
Abstract
The Ryukyu Islands of Japan are a biodiversity hotspot due to geographical and historical factors. Tricyrtis formosana is a perennial herbaceous plant that commonly found in Taiwan. But only a few populations have been identified in a limited habitat on Iriomote Island, while populations of unknown origin occur near human settlements in an area on the main island of Okinawa. To better understand these populations of the phylogenetic uniqueness and intrinsic vulnerability, we conducted comparative analyses including (1) phylogeny and population structure with MIG-seq data, (2) photosynthesis-related traits of plants grown under common conditions and (3) transcriptome analysis to detect deleterious variations. Results revealed that T. formosana was split into two clades by the congeners and that Iriomote and Okinawa populations independently derived from ancestral Taiwanese populations in each clade. Photosynthetic efficiency was lowest in the Iriomote population, followed by Okinawa and Taiwan. Transcriptome analysis showed that the Iriomote population accumulated more deleterious variations, suggesting intrinsic vulnerability. These results indicate that each T. formosana population in Japan is phylogenetically unique and has been independently dispersed from Taiwan, and that the Iriomote population presents a high conservation difficulty with a unique photosynthesis-related characteristic and a larger amount of deleterious variations.
Similar content being viewed by others


Introduction
The Ryukyu Archipelago and Taiwan, located along the western rim of the Pacific Ocean, comprise of approximately 140 subtropical islands and are home to many endemic species, including those listed as CR (Critically Endangered), EN (Endangered), or VU (Vulnerable) on the ICUN Red List, as well as many rare plants. The diverse floristic composition of the Ryukyu Archipelago can be attributed to multiple factors. This region overlaps with the migration routes of birds between the northern and southern hemispheres and is impacted by the Kuroshio Current, a warm current known to facilitate overseas dispersal of plant species1,2. Additionally, the Ryukyu Archipelago is regularly exposed to strong tropical storms originating from the east of the Philippines. The diverse flora of the Ryukyu Archipelago is the result of multiple factors, including the introduction of various plant taxa through different migration routes and vectors. The complexity of its geological history also plays a role in its diversity. The island arc was separated from the eastern edge of the continent during the late Miocene and early Pleistocene and underwent repeated separations and connections with neighboring islands due to marine regression-transgression associated with the glacial and interglacial cycles of the Pleistocene3,4,5,6,7. The geological history played a key role in shaping its diverse flora of the Ryukyu Archipelago, and made the area a habitat of species that have survived under isolated conditions8 as well as new endemic species that have emerged as a result of adaptive differentiation9.
Iriomote Island, locating in the southern part of the Ryukyu Archipelago, houses 1,165 species of vascular plants over an area of 289.61 km2, with 59 species (5%) being endemic10. Despite the lower proportion of plant endemicity compared to Hawaii archipelago (90%) and Galapagos Archipelago (approx. 35%)11. Iriomote shows a larger number of endemic species per area (4.02 km−2) compared to Hawaii (0.06 km−2) and Galapagos Archipelago (0.022 km−2)10,11. This highlights the high biodiversity of Iriomote and the variety of species that thrive on the small island.
This study focused on a perennial herbaceous plant, Tricyrtis formosana Baker (Lilliaceae), as a representative example of plant differentiation influenced by the complex environmental and geological conditions of the Ryukyu Archipelago. The genus Tricyrtis commonly known as ‘Toad lily’ and is endemic to eastern Asia, including the Himalayas, China, Japan, Taiwan, and the Philippines12. There are four species of Tricyrtis in Taiwan, of which only T. formosana is found on Iriomote Island and Okinawa Island of the Ryukyu Archipelago in Japan13. In Taiwan, T. formosana is a widespread and commonly found plant13,14, and is widely cultivated for horticultural purposes in Japan. In contrast, only a few wild populations are known in Iriomote and Okinawa Island.
Based on the distinct habitat and petal colour, T. formosana individuals in the Iriomote population can be distinguished from T. formosana that grows in Taiwan. The Iriomote population is known to grow in limited habitats, primarily near waterfalls under constant spray conditions, with only a small number of individuals estimated to be about one hundred found in five locations. Due to its restricted distribution, this species is listed on the Red List of the Japanese Ministry of the Environment15.
Meanwhile, the population on Okinawa Island, the central Ryukyu Archipelago, is distributed near human settlements, and its population size is much larger than that of Iriomote. Okinawa Island, located 400 km northeast of Iriomote Island, represents the northern marginal distribution of this species. Although being more distant from the Taiwan mainland than Iriomote Island, the growing environment and petal colour of the Okinawa population are similar to those of the Taiwanese population. This raises the possibility that the Okinawa population was introduced from the Taiwanese population by human activities, and the conservation value of this population has not yet been determined.
