engineering is a one-of-a-kind technique for inserting disulphide links within the target protein structure. Disulphide bonds include covalent interactions that aid in protein stability and the study of protein dynamics and interactions. To improve stability, the vaccine structure was uploaded to the Disulphide by Design (DbD) 2.12 tool51. A series of parameters was used to identify potential residue pairs (2.2 energy value and -87 to + 97 chi3 value), which were then altered with a cysteine residue.
Molecular docking analysis
For the protein-protein interaction, the HADDOCK-v-2.4 server was used52. HADDOCK was used to create a model of the biomolecular complexes. The docked complexes were visualized using PyMOL-v.1.353. PDBsum, an online service, was utilized to evaluate interactions between docked complexes54. Docking analysis was used to evaluate the binding potency of vaccine constructs with immune cell receptor (TLR4). Molecular dynamics simulations were then performed on docked complexes with high binding affinities.
Molecular dynamics simulation
Using the Schrodinger suit's Desmond software, MD simulations were run at 100 ns55. MDS was employed to analyze a dynamic interaction of protein-protein complexes generated by molecular docking. The complex was preprocessed, then optimized and minimized. The forcefield OPLS_2005 was employed in the minimization procedure56. Transferable intermolecular potential with a three point (TIP3P) solvent model along with an orthorhombic box (10 × 10 × 10) was added using the system builder tool57. The model was neutralized by adding counterions as necessary, and 0.15 M of NaCl salt was added to simulate the physiological state. Conditions for the NPT ensemble were 1 atm pressure and 300 K temperature. Prior to the simulation, the complex was relaxed. The trajectory was saved at 50 ps for simulation results analysis.
MMGBSA analysis
Binding free energies are frequently calculated using the empirical equation-based MMGBSA approach. The accuracy of this approach exceeds that of many molecular docking score functions58,59. The ante-MMPBSA.py module examined and produced the complex's initial prompt files. Using the receptor, complex, and vaccine energies variation, free energy was determined:
Codon optimization and in silico cloning
The increased expression rates may be a result of a unique codon adaptation method designed for E. coli K12. The main sequence for the subunit vaccine was expressed more frequently in the host cell type E. coli K12 using the technique, and it was then submitted to the JAVA Codon Adaptation Tool (Jcat) for modification60. The restriction enzyme cleavage site, the RHO independent transcription termination site, and the bacterial ribosome binding site were all avoided. Using the GenSmart design tool, the optimum nucleotide sequence for the developed multi-subunit vaccine was cloned between restriction sites within the E. coli pET-28a (+) vector61.
Immune simulation
The C-ImmSim server was utilized to run in silico immune simulations to test the immunological response and immunogenicity of the S. gallolyticus vaccine62. The lowest time interval between two vaccine doses that have been scientifically advised is one month. One-time, 84-time, and 168-time step variables were developed for three inoculations that were given at one-month intervals since a one-time step equals 8 hours of daily life. All other triggering parameters retained their preset levels.
Results
Core proteome retrieval and vaccine candidate prioritization
Seven well-known pathogenic strains of S. gallolyticus were taken into consideration for the development of a vaccine. Core proteome analysis was performed, and 9576 proteins were retrieved. After being uploaded to the Geptop server for essential protein prediction, 325 of the 9576 proteins were determined to be essential genes. Blast P was used to find essential proteins that are not similar to humans. A total of 138 non-homologous proteins were discovered out of 325 important proteins. The prediction of subcellular localization was utilized to determine how certain proteins fulfill their roles. 121 of the total 138 targets were anticipated to be cytoplasmic and were therefore omitted from the research. The remaining 17 cytoplasmic membrane proteins were considered for future analysis. These proteins were also tested for antigenicity, allergenicity, and stability. Six proteins have been identified as antigenic, allergenic, and stable. Additionally, their transmembrane helices were evaluated. Two antigenic, allergenic, and stable proteins having 0 transmembrane helices were selected as vaccine candidates: penicillin-binding protein and ATP synthase subunit.
