
Abstract
The Beas River is one of the important rivers of the Indus River system located in Himachal Pradesh, India, that harbors a diverse range of freshwater fish species. The present study employed COI gene to investigate the ichthyofaunal diversity of river Beas. Through the sequencing of 203 specimens from Beas River, we identified 43 species, belonging to 31 genera, 16 families, and 10 orders. To analyze the genetic divergence and phylogeny of identified species, 485 sequences of Indian origin were retrieved from BOLD, resulting in a dataset of 688 sequences. Our findings consistently revealed a hierarchical increase in the mean K2P genetic divergence within species (0.80%), genus (9.06%), and families (15.35%). Automated Barcode Gap discovery, Neighbour Joining, and Bayesian inference consensus tree methodologies were employed to determine the putative species and their phylogeny, successfully delimiting most of the species with only a few exceptions. The results unveiled six species exhibiting high intra-species divergence (> 2%), suggesting the presence of sibling species and falsely identified sequences on online databases. The present study established the first DNA barcoding-based inventory of freshwater fish species in the Beas River providing comprehensive insights into economically exploited endangered and vulnerable species. In order to ensure the sustainable use of aquatic resources in the Beas River, we recommend the implementation of species measures to protect biodiversity and genetic resources.
Similar content being viewed by others

Introduction
Accurate species identification plays a crucial role in understanding how evolutionary history, biodiversity patterns, and ecosystems change over time. Precise identification enables the detection of invasive, elusive, endangered, or threatened species, and helps reduce various ecosystem risks by facilitating the habitat for conservation. To effectively manage the genetic resources of fish species sustainably, a comprehensive understanding of the taxonomy and systematics of fish species is very essential. DNA-based identification techniques have greatly improved the reliability of species identification, particularly for cryptic species and larval fishes1,2. DNA barcoding utilizes a standardized gene called DNA barcode, which offers a reliable method for species identification while providing valuable insights into evolutionary relationships3,4. The flanking regions of barcode sequences should exhibit minimal sequence variations for efficient amplification, and the mutation rate of the genes used for taxonomic identification of closely related species should be below 2%5. The mitochondrial cytochrome c oxidase subunit-I gene (COI) is the preferred DNA barcode due to its ability to provide a wide range of phylogenetic signals, robust primers, and a high occurrence of base substitutions in third-position nucleotides, resulting in accelerated molecular evolution4. The COI gene sequence finds extensive applications in various fields, such as forensic science, food authentication, and biotechnology-based industries, owing to its prevalence in mitochondrial DNA2. COI has revealed divergent patterns of evolution and possible hidden variations from the present taxonomic classifications6,7,8,9,10. Understanding the processes of community construction can be gained from well-resolved phylogenies produced from DNA barcodes when combined with functional characteristic data of the same species11. Reconstructing the phylogenetic relationships from large groups of species is crucial for taxonomic studies to address various ecological problems. One of the current problems in taxonomic identification is the presence of cryptic species, which refers to species with minor physical variability but significant genetic variations12. Cryptic species might result by means of parallel evolution from recent divergence in closely related taxa or distant taxa in a comparatively short amount of time13. To represent genetic differentiation within a species, cryptic species sensu stricto and sensu lato terms are also employed14,15,16. The former represent genetically distinct species that have been shown to lack diagnostic morphology, while the latter represent genetically distinct species that are hard to visually identify but possess some distinctive phenotypic trait(s)13,17. The existence of cryptic species has been effectively established by a number of studies that have employed DNA barcoding to taxonomically identify fish species18,19,20,21,22,23,24. The COI gene also serves as a valuable genetic marker to evaluate population genetic structure of metazoans through the identification of distinct haplotypes across various geographical regions25,26,27,28,29.
India is boasted for its a rich diversity of aquatic species, including over 1045 freshwater fish species30. The Beas River, spanning 470 km through the Himalayan region of Northern India, represents a significant aquatic resource. Originating from Rohtang Pass in Himachal Pradesh and merging with Satluj River at Harike Wetlands in Punjab, the Beas River has undergone substantial modifications that have impacted ecology and fish diversity. A report from the Central Inland Fisheries Research Institute, Barrackpore has identified a total of 54 fish species in the river Beas31. According to survey, Indian major carp comprised 28.28% of fishery in the Beas, with minor carps ranking second at 22.44% followed by common carp at 22.02%, and other species at 17.75%. Catfish, on the other hand, were the least prevalent, accounting for 8.54% of the fishery. Kumar32,33 found only six fish species from the upstream of the Beas River, indicating poor faunal biodiversity. Harike Wetlands, located at the confluence of the Beas and Satluj river systems and designated as one of India's Ramsar Sites (internationally recognized wetlands under the Ramsar Convention), showcase a diverse range of fish species. In 2009, Dua34 identified 61 fish species in the Harike Wetlands, while Kour35 reported 37 species in 2017. These findings indicate the highest number of species from the Cyprinidae family, encompassing fish species from both the River Beas and Satluj. While limited research has been conducted on the ichthyofaunal diversity of the Beas River system in the past decade, only a handful of ichthyologists have employed morphological approaches to investigate fish diversity in the system which has resulted in a deficiency in our comprehension of the fish fauna in the region.
The primary objective of the present study was to establish a DNA barcoding-based inventory of freshwater fish species in Beas River and gain a deeper understanding of their genetic variation and distribution pattern. The results of the current study offer significant data that can enhance monitoring and conservation efforts aimed at efficient management of the Beas River. The study incorporates online available DNA barcodes of Indian origin to analyze and authenticate DNA barcoding and explore their genetic diversity. The current study provides new insights into the taxonomic affiliation and divergent patterns of evolution through the application of the COI gene to examine the haplotype diversity and population genetic structure of multiple fish species.