The objective of this study was to investigate the intraspecific phylogeny and physiological traits of populations T. formosana. The focus was on understanding the origin and assessing their conservation value based on the uniqueness of the rare populations known in the isolated and marginal distribution range of the species in Japan (Fig. 1). T. formosana comprises populations with varying physiological characteristics: the commonly found Taiwanese populations growing in forests, the Okinawa population growing along farm roads and in forests near human settlements, and the Iriomote population growing only near waterfalls in forests exposed to constant spray. These differences in growth conditions suggest that each population has different physiological and genomic characteristics. Some endangered species exhibit intrinsic vulnerability due to genomic factors, such as a high accumulation of deleterious mutations and a smaller ratio of duplicated genes, compared with non-endangered congener species, which increase their conservation difficulty16. To guide the appropriate direction of conservation strategies, we integrated different sources of information of phylogeny, physiological characteristics and genomic traits.

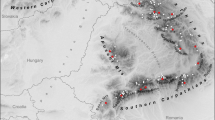
Locations of sampling sites of T. formosana with a legend indicating the number of samples per locations for MIG-seq. The gray shading in Taiwan represents areas with elevations of 2000 m or higher, which may act as barriers to the distribution of T. formosana. Shorelines were extracted from GSHHG17, and the contour lines of 2000 m were generated from Digital Elevation Model of ETOPO 202218. These vector data were integrated, and the map was created using QGIS 3.2219.
Results
Phylogenetic analysis and population structure analysis
A taxon that has been considered T. formosana s. l. was clearly divided into two clades by the intervention of congener species: T. ravenii and T. lasiocarpa (Fig. 2). The first clade consists of samples from populations in the eastern Taiwan, Orchid Island, and Iriomote Island and the other clade consists of those in Okinawa Island, the northern Taiwan and the middle Taiwan populations. The populations in Iriomote and Okinawa, Japan were found to be distantly related, with the Okinawa population being closer to northern Taiwan and distinct from both the Iriomote population and a cultivar commonly sold as T. formosana in Japan. For the clade consisting of A, C, D, G, H, and E, T. lasiocarpa was an outgroup, and population A on Okinawa Island was in the most derived position. For the clade consisting of B, F, and I, T. ravenii was an outgroup, and Iriomote population B was in a more derived position than Taiwanese population E.

ML phylogenetic tree of T. formosana based on 233 SNP loci obtained by MIG-seq analysis using TVMef model. Labels (A–I) correspond to those in Fig. 1.
The long branch separating Okinawa from northern Taiwan and long inner branching of the Okinawa population suggest it has a natural, long-standing presence on the main island of Okinawa. Conversely, the Iriomote population is closely related to the Orchid Island population and the inner branches are short (Fig. 2).
The STRUCTURE HARVESTER showed that the delta K was highest at K = 2, followed by K = 3, 7, and 4 (Supplementary Fig. S1). The results of the STRUCTURE analysis indicated that at K = 2, the Okinawa population and the Iriomote population belonged to distinct clusters. As the number of clusters increased, the population genetic structure became more intricate, but these patterns corresponded to the internal structure of the phylogenetic tree shown in Fig. 2. The results of the STRUCTURE and phylogenetic analyses were consistent and did not contradict each other. At K = 3, the FST values were as follows: Cluster 1 = 0.5319, Cluster 2 = 0.2824, and Cluster 3 = 0.6326. The fact that Cluster 1 and Cluster 3 including Iriomote and Okinawa populations, respectively, have values greater than that of Cluster 2, which corresponds to Middle Taiwan, indicates that the two Japanese populations are both distinct and derived from Taiwan.
The observed heterozygosity at all base positions calculated with populations in Stacks 2.55 was lower in the Japanese populations; 1.0 × 10–4 in the Iriomote population, 1.4 × 10–4 in the Okinawa population and 3.2 × 10–4 in the Taiwanese population.
Comparative analysis of photosynthesis-related traits
The results of the linear mixed model indicated a positive correlation between SPAD and cross-sectional length (LMM, coefficient = 56.65, t = 4.76). Additionally, the results of the GLM multiple regression analysis revealed a significant positive effect for cross-sectional length (coefficient = 61.6610, t = 9.114, p < 0.001) while the effect for the population was not significant (Fig. 3).

SPAD (chlorophyll content) and cross-sectional leaf length.
Multiple comparisons revealed a significant difference in ETR (electron transportation rate in photosystem II) between the Northern Taiwan and the Iriomote populations at PAR ≥ 380 mol quanta m−2 s−1 (p < 0.05) (Fig. 4). Despite the absence of a significant difference, the overall ETR was the highest in the Taiwanese population, followed by the Okinawa and Iriomote populations.

ETR (electron transportation rate) calculated at each PAR (photosynthetically active radiation) = 130–1640 mol quanta m−2 s−1. NT Northern Taiwan, MT Middle Taiwan, ET Eastern Taiwan, IR Iriomote, OK Okinawa population.
Estimation of conservation difficulty by transcriptome analysis
The evaluation of genetic diversity based on the number of heterozygous synonymous SNVs in the transcripts per kb revealed that individuals in the Iriomote population exhibited significantly lower diversity compared to other populations (p < 0.05) (Fig. 5A). Individuals in the Iriomote population exhibited significantly higher values in three indices of deleterious variations; the ratio of deleterious amino acid variations in heterozygous SNVs (Fig. 5B), the proportion of non-synonymous variants for each coding sequence (Fig. 5C), and the proportion of nonsense SNVs to total non-synonymous SNVs (Fig. 5D), compared to the Taiwan and Okinawa population. Individuals of the Taiwan and Okinawa populations exhibited lower rates of detrimental mutations, which corresponds to the fact that these individuals are robust and adapted to a wider range of environments in the wild.