Epitopes selection phase
CTL, B-cell, and HTL epitopes of specific antigenic proteins were forecasted. A large number of epitopes were forecasted, and the top seven CTL epitopes for vaccine formulation were chosen because they were non-toxic, immunogenic,antigenic, and non-allergenic (Table 1). Similarly, the top three non-allergenic, antigenic, immunogenic, IFN-gamma inducing, and non-toxic HTL epitopes were chosen for vaccine design (Table 2). Likewise, the top five antigenic, immunogenic, non-allergenic, and non-toxic LBL epitopes were picked for vaccine design (Table 3). Toxicity of the final epitopes were predicted and mentioned in Additional File 1: Table S2.
Population coverage analysis
A population coverage analysis was conducted on specific epitopes (CTL and HTL) used in the development of the multi-epitope vaccine. The chosen epitopes, according to the statistics, encompass 96.15% of the global population. Figure 2 shows the population of various countries.

S. gallolyticus vaccine population coverage map, 96.15% of the global population is covered.
Designing and structural analysis of vaccine
To construct the vaccine, 7 CTL, 3 HTL, and 5 B-cell epitopes were coupled with the appropriate adjuvant and linkers. The adjuvant, cholera enterotoxin subunit B (236 amino acids), was linked to the N-terminus of the vaccine via the EAAAK linker. Following that, AAY linkers, GPGPG linkers, and KK linkers were employed to connect CTL, HTL, and B-cell epitopes, respectively (Figure 3). The final vaccine has 360 amino acids (Figure 4A).

The S. gallolyticus vaccine construct is shown schematically.

Vaccine Construct Prediction and Validation. (A) The adjuvant sequence is shown in maroon. The EAAAK linker in red, the epitope sequence is black, the AAY linker in golden, the GPGPG connectors in blue, and the KK linkers in green (B) 3D structure of S. gallolyticus vaccine (C) Ramachandran plot analysis of the structure which shows that 9.0% and 86.9% of all amino acids were positioned in the permitted and most favorable regions, respectively, whereas 2.2% were found in the disallowed region.
The 3Dpro server was used to anticipate the 3D structure of the vaccine (Figure 4B). Additionally, the Galaxy refine server was used to refine it. The model's accuracy was confirmed by a Ramachandran plot study, which showed that 9.0% and 86.9% of all amino acids were positioned in the permitted and most favorable regions, respectively, whereas 2.2% were found in the disallowed region (Figure 4C). Further analysis revealed that the final vaccine structure, with a quality factor of 77.632 and a Z-score of -3.06 does not contain any poor rotamers.
Post analysis of vaccine
The ProtParam program was used to analyze the stereo-chemical properties of the generated structure, which indicated that the vaccine structure had an isoelectric point of 9.11 and a molecular weight of 39371.79 daltons, demonstrating the basic nature of the constructed vaccine structure. There are 39 positive amino acids (arginine, lysine) and 32 negative amino acids (glutamic acid, aspartic acid). The proposed structure's instability index is 21.74 and the aliphatic index is 99.33, both of which show that the vaccine's host-designed structure is stable. The GRAVY value was 0.199, showing that the vaccine structure is hydrophobic. The half-life of this vaccine design was estimated to be 30 hours in mammals (in vivo), > 20 hours in yeast (in vivo), and > 10 hours in E. coli (in vivo). Based on the SOLpro server results, it was predicted that the proposed vaccine structure would be soluble with a probability of 0.684246, allowing for simple access to the host. The structure of non-allergenic, non-toxic, and antigenic vaccine was also predicted. According to Vaxijen, the antigenicity result was 0.8738. SOPMA was used to investigate the vaccine's secondary structure. Among the 359 amino acids, 174 are involved in the formation of the α-helix (48.33% of the overall vaccine structure), 80 are engaged in the extended strands (22.22%), and 80 are involved in the coils (22.22%) formation. The Ellipro server predicted 6 continuous/linear B cell epitopes (Table 4) and 6 discontinuous/conformational B cell epitopes in the vaccine model (Table 5).
Disulfide engineering
The vaccine sequence was examined using the Disulfide by Design 2.13 server, and a total of twenty potential residue pairs that could form a disulfide bond were found. After accounting for the bond energy and the X3 parameters, one pair of residues (113THR-116ALA) was chosen because its results fit the requirements specified earlier (Figure 5).

Disulphide engineering of vaccine structure to enhance stability. The red color represents one mutated pair selected based upon energy and X3 value.