Results
DNA barcoding of fishes of Beas River
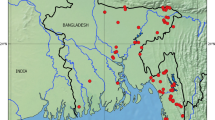
A total of 320 fish specimens were collected from 15 different sites, including major commercial fish landing stations on Beas River (Fig. 1). Out of these, sequences were successfully generated for 203 specimens, resulting in 100% amplification rate of COI barcode. All the amplified sequences represented functional COI sequences of length > 600 bp without any deletion, insertion, or stop codon. Similarity search was performed using the BOLD identification engine and NCBI nucleotide blast. The similarity percentage for all the sequences ranged from 98 to 100%. The overall GC content was 45.49% (SE = 0.10). Table 1 provides the nucleotide composition, GC content, and GC% at codon positions 1, 2, and 3 for all orders. The dataset of 203 sequences represented 43 species, 31 genera, 16 families, and 10 orders. The order Cypriniformes was the most diverse, comprising 135 specimens (66.5%) followed by Siluriformes (14.7%), Anabantiformes (5.4%), Salmoniformes (3.4%), Ovalentaria (3.4%), Beloniformes (1.9%), Clupeiformes (1.9%), Osteoglossiformes (1.4%), Cichliformes (0.4%), and Synbranchiformes (0.4%). The Cyprinidae family was most abundant comprising 21 species, followed by Danionidae with 5 species. The Bagridae, Channidae, and Siluridae families each were represented by 2 species, while the Ailidae, Ambassidae, Belonidae, Cichlidae, Clupeidae, Heteropneustidae, Mastacembelidae, Nandidae, Notopteridae, Salmonidae, and Sisoridae, families were represented by 1 species each. The classification, number of specimens barcoded, and the IUCN status of the fish species are presented in the Table 2.

Map representing sampling sites in River Beas and its tributaries in Himachal Pradesh and Punjab, India. Created in QGIS v 3.26.2 (https://www.qgis.org/en/site/).
Sequence composition and genetic divergence
To infer the genetic divergence, the 203 sequences obtained from Beas River were combined with 485 records from the BOLD database (Supplementary Table S1). The mean K2P genetic divergence within species (0.80%), genus (9.06%), and family (15.35%) showed a consistent hierarchical increase in genetic diversity with rising taxonomic levels (Table 3). The average congeneric divergence was 11 times higher than the conspecific divergence. The K2P genetic divergence was calculated within and between families. Family Danionidae and Cyprinidae showed the highest genetic divergence (0.21 ± 0.02 and 0.14 ± 0.01 respectively), whereas Sisoridae showed the lowest (0.001 ± 0.0005). Cichlidae, Clupeidae, and Salmonidae showed no genetic divergence due to the presence of a single species. Estimates of evolutionary divergence over sequence pairs between families showed the highest divergence between Bagridae and Nandidae (0.41 ± 0.05), while the minimum was observed in Heteropneustidae and Ailiidae (0.19 ± 0.02). K2P genetic divergence was also calculated between and within all the species. The inter-species genetic divergence was also calculated, revealing the highest K2P genetic divergence (44%) between Nandus nandus and Sperata seenghala, the lowest (0.4%) between Bangana dero and Labeo boggut, and negligible between Schizothorax plagiostomus and Schizothorax richardsonii.
Phylogenetic analysis and species delimitation
The ABGD analysis of 688 COI sequences identified a cluster of 8 molecularly defined operational taxonomic units (MOTUs). The prior maximal distance (p) of 0.0077 was sufficient to distinguish fish species in present study. The overall dataset of 688 sequences belonging to 43 species was clustered into 56 groups (Supplementary Fig. S1). Most of the species were clearly delimited into putative species, while Schizothorax sp. (S. richardsonii and S. plagiostomus) and Tariqilabeo sp. (T. adiscus and T. latius) could not be delimited properly. A few sequences belonging to species Cirrhinus cirrhosis, Bangana dero, Tor tor, Cirrhinus reba, and Labeo bata were grouped together with their respective nearest neighbor (Supplementary Table S2). Some groups in the partition were represented by single sequences, which indicate geographically isolated or genetically distinct haplotype of their respective species. To enhance species delimitation, Neighbor-joining (Supplementary Fig. S2) and Bayesian Inference (BI) (Fig. 2) was also conducted using the complete dataset of 688 sequences and the resulting trees showed consistent findings with ABGD species delimitation.

Bayesian inference (BI) consensus tree based on 688 COI barcodes under the GTR + I + Γ reversible evolution model. The number on branches depicts probabilities and the color illustrates probability percentage.
Genetic diversity and haplotype networks of specific taxa
The intra-species diversity observed from all the species ranged from 0 to 6%. The highest intra-species diversity was observed among Labeo gonius sequences (6.6%) and the lowest were observed in Systomus sarana (1.2%), Barilius vagra (0.9%) and Tor tor (0.9%) (Fig. 3). Significantly high intra-species diversity (> 2%) was observed in L. gonius (6.6%), O. cotio (6.5%), P. conchonius (5.7%), O. pabo (5.2%), C. nama (4.6%), C. marulius (2.7%) and C. prateri (2.4%). Distinct clusters of haplotypes can be observed in median-joining haplotype network analysis (Fig. 4). All the results showed similar partitions in ABGD species delimitation, indicating the possibility of presence of geographically divergent or sibling species. Moreover, genus Channa (12%), Cirrhinus (9%), and Labeo (7%) showed notable congeneric divergence.

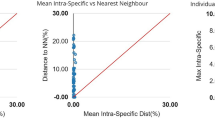
Barcode gap analysis: (a) overlap of the max intra-species distances vs the inter-species (nearest neighbor) distances; (b) overlap of the mean intra-species distances vs the inter-species (nearest neighbor) distances; (c) the number of individuals in each species against their max intra-species distances, as a test for sampling bias.

Median-joining networks and haplotype diversity indices of species with intra-species diversity > 2%; (a) Labeo gonius, (b) Osteobrama cotio, (c) Pethia conchonius, (d) Ompok pabo, (e) Chanda nama, (f) Clupisoma prateri, and (g) Channa marulius. The size of the circle represents the relative frequency of the associated haplotype over the entire dataset of respective species, and the colors represent the related state/province in India. The number of mutational changes between two haplotypes is represented by the black lines on the branches. Values for number of sequences (N), haplotype diversity (Hd), number of haplotypes (H), and Intra-species diversity (Intra-D) are given along with each haplotype network.