Box plots of genetic characteristics of the Taiwanese (northern, middle and eastern), Iriomote and Okinawa populations based on transcriptome analysis. Different alphabets indicate significant differences. (A) Genetic diversity based on the number of heterozygous synonymous SNVs per kb on the longest coding sequences, (B) ratios of deleterious amino acid variations in heterozygous SNVs estimated using PROVEAN, (C) ratios of non-synonymous SNVs calculated for each gene, (D) ratio of nonsense SNVs in the total non-synonymous SNVs.
Discussion
As for the taxonomic treatment of Trycyrtis in Taiwan, The 1st edition of the Flora of Taiwan recognizes a total of 5 species and 2 varieties of Trycyrtis plants in Taiwan20, which include T. formosana var. formosana, T. formosana var. glandosa, T. stolonifera, T. ovatifolia, T. lasiocarpa, and T. suzukii, while in the 2nd edition of the Flora of Taiwan, two species and five varieties of Tricyrtis are recognized21, including T. formosana var. formosana, T. formosana var. glandiflorum, T. formosana var. stolonifera, T. formosana var. ovatifolia, T. formosana var. lasiocarpa, and T. suzukii. Subsequently, a taxonomic reevaluation of the varieties of T. formosana had classified evergreen plants with traits of glabrous ovaries/capsules, primarily found in low-elevation areas, as T. formosana, and also introduced a new species, T. ravenii, for plants primarily distributed in high-elevation areas exhibiting deciduous characteristics and hairy ovaries/capsules13. Furthermore, due to distinct differences in leaf morphology, flower and fruit characteristics, T. formosana var. lasiocarpa was recognized as an independent species, T. lasiocarpa. Following the revisions, the Tricyrtis genus in Taiwan is currently recognized to consist of four species: T. formosana, T. ravenii, T. lasiocarpa, and T. suzukii13,22.
In this study, ML phylogenetic tree revealed that T. formosana was divided into two distinct clades by the intervention of two congener species, and each of them may potentially represent separate species (Fig. 2). However, we have not identified any distinguishing morphological characteristics to differentiate between these two clades. The taxonomic treatment of these two clades, and integrated understanding of phylogenetic and morphological differentiation are matters that should be addressed in the future. The phylogenic clusters of the Taiwanese population of T. formosana corresponded to the geographic distribution; however, the two populations in Japan did not have the closest phylogenetical relationship to each other; deriving from different clades in Taiwan. Both the Iriomote and Okinawa populations had a long branching from the most related Taiwanese population, indicating their phylogenetic uniqueness.
In accordance with the phylogenetic tree, the STRUCTURE analysis split the ancestral genetic cluster into a consistent pattern (Fig. 6), suggesting that the Okinawa population was derived from the northern Taiwan population, and the Iriomote population has the closet phylogenetic relationship with the eastern Taiwan and Orchid Island population. Based on the mean Fst values at K = 3 in STRUCUTRE, the ancestor of T. formosana (Fst = 0.2824 for Cluster 2 in the panel of K = 3, Fig. 6) is likely to have originated from a population that has diverged from congener species in the middle of the Taiwan mainland. Then the population have expanded the distribution to the northern (Fst = 0.6326 for Cluster 3 in the panel of K = 3, Fig. 6) and the eastern (Fst = 0.5319 for Cluster 1 in the panel of K = 3, Fig. 6) areas, followed by dispersal to Okinawa Island from the northern area, as well as Iriomote Island from the eastern area. It is noted that T. formosana is distributed from 100 to 1500 m sea level in Taiwan mainland15, and the populations of Northern Taiwan, Eastern Taiwan and Middle Taiwan in this study are separated from each other by 2000–3000 m mountainous ranges that can act as geographical barrier.

Population structure of the 28 populations of T. formosana estimated by STRUCTURE analysis. The mean Fst at K = 3; Cluster 1 = 0.5319, Cluster 2 = 0.2824, Cluster 3 = 0.6326.
The two Japanese populations were phylogenetically distinct (Fig. 2), and it is likely to have arrived in Japan independently through separate and distinct routes; from the northern area in Taiwan to Okinawa Island, and from the eastern area in Taiwan to Iriomote Island via Orchid Island. Thus, the Japanese populations of T. formosana are distinctive from each other in their phylogenetic position as well as in their migratory pathways, and each has separate conservation values.
The population of T. formosana in Okinawa presents a case of disjunct distribution, as it is inferred to have originated from northern Taiwan, located approximately 600 km away. While the region experiences high-intensity tropical storms, which could provide strong winds capable of seed dispersal, the seeds lack the aerodynamic structure necessary for successful dispersal through wind. Dispersal by sea currents via rafting is considered unlikely due to the lack of floatability and saltwater tolerance of the seed, which are necessary for long-distance dispersal by sea currents. The small and flat seeds of T. formosana, measuring less than 2 mm square, make it an ideal candidate for dispersal by birds. Phylogenetic studies of Solenogyne mikadoi (Asteraceae) and Lobelia loochooensis (Campanulaceae) support the hypothesis of avian-mediated dispersal, as the antitropical distribution of these species suggests the presence of migratory birds in more than 7000 km distance from Australia to the Ryukyu Archipelago23,24.