Molecular docking
To elicit an effective immune response, a vaccine must have a significant affinity for the host's immunological receptors, notably toll-like receptors. The protein-protein docking investigation between the human TLR4 and the proposed vaccine was performed using the HADDOCK v. 2.4 server. According to the docking research, the vaccine and TLR4 exhibit strong binding interactions. The TLR4-vaccine binding score was 698.1 +/- 21.8 kcal/mol. The statistics from the docking assay are depicted in Table 6. In Figure 6, a docked complex is displayed. Cyan is used to symbolize TLR4, while pink is used to represent the vaccine. The PDBsum server was utilized to obtain in-depth knowledge of the binding interactions between TLR4 and vaccine in order to create the interactions map among the docked-complexes. Four hydrogen bonds between the vaccine and TLR4 in the range of 2.91Å were detected.

TLR-4 receptor and vaccine docked together. TLR4 is displayed in cyan, while the vaccine is displayed in pink. Red lines between the residues represents salt bridges, yellow line represents disulphide bonds, blue line represents hydrogen bonds, and orange line represents non-bonded contacts.
MD simulation
Molecular Dynamics (MD) simulations are commonly used to analyze the dynamics and behavior of protein molecules at an atomic level. In MD simulations, the RMSD can be used to monitor the stability and conformational changes of a protein over time. To calculate the RMSD of a protein in an MD simulation, the trajectory of the protein's atomic coordinates is first extracted from the simulation output. Then, a reference structure is chosen, which can be either an experimental structure or an initial structure used in the simulation. The trajectory frames are superimposed onto the reference structure, and the RMSD is calculated for each frame. The RMSD values can be plotted as a function of time to visualize the protein's conformational changes and to determine the stability of the protein structure over the course of the simulation. Additionally, the RMSD values can be used to compare different simulations or to evaluate the accuracy of a computational model of a protein structure. In Figure 7 A, it has been shown that the vaccine-receptor docked complex was stable during the 100 ns time period of simulation. During 40 ns it was between 3.2 and 4.8 Å and after 40 ns until 100 ns, it remained between 4.8 Å and 5.6 Å which was quite acceptable. RMSD was slightly higher than an ideal value, but this was due to the loop region in the complex and it will not affect the function of the vaccine construct. RMSF (Root Mean Square Fluctuation) is a measure of the flexibility of a protein molecule. During an MD (Molecular Dynamics) simulation, the RMSF of a protein can be calculated by measuring the deviation of each atom in the protein from its average position over the course of the simulation. The RMSF profile provides a useful insight into the flexibility of different regions of the protein, which can help to understand the protein's function and how it interacts with other molecules. Similarly, Figure 7B revealed the structure compatibility during the 100 ns, but at some residues, RMSF was higher due to other secondary structures instead of alpha helices and beta sheets. RMSF was also in the range of an ideal value. It was between 3Å and 4Å.

Molecular dynamics simulation-based statistical analysis to evaluate the inter-molecular stability and dynamics of the complexes: (A) Root Mean Square Deviation (RMSD) of the vaccine-receptor complex. (B) Root Mean Square Fluctuation (RMSF) of the vaccine-receptor complex.
MMGBSA analysis
Using the MMGBSA, the binding free energy of the docked complex was determined and is shown in Table 7. ΔGtotal value is typically used to calculate the protein-ligand complex's stability63. The stability of the complex is indicated by lower values of ΔGtotal, and vice versa. Van der Waals energy (EvdW), electrostatic energy (Eele), and GGB (electrostatic contribution to solvation-free energy via Generalized Born) are some of the protein-ligand interactions that contribute to the total binding free energy calculated using the MM/GBSA model.
Immune simulation
The immunogenic characteristics of potential S. gallolyticus vaccine candidate was ascertained using the C-IMMSIM server. Elevated IgM concentrations were found during the initial reaction, as predicted by the modeling results. Secondary and tertiary responses both demonstrated a typical increase in immunoglobulin activity, with accompanying antigen depletion (IgM, IgG + IgM, IgG1 + IgG2, and antibodies) (Figure 8A). Additionally, there were increased amounts of cytokines, including IFN- and IL-2, which are crucial for reducing cellular immunity and viral replication. The vaccine's immune-stimulating qualities ensured that it would be efficacious in human subjects (Figure 8B).