(a) Channa Species: The genus Channa comprised a total of 38 sequences belonging to C. marulius (20) and C. punctata (18). Genetic divergence among genus Channa was highest (12%) among all other genera. C. marulius showed intra-species diversity of 2.7%, haplotype diversity (Hd) of 0.78 and 6 haplotypes (H), whereas C. punctata revealed lower intra-species diversity (0.7%), comparatively lower haplotype diversity (0.69) and higher number of haplotypes (7). The inter-species divergence between C. marulius and C. punctata was 2.9%. The NJ tree of haplotypes and median-joining network of genus Channa shows haplotype 12 is separated by haplotype 7 by 44 mutations, which reveals highly divergent population of C. marulius (Fig. 5a). The ABGD species delimitation also reveals that all the sequences of haplotype 12 are forming a separate group at optimal threshold (P = 0.0077), suggesting the formation of a new putative species of Channa. (b) Cirrhinus Species: In the present study, there were a total of 51 sequences from the Cirrhinus genus, including sequences from C. reba (19), C. mrigala (27), and C. cirrhosis (5) revealing 9% of genetic divergence within genus. The K2P divergence was 19% between both C. reba and C. mrigala; and between C. reba and C. cirrhosis. The K2P divergence was negligible between C. mrigala and C. cirrhosis. C. reba and C. mrigala had nearly equal intra-species divergence (0.35%), however C. mrigal (H = 10; Hd = 0.68) had a greater number of haplotypes and higher haplotype diversity than C. reba (H = 4; Hd = 0.38). On the other hand, C. cirrhosis showed only one haplotype and no intra-species diversity. Two distinct sets of haplotypes can be seen in the median joining network of haplotypes and NJ tree, one belonging to C. reba (Hap 3, 5, 9, and 11), and the other to C. mrigala and C. cirrhosis (Hap 1, 2, 4, 6, 7, 8, 10, 12, 13, and 14) (Fig. 5b). Haplotype 1 consisted of 15 sequences of C. mrigala and 5 sequences of C. reba, which demonstrated the possibility of falsely identified lineages on online database or presence of introgressive hybridization between them. (c) Labeo Species: A total of 172 sequences constituting five species; L. rohita (51), L. calbasu (37), L. boggut (34), L. bata (20), L. catla (15), and L. gonius (9). The K2P genetic divergence observed within genus was 7%. The inter-species diversity between species of genus Labeo was highest between L. catla and L. gonius (12.5%) and lowest between L. bata and L. boggut (5%). The highest K2P divergence within species was observed in L. boggut (0.3%) and highest in L. gonius (6%). L. gonius was the only species in genus Labeo showing more than 2% intra-species divergence indicating presence of highly diverged sibling species. All the sequences of L. gonius were double checked for similarity percentage of the sequence on NCBI blast and BOLD IDs databases, which revealed 100% similarity with L. gonius. The number of haplotypes (H) and haplotype diversity (Hd) were also calculated, revealing the L. gonius haplotypes (Hap 36 and 37), which are substantially divergent (Fig. 5 c). Higher intra-species divergence (1.9%) and haplotype diversity of 0.76 (H = 7) were also observed in L. bata. Haplotype 33 of L. bata population showed a highly divergent group from the remaining cluster of haplotypes (Hap 28, 29, 30, 31, 32), possibly due to genetically isolated or geographically distant population.

Neighbour-joining (NJ) phylogenetic tree and median-joining haplotype network showing distinct lineages of species from (a) genus Channa (C. punctata and C. marulius) haplotypes, (b) genus Cirrhinus (C. reba, C. mrigala and C. cirrhosus) haplotypes, (c) genus Labeo (L. rohita, L. catla, L. boggut, L. bata, L. gonius and L. calbasu) haplotypes. The NJ phylogenetic tree is constructed with a bootstrap value of 1000 using the MEGAX software package. The size of the circle in haplotype networks represents the relative frequency of the associated haplotype over the entire dataset, and the colors represent the related state/province in India. The number of mutational changes between two haplotypes is represented by the black lines on the branches.
Discussion
DNA barcoding is a useful tool for identifying species by creating barcodes from different geographical regions. Various factors such as genetic drift, ecological factors, and developmental flexibility can lead to population-level diversification and speciation, resulting in variations in genotypic and phenotypic features of species across geographic regions36. In this study, we contributed a novel perspective on fish biodiversity and conservation by presenting the first molecular-based assessment of the fish fauna in Beas River. The study encompassed the establishment of a reference library comprising 203 DNA barcodes from 43 different species belonging to 10 orders, with Cypriniformes exhibiting the highest diversity, followed by Siluriformes. Among the 43 species investigated, 27 were previously documented in the Beas River32,33. The current study has documented 16 species for the first time including Bangana dero, Barilius vagra, Cabdio morar, Chagunius chagunio, Chanda nama, Cirrhinus cirrhosis, Clupisoma prateri, Esomus danrica, Labeo boggut, Oreochromis niloticus, Salmostoma bacaila, Salmostoma phulo, Schizothorax plagiostomus, Tariqilabeo adiscus, Tariqilabeo latius, and Tor tor. Although some of these species have been previously reported in studies based on the Harike wetlands, which encompass both the Satluj River and the Beas River, it may not be accurate to directly compare these findings when estimating the ichthyofaunal diversity specifically within the Beas River34,35,37. The base composition analysis of the 203 sequences from the Beas River indicated a richness in AT content (54.54%). A lower GC composition (45.47%) and G (18.07%) were observed, characteristics of mitochondrial DNA in fishes. The first codon GC content was notably higher than the other two positions, with the base usage bias at the third codon position being the lowest. This pattern aligns with observation in other studies where the third codon position exhibited a clear anti-G bias38,39,40,41.
To assess genetic divergence and established phylogenetic relationships on a wider scale, we aggregated 485 sequences from Indian origin on BOLD database with sequences from the Beas River. The K2P genetic divergence within the dataset of 688 sequences was 0.8% within species, escalating to 9.06% within genus and 15.35% within family. This hierarchical increase in divergence with the progression of taxonomic levels has been also observed in previous studies on fish populations including Australian marine fish (0.39%, 9.93%, 15.46%)7, Canadian freshwater fishes (0.27%, 8.37%, 15.38%)42, Indian Marine fishes (0.30%, 6.60%, 9.91%) 42, Narmada River, India (0.36%, 12.29%, 17.87%)43, Indian freshwater fishes (0.40%, 9.60%, 13.10%)39, Indo-Myanmar fishes (0.42%, 10.19% and 12.77%)39, and Ranganadi River, India (0.23%, 11.31%, 26.25%) 44. The consistent hierarchical increase in divergence across diverse studies and regions among fish populations likely reflects the accumulation of genetic differences and adaptive divergence over time. This pattern carries significant ecological and evolutionary implications, underscoring the importance of tailored conservation strategies to preserve specialized genetic adaptations. It also highlights the remarkable biodiversity within fish families that warrants recognition and protection. This consistent pattern observed across various regions suggests the fundamental nature of this phenomenon. Further research is needed to understand the genetic mechanisms behind this consistent pattern that will help better understand the evolution of species and the diversity of their genes across the different taxonomic levels.
