
Abstract
Pseudomonads are ubiquitous bacteria with importance in medicine, soil, agriculture, and biomanufacturing. We report a novel Pseudomonas putida phage, MiCath, which is the first known phage infecting P. putida S12, a strain increasingly used as a synthetic biology chassis. MiCath was isolated from garden soil under a tomato plant using P. putida S12 as a host and was also found to infect four other P. putida strains. MiCath has a ~ 61 kbp double-stranded DNA genome which encodes 97 predicted open reading frames (ORFs); functions could only be predicted for 48 ORFs using comparative genomics. Functions include structural phage proteins, other common phage proteins (e.g., terminase), a queuosine gene cassette, a cas4 exonuclease, and an endosialidase. Restriction digestion analysis suggests the queuosine gene cassette encodes a pathway capable of modification of guanine residues. When compared to other phage genomes, MiCath shares at most 74% nucleotide identity over 2% of the genome with any sequenced phage. Overall, MiCath is a novel phage with no close relatives, encoding many unique gene products.
Similar content being viewed by others

Introduction
Pseudomonas putida is a gram-negative bacteria that can be found in most soils and water and includes many members that can colonize plant roots and provide benefits to the root microbiomes1. Pseudomonas putida strains have been widely explored for use in biomanufacturing platforms, bioremediation, biocontrol, and as a factory for natural products2. Few bacteriophages have been isolated that infect P. putida3,4,5. Phages targeting P. putida are of interest not only for understanding viral ecology of Pseudomonads, but also potential biotechnology applications, such as manipulating P. putida populations in the rhizosphere6, degrading biofilms through phage-encoded depolymerase enzymes7 or potentially as vectors for delivery of large genetic cassettes into P. putida genomes.
Here we report the first phage isolated against P. putida S12, MiCath. MiCath has a myovirus morphology, is a virulent phage, and has a ~ 61 kbp dsDNA genome encoding a unique queuosine biosynthesis pathway, endosialidase, and a cas4 exonuclease. MiCath has a broad host range infecting five diverse strains with average nucleotide identity (ANI) ranging from 90.2 to 99%. Further, MiCath designates a new genus and species of phage taxonomy since the nearest phage relative only shares 2% of its genome with MiCath. Overall, MiCath represents a novel phage that could have application spaces in biomanufacturing, bioremediation, and biocontrol.
Results and Discussion
MiCath isolation, morphology, and characterization
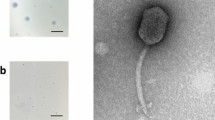
Bacteriophage MiCath was isolated from soil obtained from a home garden under a tomato plant using the enrichment plating method. MiCath produced clear, 1mm plaques on P. putida S12 after 16 h at 30 °C (Fig. 1a). A high titer lysate of MiCath was produced, grids were prepared and stained with 10% gadolinium acetate tetrahydrate and used for transmission electron microscopy (TEM) imaging. MiCath has an A1 myovirus morphology8 (Fig. 1b). The capsid measures: height 58.1 nm (± 1.5 nm), width 45.4 nm (± 2.8 nm), and the tail measures 123.5 nm (± 2.1 nm) (n = 5).

MiCath isolation and morphology. (a) MiCath 1 mm clear plaques on P. putida S12. (b) TEM image of MiCath revealing an A1 myovirus morphotype. Grids were stained with 10% gadolinium acetate tetrahydrate.
MiCath infection kinetics
We sought to explore infection kinetics typically tested for bacteriophages for MiCath. We explored burst size and lysis infection timing. The burst size for MiCath is 18 ± 5 (n = 3); this burst size is small compared to ~ 60 which is the phage average9. However, many of the phages included in the average are coliphages, which have different growth conditions than many environmental bacteria such as P. putida and different growth kinetics than phages of other bacterial genera10. MiCath begins lysis around 30 min, overt lysis is observed after 90 min, and at 240 min significant lysis of the culture is observed (Fig. 2). Despite its smaller burst size, it is competent and can quickly kill a culture within a few hours.