In silico immune simulation results obtained after administration of three injections of the vaccine. (A) Concentrations of immunoglobulins in proportion to antigen concentrations. (B) The production of various cytokines in response to the administration of c-ImmSim vaccine.
Codon optimization and in silico cloning
Because different codon usage patterns inhibit the translation of foreign genes, codon adaptation is the most efficient strategy for enhancing translational efficacy. The JCat program was used to optimize the codon usage of our developed vaccine in relation to the E. coli K12 strain. A CAI of 1 and a GC content of 48.3% in the optimized sequence indicated that it was effectively expressed in the E. coli host. As shown in Figure 9, the updated codon sequence from the vaccine construct was then inserted between the XhoI and HindIII restriction sites in the PET28a (+) E. coli expression vector. The size of the clone was 6449bp.

S. gallolyticus vaccine in silico restriction cloning into the pET28a (+) expression vector between between the XhoI and HindIII restriction sites. Vaccine construct is depicted as MCS in blue colour.
Discussion
S. gallolyticus is one of the most common causes of bacterial endocarditis, accounting for up to 15-20% of all cases. S. gallolyticus endocarditis usually occurs in people with a history of congenital heart defects, heart valve disease, or intravenous drug use. Endocarditis caused by S. gallolyticus can be a serious and life-threatening condition with a mortality rate of 10-50%. Bacteria can enter the bloodstream through the mouth, gastrointestinal or dental surgery, or other sources of infection. S. gallolyticus is known to develop antibiotic resistance, which can complicate treatment and lead to treatment failure64,65. So, an effective vaccine against S. gallolyticus needs to be developed. Vaccines can help reduce the morbidity and mortality associated with this disease. Overall, the development of a vaccine against S. gallolyticus-associated endocarditis could have significant public health benefits, especially for people who already have heart disease and other risk factors. However, vaccine development requires extensive research, clinical trials, and regulatory approval, which can be a long and difficult process. The use of bioinformatics, reverse vaccinology, and immunoinformatics approaches has recently become an alternative method for developing vaccines against various pathogens66,67. Similar techniques have previously been used to propose vaccines for Mycoplasma pneumoniae68, Klebsiella aerogenes69, Schistosomiasis70, and Campylobacter jejuni71.
We developed multiple epitope-based vaccines against S. gallolyticus using pan-genome, immunoinformatics, subtractive proteomics, molecular docking, and simulation techniques. Seven well-known pathogenic S. gallolyticus strains were considered for vaccine development, and their core proteome analysis was performed. Because core proteins are present in all or most strains of the pathogen being targeted and their presence in vaccine development offers immune protection against a broader range of pathogen species, they are now being studied in the creation of vaccines69. The core proteome underwent a subtractive proteomics procedure to reveal essential, non-homologous, antigenic, immunogenic, and allergenic proteins for a vaccine. The presence of transmembrane helices is also a significant parameter for eliminating proteins. Since purifying proteins with multiple transmembrane helices is an arduous task, it appears wise to omit such proteins from the selection process. During the vaccine development process, this method checks that a prospective vaccine is suitable for experimental validation and that the proteins have little resemblance to human proteins to lessen the possibility of eliciting an autoimmune response. Two antigenic proteins: penicillin-binding protein and ATP synthase subunit were shortlisted as vaccine candidates. The target proteins anticipated CTL, HTL, and LBL epitopes. The majority of the work for the study was processed on IEDB tools, and the application was suitable and appropriate for thorough analysis, as indicated by Dhanda et al. (2019)47. Epitopes for the multi-epitope candidate were chosen for their antigenicity, immunogenicity, allergenicity, and toxicity. For a vaccine molecule to be effective, it must offer broad-spectrum defense against a variety of populations worldwide. Therefore, when developing an epitope-based subunit vaccination, it is necessary to identify the population fractions in the desired endemic regions according to HLA genotypic frequencies. The results showed that the chosen HTL and CTL epitopes covered a large portion of the endemic S. gallolyticus population.