Several instances are recognized where different closely related taxa are distributed and have undergone differentiation across various regions of the Ryukyu Islands. Tashiroea okinawensis/T. yaeyamensis (Melastomataceae) exhibits shallow differentiation in the Ryukyu region, with closely related species found on the Chinese mainland25. Viola iwagawae/V. tashiroi (Violaceae) show minor differentiation in this area26. The globally widespread species Dodonaea viscosa (Sapindaceae) has lineages near Taiwan on Ishigaki Island in the southern Ryukyus while lineages closer to the Philippines on the islands in the northern and central Ryukyus27. While molecular phylogenetic analysis has not been conducted, taxa such as Eurya zigzag/E. yaeyamensis (Pentaphylacaceae), Adinandra ryukyuensis/A. yaeyamensis (Pentaphylaceae), and Swertia tashoroi/S. makinoana (Gentianaceae) exhibit similar distributions in this area. Comparative analyses of these taxa could provide valuable information on the formation processes of biodiversity in this region.
The discovery that the Orchid Island population is the most closely related to the Iriomote Island population, rather than the geographically proximate northern Taiwanese population, is an interesting finding. Moreover, it is intriguing to note that Freycinetia williamsii (Pandanaceae), Nothapodytes foetida (Icacinaceae), Monoon liukiuensis (Annonaceae), which are critically endangered plant species restricted to Iriomote Island in Japan, are not distributed on the main island of Taiwan but on Orchid Island. This observation raises the possibility of an unknown biological factor that could be linking Iriomote Island and Orchid Island. Further investigation is necessary to elucidate the underlying mechanisms responsible for this pattern, and such research could provide valuable insights into the distribution of rare plant species in the region.
Despite the Okinawa population being sampled within a smaller range of 150 m stratch along the rural road, compared to the Iriomote populations, which were separated more than 10 km, we found that the genetic diversity assessed by number of heterozygous synonymous SNVs per kb was about twice as high in the Okinawa population (Fig. 5A). The greater genetic diversity of the Okinawa population may be due to its larger population size compared to that of the Iriomote population. The high genetic diversity of the Okinawa population is crucial for its survival, as it may provide a foundation for the species to adapt to future environmental changes28.
The conservation value was recognized on both population of the T. formosana in Iriomote Island and Okinawa Island for their own uniqueness. However, we indicate that the Iriomote population was characterized higher vulnerability as evidenced by its the lower genetic diversity (Fig. 5A) and larger accumulation of deleterious variants (Fig. 5B–D) compared to other populations. The genome-wide reduction of genetic diversity and accumulation of deleterious variants in functional genes might result in the narrow habitat range and high vulnerability for the Iriomote population, which is limited only to the vicinity of a waterfall. Previous studies have shown that population isolation and contraction can weaken purifying selection and increase the number of deleterious mutations in wild species, such as the brown eared pheasant (Crossoptilon mantchuricum) and two subspecies of mountain gorilla (Gorilla spp.)29,30. This trend is also expected to be observed in the Iriomote population due to its narrow habitat range and small population size. However, inbreeding can sometimes lead to purging of deleterious recessive mutations in a small wild population. While it has been observed that inbreeding reduces the burden of deleterious mutation31,32, it can not necessarily apply to extremely small populations, because alleles may fall out of purifying selection, leading to population decline and the accumulation of deleterious mutations33,34. In contrast, the Okinawa population shows less loss of genetic diversity and accumulation of deleterious mutations, likely due to its larger population size. This is supported by information in the Okinawa prefecture Red List35, which states that T. formosana in Okinawa Island is widely distributed throughout the upper of mountain range in the same water catchment area.
Previous research presented a relationship between photosynthesis efficiency, as indicated by PAR (photosynthetically active radiation) absorption, and levels of net primary production36. The ETR (electron transportation rate) is a measure of light energy and the decrease leads to photoinhibition, which is characterized by damage to the reaction center chlorophyll37 and a decline in photosynthetic efficiency. Our study found that the ETR values of the Iriomote population were significantly lower than those of the Northern Taiwan population when PAR was greater than 380 (Fig. 4). Shade plants typically have lower ETR compared to sun plants, which is consistent with the fact that the Iriomote population is restricted to the vicinity of the waterfall in the forest. The Iriomote population is considered a critically endangered species on the Red List, Ministry of Environment of Japan2. To ensure the conservation of this population, care must be taken to prevent photoinhibition under intense sunlight, as shade plants are susceptible to an excess of light. On the contrary, the Okinawa population did not show a reduction in genetic diversity or an accumulation of deleterious variants, hence it is considered that the population can be conserved by maintaining the status quo, that is, relatively simple management, such as the removal of competing herbaceous species in the habitat.