Numerous studies have used the ABGD tool for the construction of phylogenetic trees and species delimitation. ABGD clusters sequences into candidate species based on the pairwise distances and barcode gap10,43,45,46. In this study, ABGD analysis successfully differentiated all species except for Schizothorax (S. richardsonii and S. plagiostomus), Tariqilabeo (T. adiscus and T. latius) and a few sequences of Cirrhinus cirrhosis, Bangana dero, Tor tor, Cirrhinus reba, and Labeo bata. These finding were corroborated through phylogenetic reconstruction using the Neighbour joining and Bayesian inference (BI) tree. The utilization of native populations in hatcheries for artificial breeding and the potential interbreeding between escaped farmed fish and wild types may have detrimental effects on the genetic integrity of the fish species17,25. The presence of introgressive hybridization is indicated by the shared mitochondrial lineage and low intra-species divergence values, particularly between S. richardsonii and S. plagiostomus as well as T. adiscus and T. latius. Mitochondrial introgression occurs when a species possesses two mitochondrial lineages, one from its own mitochondria and the other acquired from another species26. Introgression and hybridization associated with inter-species gene flow, enable the integration of novel genetic information into the parent species' genome, resulting in species harboring both mitochondrial lineages27. Akhtar 47 revealed genetic divergence of 0.009% between S. plagiostomus, S. esocinus, S. progastus, and S. niger suggesting the possibility of ancestral polymorphism and introgressive hybridization. The sequences for the genus Schizothorax (S. richardsonii and S. plagiostomus) in the present investigations were restricted to Beas River due to the absence of trace files for remaining sequences in the BOLD database. Reconstructing phylogenetic relationships becomes more challenging when there is ancestral hybridization and polyploidization, causing limited gene transmission between different populations or species48,49. Furthermore, comprehending the mitochondrial complexity of such species proves challenging due to limited sample size and geographically confined locations. The present study also showed Labeo boggut and Bangana dero, both belonging to family Cyprinidae and sub-family Labeoninae, in a single monophyletic clade in COI lineage. This observation aligns with previous studies where Labeo sp. and Bangana sp. formed a unified clade50,51. Yang31 demonstrated that B. dero formed a distinct lineage with L. bata, L. boggut, and three additional forms, naming the clade “Bangana sensu stricto”. The studies on the molecular phylogeny of genus Bangana are in its early stages and, its systematic status remains debatable32.
Intra-species genetic divergence exceeding 2% was observed in Labeo gonius, Osteobrama cotio, Pethia conchonius, Ompok pabo, Chanda nama, Channa marulius, and Clupisoma prateri. O. cotio exhibited a 6.5% divergence, supported by a previous study on the species, based on COI and 16srRNA gene sequences from rivers in India and Bangladesh52, suggesting genetic differentiation of different populations of Osteobrama and the possible existence of a new putative species named O. serrata. High intra-species divergence within species suggests the potential presence of geographically divergent or sibling species formed due to adaptation and high evolutionary potential of the species53. The genetic divergence values of P. conchonius in Ganga and Yamuna rivers of Uttrakhand, India, as reported by Joshi54 did not reveal higher values compared to the current study. Negi 55 reported a falsely identified sequence for Puntius chola and noted close proximity between sequences of Puntius terro and Pethia conchonius. Haplotype diversity and the number of haplotypes were also investigated for species with high genetic divergence; O. cotio, P. conchonius, O. pabo and C. marulius, showing high haplotype diversity, and L. gonius, C. nama, and C. prateri exhibiting moderate diversity. All these highly diverged species exhibit at least one haplotype with significant mutations, distinctly separated by clusters of other haplotypes. Sequences from these unique haplotypes form distinct group in the ABGD species delimitation analysis, supporting the potential formation of new putative species. Following the criteria outlined by Hebert, and Stoeckle56, if the genetic divergence between two specimens of a supposedly single species is greater than ten times the average divergence within that group, it is likely that one of them represents a new putative species. When proposing a species as a potentially new species, it is essential to conduct thorough examination of its morphological, genetic, geographical, and ecological characteristics for taxonomic validation2. Further, the study recommends the utilization of diverse computational approaches in identifying species through DNA analysis. This is crucial for assessing whether the observed genetic variation in the species is a result of formation of cryptic species or the divergence can be attributed to misidentification stemming from the reliance on a single computational method. The study specifically addresses the freshwater fishes of the river Beas that hold commercial importance to the local livelihood and protein requirements. During a survey conducted at commercial landing stations near Beas River, we identified one endangered species, six vulnerable species, and one near threatened species. Of particular concern is the endangered species Tor putitora, commonly known as Golden mahseer, which holds a high value both the table and in the commercial markets. In India, the indiscriminate fishing of Golden mahseer persist due to lack of market control and specific conservation measures, despite the species’ ecological and economic significance38. The Department of Fisheries in Himachal Pradesh, India has implemented legal measures to restrict the fishing of Golden mahseer during breeding seasons. With a well-executed grassroots strategic plan, these measures have the potential to effectively conserve the Mahseer population. Despite various conservation efforts, several vulnerable species including Cyprinus carpio, Cirrhinus cirrhosus, Schizothorax richardsonii, Schizothorax plagiostomus, Wallago attu, and Bagarius bagarius continue to be exploited for commercial purposes in the Beas River. The wild form of C. carpio is classified as vulnerable, yet it is widely considered highly invasive and has been domesticated and introduced globally39. The decline of C. cirrhosis, W. attu, and B. bagarius in the Indo-Ganges River basin is linked to various factors, including habitat degradation caused by environmental changes, human interventions disrupting spawning and feeding, depletion of aquatic environments, especially for brood fish, and insufficient management of fish catchment40,41,42. S. richardsonii along with several other cold-water fish species, has significantly declined and is categorized as vulnerable on the IUCN red list43. The irreversible