Three linkers, successfully utilized by Israr et al.72, Ahtisham et al.73, Naveed et al.74,75, and Alizadeh et al.76, were used to adjoin the selected epitopes together. The AAY (Ala-Ala-Tyr) linker is the cleavage site for proteasomes in mammalian cells. Hence, the epitopes linked by the AAY linker are efficiently segregated within cells, lowering junctional immunogenicity. The AAY linker also improves the immunogenicity of the multi-epitope vaccine77. The lysosomal protease Cathepsin B targets the KK linker (lysine linker) in order to process the antigenic peptides and prepare them for presentation on the cell surface in an MHC-II restricted antigen presentation. Additionally, it is essential for lowering junctional immunogenicity because it prevents the formation of antibodies against the peptide sequence that occurs when individual epitopes are joined linearly78. Moreover, KK linkers boost immunogenicity79.GPGPG linkers have been shown to generate TH lymphocyte (HTL) responses, which is critical for a multi-epitope vaccination. Additionally, the GPGPG linker is an effective technique for reducing junctional immunogenicity, which restores the immunogenicity of specific epitopes. Livingston et al. empirically proved this using mouse models80. As an adjuvant, cholera toxin B subunit (CTB) was combined with the EAAAK linker. Due to its salt bridge coupled to lysine and glutamic acid, the EAAAK linker creates a stable helix structure and prevents protein domains from convergence81.CTB has been investigated as a conventional mucosal adjuvant that can increase the immunogenicity of vaccines68,82. Employing the adjuvant used by Mariam et al.83 and Tamalika et al.84 helped us achieve the desired stability while not affecting the antigenicity.
The structure of the developed vaccine solution was predicted to allow easy access to the host. Furthermore, the projected findings demonstrate that the S. gallolyticus vaccine is non-toxic, antigenic, and non-allergenic. The outcomes for each of the three categories aligned with the findings of Naveed et al. (2021), who conducted antigenicity analyses all along the way75. Similar methods were used in this study, and the final epitopes were selected by processing each epitope's unique antigenicity and allergenicity. A good vaccine must have antigenic qualities that are required to elicit the host's immunological response, as well as the ability to develop long-lasting adaptive immunity through the antigen. Solubility is a crucial development characteristic because the vaccine will be delivered in a water environment inside the host body. It has been found that low solubility subunit vaccines are ineffective at producing significant amounts of viral proteins. Hence, creating vaccines with high solubility is crucial for various functional and biochemical studies. The vaccine design was analyzed to be soluble upon expression, meaning that it would have easy access to the host.
TLRs are essential for innate immune system activation and viral particle identification. TLRs have the potential to be an important target for the creation of vaccines and disease prevention85. The precise binding affinities and interactions of the multi-epitope final subunit vaccine design against TLR4 were investigated using molecular docking and simulation studies. During docking, a significant number of H-bonds were observed, as well as minor fluctuations during MD simulations. These findings suggest that the vaccine can effectively bind to TLR4.
Codon optimization is required in prokaryotic hosts for the production of eukaryotic proteins because all synonymous codons within a codon family do not express diverse proteins at the same rate in E.coli67. Therefore, to increase protein expression levels, codon optimization was carried out in E. coli strain K12. Hence, the codon compatibility index (1.0) and the GC content (48%) were determined, indicating a significant likelihood of protein expression. Furthermore, immunological simulation of the designed vaccine produced promising findings in terms of cellular and humoral immune response. According to the bioinformatics analysis of the vaccine design, this candidate may be particularly effective against S. gallolyticus. Immunoinformatics/computational methodologies were developed on the basis of experimental procedures to create raw data for research objectives. The consistency and proficiency of computer algorithms limit the accuracy of immunoinformatics predictions. Therefore, in vitro and in vivo experiments are needed to confirm the real potential of the proposed anti-S. gallolyticus vaccine.
Conclusion
The development of a proteomics-based approach to identifying vaccine targets against antibiotic-resistant S.gallolyticus is a promising avenue for combating this difficult pathogen. A comprehensive analysis of the bacterial proteomes of seven strains revealed two antigenic proteins as vaccine candidates and identified potential epitopes, laying the groundwork for the development of a multi-epitope vaccine. A safe and latent vaccine that may trigger humoral, cellular, and innate immune responses was developed using immunoinformatics and molecular docking approaches. By targeting multiple epitopes, such a vaccine could improve immune recognition and address the issue of antigenic variation, potentially providing broad protection against various strains of S. gallolyticus. This novel strategy has great promise for combating the growing threat of antibiotic resistance and improving public health outcomes. However, more research and clinical trials are required to validate the efficacy and safety of this vaccine candidate before it can be widely used as a preventive measure against S. gallolyticus infections.