In conclusion, our study has shown that the two Japanese populations of T. formosana are phylogenetically distinct and have high conservation values due to their independent migration from Taiwan. In Japan, the Act on Conservation of Endangered Species of Wild Fauna and Flora calls for maximum conservation efforts for species that is designated as nationally rare and endangered. While T. formosana in Japan has not yet been designated as such, our results suggest that the Iriomote population deserves this recognition. Furthermore, our findings indicate that the Iriomote population is more difficult to conserve compared to other populations, due to its photophysiological traits and accumulation of deleterious mutations in the genome. These results provide a basis for future conservation and breeding programs and highlight the need for more careful management of plants from the Iriomote populations under ex-situ conditions, including protection measures to prevent photoinhibition. Our study provides insight into the phylogenetic origin and uniqueness of endangered T. formosana in Japan and evaluates the conservation difficulty of each population. The distribution of plant species and administrative divisions, such as national borders, are independent. Many plant species have a broad geographic range that spans multiple countries and, as a result, may be deemed rare in a particular country or region. By correctly evaluating the phylogenetic distinctiveness and genomic features of plant populations in each region through methods like those used in this study, we can assess the conservation status of these species and develop effective conservation strategies.
Materials and methods
Sample collection and DNA extraction
Leaf samples were collected from Okinawa Island, Iriomote Island, Taiwan mainland and Orchid Island. One marketed cultivar and two outgroups (one each of T. lasiocarpa and T. ravenii) were also used for the analysis (Supplementary Table S1). The collection and use of plant materials in this study were carried out in compliance with relevant institutional, national, and international guidelines and legislation. Necessary permissions and licenses were obtained from the relevant authorities (Ministry of the Environment and Forestry Agency for Japanese samples, and Academia Sinica for Taiwanese samples). The plant specimens were identified by two of the current authors, Akiyo Naiki, an associate professor at University of the Ryukyus and Takuro Ito, an assistant professor at Tohoku University and voucher specimens were deposited at the Herbarium of the Faculty of Science, University of the Ryukyus (RYU; Naiki 18254 and Naiki 18246) and the Herbarium of Tohoku University (TUS; T. Ito 4674, T. Ito 4915, T. Ito 4928, T. Ito 5019, T. Ito 5021, T. Ito 6126 and T. Ito 8800). Genomic DNA was extracted from a total of 28 leaf samples using a modified CTAB method38.
SNP genotyping by MIG-seq
DNA samples were sequenced by MIG-seq method following the protocol of Suyama and Matsuki39. MIG-seq library were constructed through two PCR steps. First, multiple nonrepetitive regions from various ISSRs were amplified by multiplexing with tailed ISSR primers. PCR was performed in an AB Veriti™ 96-Well Thermal Cycler (Thermo Fisher Scientific, Applied Biosystem, CA, USA) under the following thermocycler conditions: initial denaturation at 94 °C for 1 min; followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 38 °C for 1 min and extension at 72 °C for 1 min. A final extension at 72 °C for 10 min completed the first PCR. Second, we cleaned and normalized each 1st PCR product using Short read eliminator (SRE) kit (Pacbio, London, UK), followed by a second PCR according to the original protocol. We did purification and size selection (400–800 bp) with SPRIselect (Beckman Coulter, Brea, CA, USA). We confirmed successful library preparation using the Microchip Electrophoresis System for DNA/RNA Analysis MCE®-202 MultiNA with the DNA-2500 Reagent Kit (Shimadzu, Kyoto, Japan) following the manufacturer’s protocol. Approximately 12 pM of each library was sequenced on an Illumina MiSeq platform (Illumina, San Diego, CA, USA).
Raw reads were trimmed, and quality filtered (CROP: 150, HEADCROP: 20, SLIDING WINDOW: 4: 15, MINLEN: 130) with trimmomatic-0.38 software40.
De novo SNP calling was conducted with denovo_map.pl pipeline in Stacks 2.55 software41. The optimization parameters were set as follows: minimum depth of coverage = 3, number of mismatches allowed between stacks within individuals = 2. Population structure analysis was performed using the populations pipeline with following parameters; the number of populations = 1, the minimum percentage of individuals in a population = 0.1, minimum minor allele frequency = 0.05, and maximum observed heterozygosity = 0.5. We calculated observed heterozygosity as an output option of the populations pipeline. The SNP data was filtered using Tassel 5 software42, selecting SNPs shared by at least 60% of individuals and with a probability of 10% of being shared by these individuals. For each sample, we obtained 59,876 to 156,382 reads (average: 113,113), 3,194 to 6,693 loci (average: 5162) and 889 to 2031 (average: 2031) SNPs.
Phylogenetic and population structure analyses
We employed ModelTest-NG software43 to determine the optimal nucleotide substitution model for constructing a maximum likelihood (ML) phylogenetic tree. Phylogenies were inferred using RAxML-NG 0.9.0 software44, with 100 bootstrap replicates to evaluate reliability of the topology. The result was visualized using FigTree 1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/).
We analyzed population structure of T. formosana using STRUCTURE v.2.3.4 software45. The population model was configured to allow admixture and the correlation of allele frequencies between clusters. Ten independent simulations were conducted for each K (K = 1–15) with 50,000 burn-in steps followed by 100,000 Markov Chain Monte Carlo steps. The Optimal value of K was estimated using STRUCTUR EHARVESTER46.