Lysis timing for MiCath. Lysis assays were completed in triplicate for MiCath using P. putida S12 as the host. MiCath begin lysis around 30 min with nearly complete lysis observed by 240 min. Error bars represent one standard deviation. N = 3.
MiCath host range
To determine the host range of MiCath, seven additional P. putida strains and P. aeruginosa PAO1 were obtained, tenfold dilutions of MiCath were spotted onto a lawn of each host tested, and efficiency of plating (EOP) was calculated. MiCath infects 5 strains in two different GTDB11 species, Pseudomonas_E hunanensis and P. putida, suggesting MiCath can infect multiple strains of P. putida, including those traditionally used in biomanufacturing applications (Table 1). Pseudomonas putida EM383 is a prophage-null mutant12 of P. putida KT2440. Notably, MiCath can infect both P. putida KT2440 and P. putida EM383, yet this occurs at different EOPs. There are at least two possible reasons MiCath infects the prophage-laden KT2440 strain more efficiently. There could be unknown defense mechanisms in EM383 that are either repressed by the prophages found in KT2440 or broken by prophage integration. Alternatively, prophage encoded proteins in KT2440 may enhance the lytic capabilities of MiCath. Further work would be needed to reveal more detail of interactions between the prophages in KT2440 and the incoming MiCath DNA.
MiCath genome characterization
Sequencing and de novo assembly revealed MiCath has a double-stranded DNA genome of 60,958 bp and has 97 predicted open reading frames (ORFs) (Fig. 3). The function of 48 ORFs was predicted by comparing the protein sequences against previously annotated phages using BLASTP13,14 and against known hidden Markov Models (HMMs) using the HHpred server15,16. There were 49 ORFs (50.5%) for which function could not be predicted using these annotation tools.

Genomic characterization of MiCath. MiCath has a 60,958 bp dsDNA genome that encodes 97 ORFs. The families of functional protein calls are colored coded by function as denoted in the figure.
MiCath does not encode an integrase gene and forms clear plaques suggesting it is a virulent phage not capable of integration. We observed ORFs with high percent amino acid identity to other traditional phage genes that encode tail, capsid, terminase, and lysis proteins. MiCath also encodes an entire cassette dedicated to queuosine biosynthesis, suggesting it is capable (in certain contexts dependent on expression mechanisms) to alter its DNA nucleotides, as seen in other phages, protecting its genome from host restriction systems17. MiCath also encodes an endosialidase, which may be responsible for the broad host range observed18. Lastly, MiCath has a predicted ORF with a high percent amino acid identity to cas4 exonucleases. This is not uncommon in phage genomes: previous work reports cas endo/exonucleases in many genomes19; however, the exact function of cas endonucleases in phage genomes are still unknown. All proteins and putative functions are listed in Supplemental Table 1.
MiCath phylogenetic analysis
MiCath has nearly no nucleotide sequence similarity to any previously sequenced phage. The closest relative is a Xanthomonas phage vB_Xar_IVIA-DoCa3 (Genbank accession number: ON911540) with 74.9% identity over 2% of the genome. There are three regions of identity found with other phages including parts of the queuosine biosynthesis pathway, the major capsid protein, and a hypothetical protein. Overall, MiCath only shares any nucleotide identity with 13 additional phages infecting bacteria in the genera: Pseudomonas, Pantoea, Escherichia, and Sphingomonas and three partial phage metagenome assemblies genomes from humans (Genbank Accession Numbers: BK020071.1, BK039317.1, and BK047545.1).
Since the full nucleotide sequence was not similar to any phages, we performed phylogenetic analysis on the major capsid protein (GenBank: WAX22437.1), which is identified in most phage genomes and has been used previously for phylogenetic analysis20,21. There were 62 additional phage proteins with high percent amino acid identity to the major capsid protein encoded by MiCath. We aligned these homologous proteins and created a phylogenetic tree using FastTreeDBL22. This analysis revealed that MiCath encodes a similar major capsid protein to other Pseudomonas phages but is the most distant member of the Pseudomonad clade (Fig. 4a; Supplemental Fig. 1). Pairwise average nucleotide analysis with the phages encoding a major capsid protein in the same clade as MiCath was computed using VIRIDIC23 (Fig. 4b) and shows MiCath shares little nucleotide relatedness with these phages, similar to the result from the entire phage landscape. The criteria from the International Committee on Taxonomy of Viruses states the genera cut off is 70% nucleotide identity and species is 95% nucleotide identity, suggesting MiCath denotes a new phage genus and species within the class Caudoviricetes and subfamily Queuovirinae24,25.