Data availability
The datasets analyzed during the current study are available in the NCBI database and IDs of the datasets are mentioned in Additional File 1: Table S1.
References
Sitthicharoenchai, P. et al. Streptococcus gallolyticus and Bacterial Endocarditis in Swine, United States, 2015–2020. Emerg. Infect. Dis. 28(1), 192 (2022).
Hinse, D. et al. Complete genome and comparative analysis of Streptococcus gallolyticus subsp. gallolyticus, an emerging pathogen of infective endocarditis. BMC Genomics 12(1), 1–13 (2011).
Chamat-Hedemand, S. et al. Prevalence of infective endocarditis in streptococcal bloodstream infections is dependent on streptococcal species. Circulation 142(8), 720–730 (2020).
Arregle, F. et al. Western immunoblotting for the diagnosis of Enterococcus faecalis and Streptococcus gallolyticus infective endocarditis. Front. Cell. Infect. Microbiol. 9, 314 (2019).
Baddour, L. M. et al. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: A statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 111(23), e394–e434 (2005).
Firstenberg, M.S., Contemporary Challenges in Endocarditis. 2016: BoD–Books on Demand.
Cãruntu, F. et al. Streptococcus gallolyticus spontaneous infective endocarditis on native valves, in a diabetic patient. Med. Evol 3, 323–328 (2014).
McDonald, J. R. Acute infective endocarditis. Infect. Dis. Clin. North Am. 23(3), 643–664 (2009).
Hensler, M. E. Streptococcus gallolyticus, Infective Endocarditis, and Colon Carcinoma: New Light on an Intriguing Coincidence 1040–1042 (Oxford University Press, 2011).
Bartolomé, J.A.S. and M.A. Sanz, Streptococcus gallolyticus: a new name for a well-known old organism. Archiv. Med. 2009. 1.
Grubitzsch, H. et al. Surgery for prosthetic valve endocarditis: Associations between morbidity, mortality and costs. Interact. Cardiovasc Thoracic Surg. 22(6), 784–791 (2016).
Thomas, S. & Luxon, B. A. Vaccines based on structure-based design provide protection against infectious diseases. Expert Rev. Vaccines 12(11), 1301–1311 (2013).
Kazi, A. et al. Current progress of immunoinformatics approach harnessed for cellular-and antibody-dependent vaccine design. Pathogens Global Health 112(3), 123–131 (2018).
Dey, J., et al., Designing of multi-epitope peptide vaccine against Acinetobacter baumannii through combined immunoinformatics and protein interaction–based approaches. Immunologic Research, 2023: p. 1–24.
Dey, J. et al. Designing a novel multi-epitope vaccine to evoke a robust immune response against pathogenic multidrug-resistant Enterococcus faecium bacterium. Gut Pathogens 14(1), 1–20 (2022).
Dey, J. et al. Exploring Klebsiella pneumoniae capsule polysaccharide proteins to design multiepitope subunit vaccine to fight against pneumonia. Expert Rev. Vaccines 21(4), 569–587 (2022).
Mahapatra, S. R. et al. The potential of plant-derived secondary metabolites as novel drug candidates against Klebsiella pneumoniae: molecular docking and simulation investigation. South African J. Bot. 149, 789–797 (2022).
Narang, P. K. et al. Functional annotation and sequence-structure characterization of a hypothetical protein putatively involved in carotenoid biosynthesis in microalgae. South African J. Bot. 141, 219–226 (2021).
Mahapatra, S. R. et al. Immunoinformatics-guided designing of epitope-based subunit vaccine from Pilus assembly protein of Acinetobacter baumannii bacteria. J. Immunol. Methods 508, 113325 (2022).
Caputo, A., Fournier, P.-E. & Raoult, D. Genome and pan-genome analysis to classify emerging bacteria. Biol. Direct 14(1), 1–9 (2019).
Narang, P. K. et al. Genome-based identification and comparative analysis of enzymes for carotenoid biosynthesis in microalgae. World J. Microbiol. Biotechnol. 38, 1–22 (2022).