Morphological and physiological traits
We evaluated chlorophyll content, cross-sectional length and electron transportation rate, as for traits related to photosynthetic capacity in this study. Chlorophyll content has been shown to be a reliable proxy for the maximum rate of carboxylation (Vcmax), which is a critical determinant of photosynthetic capacity47. Cross-sectional leaf length influences leaf robustness and is positively correlated with photosynthetic output and solar radiation per area48, making it a useful indicator of leaf structural durability and photosynthetic output49. ETR measures the consumption of light energy by electrons and a decrease in ETR results in an excess of light energy, causing photoinhibition and decreased photosynthetic efficiency37.
The geographical locations of T. formosana were classified into five categories: Okinawa Island (designated as A in Fig. 1), Iriomote Island (B), Northern Taiwan (C, D, E), Eastern Taiwan (F), and Middle Taiwan (G, H). These locations were used to evaluate photosynthesis-related traits, including chlorophyll content, cross-sectional length of leaves and electron transportation rate (ETR) in photosystem II. In addition chlorophyll content was indirectly inferred from SPAD values, which serve as an index of chlorophyll content50,51.
Cross-sectional leaf length and chlorophyll content
Healthy, fully expanded leaves of individual plants from each location grown under common light intensity (ca. 300 mol quanta m−2 s−1), temperature (20 °C), daylength (10 h) and nutrition conditions in the laboratory for a minimum of two and half months were used for analysis (Table S2).
The SPAD Chlorophyll Meter (SPAD-502 Plus, CONICA MINOLTA) was used to measure the SPAD value on healthy portions of leaves. Three to five measurements were taken per leaf, and the average was used as the SPAD value for each leaf. The cross-sectional length of the leaves was determined by measuring the thickness of the same portion of the leaves using a thickness gauge.
The statistical analysis of the relationship between SPAD value and cross-sectional leaf length was performed using R v. 4.2.1. A linear mixed model regression analysis was carried out using lme4 package, with SPAD as the objective variable and cross-sectional length (fixed effect) and population (variable effect) as explanatory variables. Subsequently, a multiple regression analysis was performed using the GLM function with SPAD as the objective variable and population and cross-sectional length as explanatory variables.
Electron transportation rate based on pulse amplitude modulated fluorometry method
Chlorophyll fluorescence analysis is a widely utilized technique for evaluating plant physiology as it provides a measure of the electron transport rate and overall photosynthesis through the proportion of light absorbed by chlorophyll52. We classified the same individuals at the same category as used in 5.2.2; Iriomote Island, Okinawa Island, Northern Taiwan, Eastern Taiwan and Middle Taiwan (Supplementary Table S2), and fully expanded healthy leaves from the upper part of the plants were selected for analysis. To make the electron accepter in reaction center chlorophyll of photosystem II an oxidized state, the leaves were pre-darkened by completely wrapping them in aluminum foil for at least 30 min. Chlorophyll fluorescence and electron transport rate were then measured using a PAM fluorometers (MINI-PAM-II, Walz).
Each sample was sequentially irradiated with photosynthetically effective radiation (PAR) of 0, 50, 90, 130, 180, 250, 380, 570, 840, 1250, 1640 mol quanta m−2 s−1, and the electron transfer rate (ETR)was calculated at each PAR. For the ETR values of the five categories, an analysis of variance was performed using ANOVA function of R for each PAR phase, and in the PAR phase where the results were significant, a multiple comparison test (correction for holm p-values) was then performed to determine which populations had significant differences between them.
Estimation of conservation difficulty by transcriptome analysis
RNA extraction and de novo assembly
We extracted RNA of 19 samples from 2 Japanese and 3 Taiwanese populations (Supplementary Table S3) using an Agilent Plant RNA Isolation Mini kit (Agilent Technologies, Santa Clara, CA, USA), in accordance with the manufacturer’s protocols. Total RNA was sequenced on an Illumina NovaSeq 6,000 sequencer at 100 nucleotide paired-end (PE) reads. Low-quality reads, defined as those with more than 10% of the bases having a quality score below 30, were filtered using the FASTQ quality filter in the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Five million PE reads were collected from each individual to minimize the influence of the difference in the number of reads among individuals, and they were used for de novo RNA-seq assembly by Trinity ver. 2.11.053. In cases where multiple splicing isoforms for transcripts were obtained, the longest one with the same accession among the different isoforms in the Trinity assembly output was adopted.
Identification of heterozygous SNVs and calculation of genetic diversity
To identify heterozygous single nucleotide variants (SNVs) in each individual, the five million PE reads for each individual were mapped to the reference constructed by Trinity, using BWA version 0.7.1354. SNVs were then identified using SAMtools version 1.355. The number of heterozygous synonymous SNVs with a minimum read depth of 10, which were deemed to be putative neutral variations indicative of genetic diversity, was calculated per kb of the longest transcript. The obtained values were compared among populations (i.e., Okinawa, Iriomote and Taiwan) using Kruskal–Wallis test (one-way ANOVA), and multiple comparisons were conducted as a post-hoc test, under Holm’s correction.