alterations in their native population structure and increase in unnatural deaths can be attributed to intense human intervention and the introduction of exotic species44. In the cold waters fish of the Beas, S. plagiostomus, a fish known as “game-food fish” is commercially exploited by locals, despite its vulnerable status 45. The majority of identified species are native to India, while three species; Cyprinus carpio, Oreochromis niloticus, and Salmo trutta fario are introduced species. Notably, the globally cultivated exotic fish Oreochromis niloticus, has been reported in the Beas River for the first time. Although most of the introduced species seem to have a minimal impact on native biodiversity, their global spread poses a potential threat of becoming highly invasive in certain regions which may lead to significant negative impacts on some of the native species, and eradication of their genetic resources57. India has witnessed the consequences of climate fluctuation and dam constructions on freshwater river systems, evident in changes to the geographic distribution, breeding season, life cycle, and physiological behavior of inland fishes47,48. The Pong Dam, constructed on the Beas River, functions as both a wetland and a wildlife sanctuary. However, the excessive utilization of ecosystem services has resulted in the spread of invasive species, obstacles to fish migration, and decline in fish diversity. Additionally, overfishing and illegal trade of fishes by the local population are adversely impacting fish biodiversity in the Beas River58,59. Furthermore, the contamination of water resources from chemical effluent leakage from sugar mill has resulted to numerous fish mortality60. The uncontrolled discharge of municipal solid waste and sewage into the Beas River and its tributaries poses a significant concern. To effectively manage the aquatic biodiversity of freshwater fishes, it is crucial to monitor waste disposal in the river system and regulate the overfishing of fish fauna, especially for endangered, vulnerable, threatened, and rare species.
Conclusion
Despite the significant importance of freshwater fish to biodiversity and as a natural resource, there is still limited information on the genetics of most species. Comprehending the contribution of diverse species to ecological systems and effectively conserving biodiversity demands a profound understanding of species diversity and the composition of distinct geographic regions. DNA barcoding extends beyond species identification, finding application in various areas such as food authentication, identification of cryptic species, conservation biology, evolutionary biology, and providing essential genetic insights for assessing genetic divergence within a species. Moreover, the ichthyofaunal population is undergoing a reduction in genetic diversity due to widespread release of allochthonous fish which may lead to a significant decline or even disappearance of native populations. DNA barcoding holds the potential to play a crucial role in formulating effective conservation plans for wild fishes, ensuring the maintenance of their genetic diversity. To establish strategies and practical measures that ensure the proper identification and sustainable use of aquatic resources of freshwater river systems, we recommend integrating DNA barcoding with different species delimitation methods and accurate geographic distribution information. We further propose incorporating both morphological identification and DNA barcoding as an integral component of regular monitoring practices for genetic diversity, a vital tool for conservation of biodiversity and their genetic resources.
Materials and methods
Ethical statement
In the current study, fish sampling was conducted at all commercial landing stations adjacent to Beas River. Prior to sampling, the necessary approval was obtained from licensing authority of Himachal Pradesh State Biodiversity Board, Himachal Pradesh, India vide letter no. HIMCOSTE/HPSBB/2019/358 dated 05/12/2019. For this study, we exclusively utilized non-invasive tissue samples for DNA barcoding, which do not require any animal ethical clearance. Nevertheless, all procedures for handling the fish samples were carried out in accordance with the guidelines established by the Institutional Animal Ethical Committee (IAEC).
Sample collection
Fish specimens were obtained using two methods. The first method involved capturing fish using casting nets and local punga jaal (hand nets). The method involved catching fish with the help of local fishermen, taking scales or fins for non-invasive sampling, and then releasing them back into the water. The second method involved collecting tissue samples from fish at nearby landing centers of river Beas. A total of 320 fish specimens were collected from February 2020 to September 2022 from fifteen sites within the river system (Fig. 1). The river system was divided into three major areas. The first area, upstream of the Beas River system comprised of three locations in Himachal Pradesh namely Gammon bridge (stretch along Ramshila road, Manali), Old Manali road (Siyal), and Bajaura. The second area, the midstream in the river system in Kangra district of Himachal Pradesh included Khatiyar fish landing station, Barnali fish landing station, Gajj, Jwali Khad, Pong dam reservoir, and Sthana landing station. The third location was in Punjab, downstream of the river system and constituted Kiriyan, Talwandi, Jogyawal Maza walla, the Harike wetland landing station, Beas mand area, and Kapurthala landing station. Initially, all specimens were identified up to genus level based on morphological characteristics using identification keys 61. Tissue and fin clippings from the specimens were preserved in absolute ethanol and transported to the laboratory. Samples were then stored at − 20 °C until DNA isolation.
DNA extraction and COI amplification
Total genomic DNA was isolated from muscle tissue or fin clipping of 203 samples using the standard phenol–chloroform protocol62. A NanoDrop 1000 Spectrophotometer was used to quantify the isolated DNA, followed by visualization in 0.8% agarose gel electrophoresis. The amplification of universal COI primers FishF1 (5′-TCAACCAACCACAAAGACATTGGCAC-3′) and FishR1 (5′-TAGACTTCTGGGTGGCCAAAGAATCA-3′) was performed using a Polymerase chain reaction (PCR)7. The PCR reaction mixture (50 µl) containing 1.5 mM 10× buffer, 25 mM MgCl2, 10 mM dNTPs, 1.5 µl DMSO, 3U Taq polymerase, 2 pmol of each primer, 2 µl (~ 50 ng) DNA template and deionized water. The amplification protocol included an initial denaturation step at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s, extension at 72 °C for 45 s, and final extension at 72 °C for 7 min. Amplified products were separated on a 1.5% (w/v) agarose gel by loading 50 µl of PCR product and 3 µl loading dye. The bands of amplification were visualized under UV light with ethidium bromide and eluted using the QIAquick Gel Extraction Kit by Qiagen. The eluted PCR products were sent to Apical Scientific Laboratory (1st Base, Selangor, Malaysia) for Sanger sequencing.