Chaudhari, N. M., Gupta, V. K. & Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6(1), 24373 (2016).
Wei, W. et al. Geptop: A gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PloS One 8(8), e72343 (2013).
Qi, J., Luo, H. & Hao, B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res.arch 32(suppl_2), W45–W47 (2004).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990).
Goyal, M. & Citu, S. In silico identification of novel drug targets in Acinetobacter baumannii by subtractive genomic approach. Asian J. Pharm. Clin. Res. 11(3), 230–236 (2018).
Savojardo, C. et al. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 46(W1), W459–W466 (2018).
Doytchinova, I. A. & Flower, D. R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8(1), 4 (2007).
Dimitrov, I. et al. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 20, 1–6 (2014).
Garg, V. K. et al. MFPPI–multi FASTA ProtParam interface. Bioinformation 12(2), 74 (2016).
Chen, Y. et al. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 14, 859–865 (2003).
Wang, P. et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 11(1), 1–12 (2010).
Saha, S. & Raghava, G. P. S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 65(1), 40–48 (2006).
Doytchinova, I. A. & Flower, D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8(1), 1–7 (2007).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PloS One 8(9), e73957 (2013).
Moutaftsi, M. et al. A consensus epitope prediction approach identifies the breadth of murine T CD8+-cell responses to vaccinia virus. Nat. Biotechnol. 24(7), 817–819 (2006).
Dhanda, S. K., Vir, P. & Raghava, G. P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 8(1), 1–15 (2013).
Bui, H.-H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 7(1), 1–5 (2006).
Dhanda, S. K. et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 47(W1), W502–W506 (2019).
Li, W. et al. Peptide vaccine: Progress and challenges. Vaccines 2(3), 515–536 (2014).
Dong, R. et al. Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front. Immunol. 11, 1784 (2020).
Cheng, J. et al. SCRATCH: A protein structure and structural feature prediction server. Nucleic acids Res. 33(2), W72–W76 (2005).
Heo, L., Park, H. & Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41(W1), W384–W388 (2013).
Lovell, S. C. et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 50(3), 437–450 (2003).
Lengths, M. & Angles, M. Limitations of structure evaluation tools errat. Quick Guideline Comput. Drug Des. 16, 75 (2018).
Wiederstein, M. & Sippl, M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35(s2), W407–W410 (2007).
Dhanda, S. K. et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 47(W1), W502–W506 (2019).
Magnan, C. N. et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 26(23), 2936–2943 (2010).
Geourjon, C. & Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11(6), 681–684 (1995).
Ponomarenko, J. et al. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9(1), 1–8 (2008).
Craig, D. B. & Dombkowski, A. A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 14(1), 1–7 (2013).
Van Zundert, G. et al. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428(4), 720–725 (2016).
Yuan, S., Chan, H. S. & Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscipl. Rev. Comput. Mol. Sci. 7(2), e1298 (2017).
Laskowski, R. A. PDBsum new things. Nucleic Acids Res. 37(suppl_1), D355–D359 (2009).
Bowers, K.J., et al. Scalable algorithms for molecular dynamics simulations on commodity clusters, in Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. 2006.
Shivakumar, D. et al. Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J. Chem. Theory Comput. 8(8), 2553–2558 (2012).
Price, D. J. & Brooks, C. L. III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 121(20), 10096–10103 (2004).
Crean, R. M. et al. Reliable in silico ranking of engineered therapeutic TCR binding affinities with MMPB/GBSA. J. Chem. Inf. Model. 62(3), 577–590 (2022).
Wang, C. et al. Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 4, 87 (2018).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33(suppl_2), W526–W531 (2005).
Siddharth, R., M. Shankar, and A. Karthikeyan, An overview on “GenSmart”–A smart DNA construct design tool.
Rapin, N. et al. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PloS one 5(4), e9862 (2010).
Du, J. et al. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM–GBSA calculation. J. Comput. Chem. 32(13), 2800–2809 (2011).
Shapira, R. et al. Streptococcus gallolyticus endocarditis on a prosthetic tricuspid valve: A case report and review of the literature. J. Med. Case Rep. 15(1), 1–6 (2021).