Estimation of deleterious amino acid variation
Using Protein Variant Effect Analyzer (PROVEAN56), we examined the non-synonymous SNVs that were homologous to plant amino acid sequences to judge whether each amino acid variation might affect protein function. By default, threshold of PROVEAN score to predict deleterious variation is equal to or below − 2.5, and neutral effect is above − 2.5. However, in the present research, the non-synonymous variations with PROVEAN scores above |2.5| were regarded as deleterious because we could not distinguish derived amino acid variants from the original ones at heterogenous loci. In this way we calculated the ratio of deleterious amino acid variations in heterozygous SNVs. The proportion of non-synonymous variants for each coding sequence and the proportion of nonsense SNVs (loss-of-function SNVs) to total non-synonymous SNVs were also calculated. The obtained values were compared among populations by Kruskal–Wallis test (one-way ANOVA), and as a post hoc test, multiple comparisons were conducted under Holm’s correction.
Data availability
Raw MIG-seq data are deposited at the DDBJ Sequencing Read Archive (DRA) with accession numbers DRA015916 (Submission), PRJDB15490 (BioProject), SAMD00588474-SAMD00588501 (BioSample), DRX437804-DRX437831 (Experiment) and DRR452721-DRR452748 (Run). RNA-seq read data have been deposited at the NCBI Sequence Read Archive and are publicly available under accession number PRJDB16049.
References
Nakanishi, H. Dispersal ecology of the maritime plants in the Ryukyu Islands. Japan. Ecol. Res. 3, 163–173 (1988).
Sugai, K. et al. Extremely low genetic diversity of the northern limit populations of Nypa fruticans (Arecaceae) on Iriomote Island Japan. Conserv. Genet. 17, 221–228 (2015).
Ota, H. Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their paleogeographical implications. Res. Popul. Ecol. 40, 189–204 (1998).
Kamata, H. & Kodama, K. Tectonics of an arc-arc junction: An example from Kyushu Island at the junction of the Southwest Japan Arc and the Ryukyu Arc. Tectonophysics 233, 69–81 (1994).
Gungor, A. et al. Structural characteristics of the northern Okinawa Trough and adjacent areas from regional seismic reflection data: Geologic and tectonic implications. Tectonophysics 522–523, 198–207 (2012).
Kimura, M. Paleogeography of the Ryukyu Islands. Tropics 10, 5–24 (2000).
Osozawa, S. et al. Vicariant speciation due to 1.55 Ma isolation of the Ryukyu islands, Japan, based on geological and GenBank data. Entomol. Sci. 16, 267–277 (2013).
Tominaga, A. et al. Relict distribution of Microhyla (Amphibia: Microhylidae) in the Ryukyu Archipelago: High diversity in East Asia maintained by insularization. Zool. Scr. 48, 440–453 (2019).
Chiang, T. Y. & Schaal, B. A. Phylogeography of plants in Taiwan and the Ryukyu Archipelago. Taxon 55, 31–41 (2006).
Ministry of Environment, Japan. Amami-Oshima Island, Tokunoshima Island, Northern part of Okinawa Island, Iriomote Island. https://kyushu.env.go.jp/okinawa/amami-okinawa/description/index-en.html (2019).
Caujapé-Castells, J. et al. Conservation of oceanic island floras: Present and future global challenges. Perspect. Plant Ecol. Evol. Syst. 12, 107–129 (2010).
Kono, Y., Peng, C.-I., Chao, C.-T. & Oginuma, K. Karyomorphological study of Taiwanese Tricyrtis (Liliaceae) and the taxonomic implication. Chromosom. Bot. 10, 61–66 (2015).
Peng, C. I., Tiang, C. L. & Hsu, T. W. Tricyrtis ravenii (Liliaceae), a new species from Taiwan. Bot. Stud. 48, 357–364 (2007).
Tsai, L. C. et al. Species identification using sequences of the trnL intron and the trnL-trnF IGS of chloroplast genome among popular plants in Taiwan. Forensic Sci. Int. 164, 193–200 (2006).
Ministry of Environment, Japan. Red List Japan https://www.env.go.jp/content/900515981.pdf (2020).
Hamabata, T. et al. Endangered island endemic plants have vulnerable genomes. Commun. Biol. 2, 1–10 (2019).
Wessel, P. & Smith, W. H. F. A global self-consistent, hierarchical, high-resolution shoreline database. J. Geophys. Res. 101, 8741–8743 (1996).
NOAA National Centers for Environmental Information. 2022: ETOPO 2022 15 Arc-Second Global Relief Model. NOAA National Centers for Environmental Information. https://doi.org/10.25921/fd45-gt74. Accessed [23 Oct 2023].
QGIS Development Team. QGIS Geographic Information System. QGIS Association (2020).
Liu, T. S. & Ying, S. S. Tricyrtis in Flora of Taiwan 1st ed. 78–82 (Epoch Publishing Company, 1978)
Ying, S. S. Trycyrtis in Flora of Taiwan 2nd ed. 66–69 (National Science Council of the Republic of China, 2003).
Hsu, T. C., Chung, S. W. Trycyrtis in Illustrated Flora of Taiwan 227–229 (Owl Publishing House 2016)
Nakamura, K. et al. Molecular phylogeography reveals an antitropical distribution and local diversification of Solenogyne (Asteraceae) in the Ryukyu Archipelago of Japan and Australia. Biol. J. Linn. Soc. 105, 197–217 (2012).