Sequence-based identification and genetic data analysis
Sequence chromatograms were examined for quality using Sequencing Analysis software v5.1.1. The dataset of 203 sequences had an average length of 640 bp. Sequences were identified using NCBI Blast nucleotide and BOLD (Barcode of Life Data System) identification engine. All sequences were submitted to the BOLD (https://www.boldsystems.org/) database under the project “DNA barcoding of Fishes of Beas River” (code: BEAS) and GenBank with accession numbers (OR148039—OR148241). To estimate genetic divergence and phylogenetic relationships, 485 sequences belonging to the same species with sampling sites from India were retrieved from BOLD. The coordinates of sampling locations and corresponding specimen information for each sequence is given in Supplementary Table S1. A total of 688 sequences representing 43 species were analyzed to calculate genetic divergence at various taxonomic levels using Kimura two-parameter model (K2P)63 in BOLD v464. BioEdit (https://bioedit.software.informer.com/) was used to assemble and align all the sequences. DnaSP v565 was used to estimate haplotype frequency and diversity within different species. Nexus files were created from aligned sequences in DnaSP v5 software to build a median joining haplotype network and to determine the frequency of haplotypes in various regions. PopART66 was then used to construct the median-joining network of haplotypes using default parameters. A phylogenetic tree was constructed among sequences using the neighbor-joining (NJ) approach with the K2P model and 1000 bootstrap replicates in MEGA X (https://www.megasoftware.net/).
For species delimitation, Automatic Barcode Gap Discovery (ABGD) tool (https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html)67 was used with K2P model, Transition/transversion ratio (TS/TV) = 2.0, relative gap width (X) = 1.0, Prior Intra-species divergence from Pmin = 0.001 to Pmax = 0.1, and distance distribution (Nb bins) = 20. Phylogenetic relationships were reconstructed using Bayesian Inference (BI) analysis in MrBayes 3.2.768. The dataset was run with gamma-distribution rate variation, invariable positions (GTR + G + I), and six substitution sites (nst = 6). MrBayes was run with Monte Carlo Markov chains (MCMC) for 20,000,000 generations, sampling every 10,000 generations, and a chain temperature of 0.2. The initial 25 percent of the trees were burned in and the resulting tree file was visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/).
Data availability
Sequence data and collection information have been made available on BOLD (www.boldsystems.org), Project code: BEAS. Sequences are also deposited in GenBank (Accession numbers OR148039—OR148241). The overall dataset of sequences analyzed in the study has been made publicly visible on BOLD (Dataset Code: DS-INTF).
References
Ko, H. L. et al. Evaluating the accuracy of morphological identification of larval fishes by applying DNA barcoding. PLoS ONE 8, e53451. https://doi.org/10.1371/journal.pone.0053451 (2013).
Bhattacharya, M. et al. DNA barcoding to fishes: Current status and future directions. Mitochondr. DNA Part A 27, 2744–2752. https://doi.org/10.3109/19401736.2015.1046175 (2016).
Krishna-Krishnamurthy, P. & Francis, R. A. A critical review on the utility of DNA barcoding in biodiversity conservation. Biodivers. Conserv. 21, 1901–1919. https://doi.org/10.1007/s10531-012-0306-2 (2012).
Hebert, P. D., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. B 270, 313–321. https://doi.org/10.1098/rspb.2002.2218 (2003).
Ji, Y. et al. Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol. Lett. 16, 1245–1257. https://doi.org/10.1111/ele.12162 (2013).
Lara, A. et al. DNA barcoding of Cuban freshwater fishes: Evidence for cryptic species and taxonomic conflicts. Mol. Ecol. Resour. 10, 421–430. https://doi.org/10.1111/j.1755-0998.2009.02785.x (2010).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. DNA barcoding Australia’s fish species. Philos. Philos. Trans. R Soc. Lond. B Biol. Sci. 360, 1847–1857 (2005).
Zemlak, T. S., Ward, R. D., Connell, A. D., Holmes, B. H. & Hebert, P. D. DNA barcoding reveals overlooked marine fishes. Mol. Ecol. Resour. 9, 237–242 (2009).
Ude, G. N. et al. DNA barcoding for identification of fish species from freshwater in Enugu and Anambra States of Nigeria. Conserv. Genet. Resour. 12, 643–658 (2020).
Ali, F. S., Ismail, M. & Aly, W. DNA barcoding to characterize biodiversity of freshwater fishes of Egypt. Mol. Biol. Rep. 47, 5865–5877 (2020).
Kress, W. J., García-Robledo, C., Uriarte, M. & Erickson, D. L. DNA barcodes for ecology, evolution, and conservation. Trends Ecol. Evol. 30, 25–35 (2015).
Korshunova, T. et al. Multilevel fine-scale diversity challenges the ‘cryptic species’ concept. Sci. Rep. 9, 6732 (2019).
Shin, C. P. & Allmon, W. D. How we study cryptic species and their biological implications: A case study from marine shelled gastropods. Ecol. Evol. 13, e10360 (2023).
Fourie, A., Wingfield, M. J., Wingfield, B. D. & Barnes, I. Molecular markers delimit cryptic species in Ceratocystis sensu stricto. Mycol. Prog. 14, 1–18 (2015).
Obase, K., Douhan, G. W., Matsuda, Y. & Smith, M. E. Revisiting phylogenetic diversity and cryptic species of Cenococcum geophilum sensu lato. Mycorrhiza 26, 529–540 (2016).
Holzer, A. S. et al. ‘Who’s who’in renal sphaerosporids (Bivalvulida: Myxozoa) from common carp, Prussian carp and goldfish–molecular identification of cryptic species, blood stages and new members of Sphaerospora sensu stricto. Parasitology 140, 46–60 (2013).
Casetta, E., Marques-da-Silva, J. & Vecchi, D. From Assessing To Conserving Biodiversity: Conceptual and Practical Challenges 452 (Springer Nature, 2019).
Puckridge, M., Andreakis, N., Appleyard, S. A. & Ward, R. D. Cryptic diversity in flathead fishes (Scorpaeniformes: Platycephalidae) across the Indo-West Pacific uncovered by DNA barcoding. Mol. Ecol. Resour. 13, 32–42 (2013).