Romay, E. et al. Relationship among Streptococcus gallolyticus Subsp gallolyticus, Enterococcus faecalis and colorectal neoplasms in recurrent endocarditis: A historical case series. J. Clin. Med. 11(8), 2181 (2022).
Oli, A.N., et al., Immunoinformatics and vaccine development: an overview. Immunotargets Ther., 2020: p. 13–30.
Micoli, F. et al. The role of vaccines in combatting antimicrobial resistance. Nat. Rev. Microbiol. 19(5), 287–302 (2021).
Mahmood, M. et al. Rational design of multimeric based subunit vaccine against Mycoplasma pneumonia: Subtractive proteomics with immunoinformatics framework. Infect. Genet. Evol. 91, 104795 (2021).
Alzarea, S. I. Identification and construction of a multi-epitopes vaccine design against Klebsiella aerogenes: molecular modeling study. Sci. Rep. 12, 14402 (2022).
Onile, O. S. et al. An immunoinformatics approach for the design of a multi-epitope subunit vaccine for urogenital schistosomiasis. PeerJ 8, e8795 (2020).
Gupta, N. & Kumar, A. Designing an efficient multi-epitope vaccine against Campylobacter jejuni using immunoinformatics and reverse vaccinology approach. Microb. Pathog. 147, 104398 (2020).
Albutti, A. Proteome-wide and protein-specific multi-epitope vaccine constructs against the rift valley fever virus outbreak using integrated omics approaches. Front. Microbiol. 13, 921683 (2022).
Umar, A. et al. Development of a candidate multi-epitope subunit vaccine against Klebsiella aerogenes: Subtractive proteomics and immuno-informatics approach. Vaccines 9(11), 1373 (2021).
Naveed, M. et al. A vaccine construction against COVID-19-associated mucormycosis contrived with immunoinformatics-based scavenging of potential Mucoralean Epitopes. Vaccines 10(5), 664 (2022).
Naveed, M. et al. Design of a novel multiple epitope-based vaccine: An immunoinformatics approach to combat SARS-CoV-2 strains. J. Infect. Public Health 14(7), 938–946 (2021).
Alizadeh, M. et al. Designing a novel multi-epitope vaccine against Ebola virus using reverse vaccinology approach. Sci. Rep. 12, 7757 (2022).
Yang, Y. et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccines Immunother. 11(3), 795–805 (2015).
Yano, A. et al. An ingenious design for peptide vaccines. Vaccine 23(17–18), 2322–2326 (2005).
Li, X. et al. Design and evaluation of a multi-epitope peptide of human metapneumovirus. Intervirology 58(6), 403–412 (2016).
Livingston, B. et al. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J. Immunol. 168(11), 5499–5506 (2002).
Takamatsu, N. et al. Production of enkephalin in tobacco protoplasts using tobacco mosaic virus RNA vector. FEBS Lett. 269(1), 73–76 (1990).
Hou, J. et al. Cholera toxin B subunit acts as a potent systemic adjuvant for HIV-1 DNA vaccination intramuscularly in mice. Hum. Vaccines Immunother. 10(5), 1274–1283 (2014).
Rezaei, M. et al. In silico design and in vivo evaluation of two multi-epitope vaccines containing build-in adjuvant with chitosan nanoparticles against uropathogenic Escherichia coli. Int. Immunopharmacol. 117, 109999 (2023).
Kar, T. et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 10(1), 10895 (2020).
Khanmohammadi, S. & Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 93(5), 2735–2739 (2021).
Author information
Authors and Affiliations
Contributions
Conceptualization, X.C. ; methodology, P.C, X.Z.; software,. P.C, X.Z.; L.Z; validation, P.C, X.Z. L.Z,A.Y.; formal analysis, P.C, X.Z.;Y.W.; investigation, X.C.; data curation, P.C, X.Z.;Y.W.; writing—original draft preparation, P.C,X.Z.;, X.C; writing—review and editing, P.C, X.Z.A.Y.; supervision, X.C.; project administration, X.C.; funding acquisition, X.C. All authors have read and agreed to the publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chao, P., Zhang, X., Zhang, L. et al. Proteomics-based vaccine targets annotation and design of multi-epitope vaccine against antibiotic-resistant Streptococcus gallolyticus. Sci Rep 14, 4836 (2024). https://doi.org/10.1038/s41598-024-55372-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55372-3
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