Kokubugata, G. et al. Antitropical distribution of Lobelia species (Campanulaceae) between the Ryukyu Archipelago of Japan and Oceania as indicated by molecular data. Aust. J. Bot. 60, 417–428 (2012).
Kokubugata, G. et al. Reappraisal of Tashiroea as a genus independent of Bredia (Melastomataceae) based on molecular data. Phytotaxa 392, 75–83 (2019).
Nakamura, K. et al. Phylogeny and biogeography of the Viola iwagawae-tashiroi species complex (Violaceae, section Plagiostigma) endemic to the Ryukyu Archipelago Japan. Plant Syst. Evol. 301, 337–351 (2015).
Kokubugata et al. Three independent migrations of Dodonaea viscosa (Sapindaceae) to Japan Archipelago revealed by molecular data. Bull. Natl. Mus. Nat. Sci., Ser. B. 45, 137–145 (2019).
Jump, A. S., Marchant, R. & Peñuelas, J. Environmental change and the option value of genetic diversity. Trends Plant Sci. 14, 51–58 (2009).
Wang, P. et al. Genomic consequences of long-term population decline in brown eared pheasant. Mol. Biol. Evol. 38, 263–273 (2021).
Xue, Y. et al. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348, 242–245 (2015).
Grossen, C., Guillaume, F., Keller, L. F. & Croll, D. Purging of highly deleterious mutations through severe bottlenecks in Alpine ibex. Nat. Commun. 11, 1001. https://doi.org/10.1038/s41467-020-14803-1 (2020).
Khan, A. et al. Genomic evidence for inbreeding depression and purging of deleterious genetic variation in Indian tigers. Proc. Natl. Acad. Sci. USA. 118, e2023018118. https://doi.org/10.1073/pnas.2023018118 (2021).
Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 246, 96–98 (1973).
Akashi, H., Osada, N. & Ohta, T. Weak selection and protein evolution. Genetics 192, 15–31 (2012).
Okinawa Prefecture. Threatened Wildlife in Okinawa (Fungi and Plants) 3rd ed. (Okinawa Prefecture, 2018).
Cramer, W. et al. Comparing global models of terrestrial net primary productivity (NPP): Overview and key results. Glob. Chang. Biol. 5, 1–15 (1999).
Murata, N., Allakhverdiev, S. I. & Nishiyama, Y. The mechanism of photoinhibition in vivo: Re-evaluation of the roles of catalase, α-tocopherol, non-photochemical quenching, and electron transport. Biochim. Biophys. Acta Bioenerg. 1817, 1127–1133 (2012).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4326 (1980).
Suyama, Y. & Matsuki, Y. MIG-seq: An effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 5, 1–12 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Catchen, J. et al. Stacks: An analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Darriba, Di. et al. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294 (2020).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 7, 574–578 (2007).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361.
Croft, H. et al. Leaf chlorophyll content as a proxy for leaf photosynthetic capacity. Glob. Chang. Biol. 23, 3513–3524 (2017).
Onoda, Y. et al. Global patterns of leaf mechanical properties. Ecol. Lett. 14, 301–312 (2011).
Niinemets, Ü. Global-scale climatic controls of leaf dry mass per area, density, and thickness in trees and shrubs. Ecology 82, 453–469 (2001).
Monje, O. A. & Bugbee, B. Inherent limitations of nondestructive chlorophyll meters: A comparison of two types of meters. HortScience 27, 69–71 (1992).
Markwell, J., Osterman, J. C. & Mitchell, J. L. Calibration of the Minolta SPAD-502 leaf chlorophyll meter. Photosynth. Res. 46, 467–472 (1995).
Maxwell, K. Growth and physiological performance of aerobic and lowland rice as affected by water stress at selected growth stages. Rice Sci. 25, 82–93 (2000).
Grabherr, M. G. et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid ubstitutions and indels. PLoS One 7, e46688. https://doi.org/10.1371/journal.pone.0046688 (2012).
Acknowledgements
This research was supported by the Environment Research and Technology Development Fund, the Ministry of Environment of Japan (4-1902 (JPMEERF20194002), 4-2003 (JPMEERF20204003) and 4MF-2202 (JPMEERF20224M02)), Grant-in-Aid for JSPS Fellows (18J01069) and University of the Ryukyus Research Project Promotion Grant (17SP01302). We would like to thank Haruka Kawakita and Kana Magota for their assistance in measuring of photosynthesis-related traits, and Nahoko Sashimura for her contribution to sample collection.
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.I. and K.T. Sample collection: Y.I., T.I., M.Y., A.A., A.N., K.C. Methodology: K.T., Y.I., M.S., C.E., Y.S., A.M., H.S., T.M. Supervision: Y.I. Writing: K.T., Y.I. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tsunenari, K., Ito, T., Yokota, M. et al. Double migration of the endangered Tricyrtis formosana (Liliaceae) in Japan. Sci Rep 14, 957 (2024). https://doi.org/10.1038/s41598-024-51431-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51431-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