Hubert, N. et al. Cryptic diversity in Indo-Pacific coral-reef fishes revealed by DNA-barcoding provides new support to the centre-of-overlap hypothesis. PLoS One 7, e28987. https://doi.org/10.1371/journal.pone.0028987 (2012).
Mat Jaafar, T. N. A., Taylor, M. I., Mohd Nor, S. A., de Bruyn, M. & Carvalho, G. R. DNA barcoding reveals cryptic diversity within commercially exploited Indo-Malay Carangidae (Teleosteii: Perciformes). PLoS One 7, e49623. https://doi.org/10.1371/journal.pone.0049623 (2012).
Iyiola, O. A. et al. DNA barcoding of economically important freshwater fish species from north-central Nigeria uncovers cryptic diversity. Ecol. Evol. 8, 6932–6951. https://doi.org/10.1002/ece3.4210 (2018).
Winterbottom, R., Hanner, R. H., Burridge, M. & Zur, M. A cornucopia of cryptic species-a DNA barcode analysis of the gobiid fish genus Trimma (Percomorpha, Gobiiformes). ZooKeys 381, 79–111. https://doi.org/10.3897/zookeys.381.6445 (2014).
Hyde, J. R., Underkoffler, K. E. & Sundberg, M. A. DNA barcoding provides support for a cryptic species complex within the globally distributed and fishery important opah (Lampris guttatus). Mol. Ecol. Resour. 14, 1239–1247. https://doi.org/10.1111/1755-0998.12268 (2014).
Wang, T., Zhang, Y. P., Yang, Z. Y., Liu, Z. & Du, Y. Y. DNA barcoding reveals cryptic diversity in the underestimated genus Triplophysa (Cypriniformes: Cobitidae, Nemacheilinae) from the northeastern Qinghai-Tibet Plateau. BMC Evol. Biol. 20, 1–15. https://doi.org/10.1186/s12862-020-01718-0 (2020).
Doorenweerd, C., San Jose, M., Barr, N., Leblanc, L. & Rubinoff, D. Highly variable COI haplotype diversity between three species of invasive pest fruit fly reflects remarkably incongruent demographic histories. Sci. Rep. 10, 6887. https://doi.org/10.1038/s41598-020-63973-x (2020).
Zhao, Y. et al. Genetic diversity and variation of seven Chinese grass shrimp (Palaemonetes sinensis) populations based on the mitochondrial COI gene. BMC Ecol. Evol. 21, 167. https://doi.org/10.1186/s12862-021-01893-8 (2021).
Yan, J. et al. Genetic diversity analysis of brown marmorated stink bug, Halyomorpha halys based on mitochondrial COI and COII haplotypes. BMC Genom. Data 22, 1–16. https://doi.org/10.1186/s12863-021-00961-8 (2021).
Goodall-Copestake, W. P., Tarling, G. A. & Murphy, E. On the comparison of population-level estimates of haplotype and nucleotide diversity: A case study using the gene cox1 in animals. Heredity 109, 50–56. https://doi.org/10.1038/hdy.2012.12 (2012).
Zhang, G. et al. Genetic diversity and phylogeography of Taenioides cirratus in five geographical populations based on mitochondrial COI and Cytb gene sequences. J. Appl. Ichthyol. 2023, 4459823. https://doi.org/10.1155/2023/4459823 (2023).
Froese, R. FishBase. world wide web electronic publication. http://www.fishbase.org (2022).
Moza, U. & Mishra, D. N. River Beas Ecology and Fishery (Central Inland Fisheries Research Institute, 2007).
Kumar, A. & Khanna, D. R. Ichthyofaunal diversity in upper stretches of River Beas, Himachal Pradesh, India. IJRBAT 2, 269–275 (2014).
Kumar, A. Hydrological conditions of river Beas and its fish fauna in Kullu Valley, Himachal Pradesh, India. Environ. Conserv. J. 11, 7–10. https://doi.org/10.36953/ECJ.2010.110302 (2010).
Dua, A. & Parkash, C. Distribution and abundance of fish populations in Harike wetland-A Ramsar site in India. J. Environ. Biol. 30, 247–251 (2009).
Kaur, H., Datta, S. N. & Singh, A. Fish catch composition and biodiversity indices at harike wetland-a ramsar site in India. J. Anim. Res. 7, 935–941. https://doi.org/10.5958/2277-940X.2017.00142.5 (2017).
Lostrom, S. et al. Linking stream ecology with morphological variability in a native freshwater fish from semi-arid Australia. Ecol. Evol. 5, 3272–3287. https://doi.org/10.1002/ece3.1590 (2015).
Jindal, R., Singh, H. & Sharma, C. Fish diversity of Pong dam reservoir and Harike wetland. Int. J. Appl. Sci. Eng. 3, 232–240. https://doi.org/10.6088/ijaser.030100023 (2014).
Lakra, W. S. et al. DNA barcoding Indian marine fishes. Mol. Ecol. Resour. 11, 60–71. https://doi.org/10.1111/j.1755-0998.2010.02894.x (2011).
Lakra, W. S. et al. DNA barcoding Indian freshwater fishes. Mitochondr. DNA A DNA Mapp. Seq. Anal. 27, 4510–4517. https://doi.org/10.3109/19401736.2015.1101540 (2016).
Bingpeng, X. et al. DNA barcoding for identification of fish species in the Taiwan Strait. PloS One 13, e0198109. https://doi.org/10.1371/journal.pone.0198109 (2018).
Modeel, S., Joshi, B. D., Yadav, S., Bharti, M. & Negi, R. K. Mitochondrial DNA reveals shallow population genetic structure in economically important Cyprinid fish Labeo rohita (Hamilton, 1822) from South and Southeast Asia. Mol. Biol. Rep. 50, 1–9. https://doi.org/10.1007/s11033-023-08386-5 (2023).
Hubert, N. et al. Identifying Canadian freshwater fishes through DNA barcodes. PLoS One 3, e2490. https://doi.org/10.1371/journal.pone.0002490 (2008).
Khedkar, G. D., Jamdade, R., Naik, S., David, L. & Haymer, D. DNA barcodes for the fishes of the Narmada, one of India’s longest rivers. PloS One 9, e101460. https://doi.org/10.1371/journal.pone.0101460 (2014).
Pandey, P. K. et al. DNA barcoding and phylogenetics of freshwater fish fauna of Ranganadi River, Arunachal Pradesh. Gene 754, 144860. https://doi.org/10.1016/j.gene.2020.144860 (2020).
Rees, D. J. et al. Global phylogeography suggests extensive eucosmopolitanism in Mesopelagic Fishes (Maurolicus: Sternoptychidae). Sci. Rep. 10, 1–12. https://doi.org/10.1038/s41598-020-77528-7 (2020).
Chen, W., Ma, X., Shen, Y., Mao, Y. & He, S. The fish diversity in the upper reaches of the Salween River, Nujiang River, revealed by DNA barcoding. Sci. Rep. 5, 17437. https://doi.org/10.1038/srep17437 (2015).
Akhtar, T. & Ali, G. DNA barcoding of Schizothorax species from the Neelum and Jhelum Rivers of Azad Jammu and Kashmir. Mitochondr. DNA B: Resour. 1, 934–936. https://doi.org/10.1080/23802359.2016.1258337 (2016).
Baskett, M. L. & Gomulkiewicz, R. Introgressive hybridization as a mechanism for species rescue. Theor. Ecol. 4, 223–239. https://doi.org/10.1007/s12080-011-0118-0 (2011).
Tan, M. & Armbruster, J. W. Phylogenetic classification of extant genera of fishes of the order Cypriniformes (Teleostei: Ostariophysi). Zootaxa 4476, 6–39. https://doi.org/10.11646/zootaxa.4476.1.4 (2018).
Yang, L. et al. Molecular phylogeny of the cyprinid tribe Labeonini (Teleostei: Cypriniformes). Mol. Phylogenet. Evol. 65, 362–379. https://doi.org/10.1016/j.ympev.2012.06.007 (2012).
Zheng, L. P., Chen, X. Y. & Yang, J. X. Molecular phylogeny and systematic revision of Bangana sensu lato (Teleostei, Cyprinidae). J. Zool. Syst. Evol. Res. 57, 884–891. https://doi.org/10.1111/jzs.12294 (2019).
Rahman, M. M., Norén, M., Mollah, A. R. & Kullander, S. The identity of Osteobrama cotio, and the status of “Osteobrama serrata” (Teleostei: Cyprinidae: Cyprininae). Zootaxa 4504, 105–118. https://doi.org/10.11646/zootaxa.4504.1.5 (2018).
Garcia-Cisneros, A. et al. Intraspecific genetic structure, divergence and high rates of clonality in an amphi-Atlantic starfish. Mol. Ecol. 27, 752–772. https://doi.org/10.1111/mec.14454 (2018).
Joshi, B. D. et al. Understanding genetic diversity and population genetic structure of three Cyprinidae fishes occupying the same habitat from Uttarakhand, India. Mitochondr. DNA B: Resour. 4, 2956–2961. https://doi.org/10.1080/23802359.2019.1662740 (2019).
Negi, R. K., Joshi, B. D., Johnson, J. A. & Goyal, S. P. Application of computational methods in fish species identification based on mitochondrial DNA sequences. Curr. Sci. 2172–2176, 2016. https://doi.org/10.18520/cs/v110/i11/2172-2176 (2016).
Hebert, P. D. N., Stoeckle, M. Y., Zemlak, T. S. & Francis, C. M. Identification of birds through DNA barcodes. PLoS Biol. 2, e312. https://doi.org/10.1371/journal.pbio.0020312 (2004).
Ricciardi, A. & Kipp, R. Predicting the number of ecologically harmful exotic species in an aquatic system. Divers. Distrib. 14, 374–380. https://doi.org/10.1111/j.1472-4642.2007.00451.x (2008).
Sharma, I. & Dhanze, R. Length-weight relationship of Schizothorax richardsonii (Gray) from Indus (beas river system, HP) India. Rec. Zool. Surv. India 111, 63–70. https://doi.org/10.26515/rzsi/v111/i1/2011/158892 (2011).
Ibrahim, A. et al. Skin marks on the Indus River Dolphin (Platanista minor) and their implications for conservation. Pak. J. Zool. 54, 2329–2336. https://doi.org/10.17582/journal.pjz/20210828090819 (2021).
Singh, S. Punjab: Large number of fish found dead in Beas river due to release of sugar mill fluid. Hindustan Times. https://www.hindustantimes.com/punjab/punjab-large-number-of-fish-found-dead-in-beas/story-0XDIbfquUB0h2evGF9qskN.html (2018).
Talwar, P. K. & Jhingran, A. G. Inland Fishes of India and Adjacent Countries (CRC Press, 1991).
Sambrook, J., Fritsch, E. F. & Maniatis, T. Molecular Cloning: A Laboratory Manual (eds. Inglis, J. et al.) 11–71 (Cold spring harbor laboratory press, 1989).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. https://doi.org/10.1007/BF01731581 (1980).
Ratnasingham, S. & Hebert, P. D. BOLD: The barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x (2007).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Leigh, J. W. & Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. https://doi.org/10.1111/2041-210X.12410 (2015).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x (2012).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Acknowledgements
SM thanks Science and Engineering Research Board (SERB) for providing research fellowship. RKN acknowledges Science and Engineering Research Board and Institution of eminence (IoE) for providing research grants. RKN and SM appreciates Dr. Pankaj Patiyal and Mr. Ankush Dhiman for assistance during the sampling process.
Funding
This work were funded by Science and Engineering Research Board (Grant F. No. EEQ/2019/000214).
Author information
Authors and Affiliations
Contributions
S.M. and R.K.N. designed the study. S.M. and M.S. performed DNA extraction and PCR amplification. S.M., P.D., S.Y., S.S., P.Y., T.N. contributed to sampling of fish specimens. S.M. analyzed the data and wrote the paper. R.K.N. reviewed the analysis, manuscript and provided guidance throughout the research work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Modeel, S., Negi, R.K., Sharma, M. et al. A comprehensive DNA barcoding of Indian freshwater fishes of the Indus River system, Beas. Sci Rep 14, 2763 (2024). https://doi.org/10.1038/s41598-024-52519-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52519-0
This article is cited by
-
Decoding the molecular phylogenetics of ornamental catfishes (siluriformes) of North East India using DNA barcoding approach
Molecular Biology Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