Phylogenetic analysis of MiCath. (a) Protein phylogenetic tree of clade containing MiCath and other phages major capsid proteins. FastTreeDBL22 was used to calculate the tree using the Le-Gascuel 2008 model44. Image is a portion of the full tree depicted in Supplementary Fig. 1. (b) Heatmap of pairwise intergenomic relatedness adapted from VIRIDIC23 output between phage genomes found in the same major capsid protein clade (Fig. 4a). Green indicates the same genome with 100% relatedness, genus clusters are in blues (70% relatedness) and species clusters are in light blue (95% relatedness). MiCath is bolded.
MiCath DNA resists restriction enzyme digestion
Many phages have been discovered whose genomes encode queosine biosynthesis pathway genes17,26,27,28. Recently, this gene cluster was shown in Enterobacteria phage 9G to modify up to 27% of the guanine nucleotides in its genome to a hyper modfied guanine, 2′-deoxyarcheosine (dG+)29. Additionally, it was shown that 9G and additional phages containing the altered dG+ nucleotides were resistant to restriction enzymes, suggesting an anti-defense mechanisms encoded by this gene cluster17.
We sought to detemine through restriction enzyme analysis if the queuosine biosynethesis gene cluster present in MiCath might be capable of incoporating dG+ or a similar modified based into DNA. We digested MiCath DNA with a panel of restriction enzymes previously used to explore queuosine biosynthesis pathways in phages17. We used an online tool, benchling30, to virtually digest the MiCath genome with each enzyme to confirm expected results, indicating seven of the eight selected enzymes should digest MiCath’s unmodified genome (Fig. 5a). We performed restriction digestions and found that SwaI doesn’t cut MiCath’s genome as predicted. However, EcoRI, BamHI, and EcoRV also do not cut MiCath’s genome suggesting protection of the MiCath genome from restriction by these enzymes (Fig. 5b). Restriction to EcoRI is consistent with the presence of dG+ or a related modification, as previously reported17,29. Further, partial digestion was observed for BstXI, while complete digestion is observed for HaeIII, NdeI, and RsaI (Fig. 5b). The resistance to these restriction enzymes, coupled with the complete functional gene set to produce dG+ (DpdA, folE, QueD, QueE, and QueC [MiCath genome coordinates 28869-33191]) observed in other phages17 suggest MiCath is likely modifings its genome with dG+ nucleotides to prevent restriction digestion by its host. As a control we also digested commercially available Lambda DNA (Fig. 5d) with the same panel of restrition enzymes and the pattern matches that generate by virtual digestion (Fig. 5c).

Restriction patterns for MiCath DNA. (a) MiCath DNA virtual restriction digests for enzymes used, prepared by Benchling online software. (b) 0.8% agarose gel after MiCath DNA was digested for 60-min with EcoRI, BamHI, BstXI, EcoRV, HaeIII, NdeI, SwaI, RsaI restriction enzymes. (c) Lambda DNA virtual restriction digest for enzymes used, prepared by Benchling online software. (d) 0.8% agarose gel after commercially available Lambda phage DNA was digested for 60-min with EcoRI, BamHI, BstXI, EcoRV, HaeIII, NdeI, SwaI, RsaI restriction enzymes. The ladder used on both agarose gels in (b) and (d) is Invitrogen 1 Kb DNA Plus, the highest molecular weight band on the ladder is 15 kb.
Conclusions
Here we report the discovery of the first phage isolated and sequenced for P. putida S12, MiCath. To date only 23 phages have been discovered for any P. putida strains3,4 indicating a gap in phage biology of this important species, and a potential need for phages suitable for use in various applications. MiCath is a virulent phage with a novel genome not similar to any previously sequenced phages. MiCath also encodes many novel genes within its genome that may allow for broad host range, alternative DNA base usage, and insights into phage-encoded cas endonucleases. Overall, MiCath represent an interesting new phage that may also be useful for bioremediation and biomanufacturing applications.
Methods
Bacterial strains
We used P. putida strains S12, DOT-T1E, F1 (kindly gifted by Grant Rybnicky), ATCC 12633 (purchased from ATCC), JUb85 (kindly provided by Samuel Buck), EM383 (kindly gifted by Huseyin Tas), p106 (kindly provided by Carey-Ann Burnham), and KT2440 (obtained from lab stocks). An overnight culture of each P. putida was prepared in Luria–Bertani (LB) broth and incubated at 30 °C overnight shaking at 250 rpm for all experiments.
Environmental sample collection and phage filtrate preparation
Soil was collected from a backyard garden (37.93132569198107, -121.7123907950711) under a tomato plant. 3–5 mL of soil and 10 mL of SM phage buffer (SMPB) (100 mM NaCl, 8 mM MgSO4·7H2O, 25 mM Tris-HCl [pH 7.4]) were mixed vigorously by inverting. The 15 mL tube was then allowed to settle for at least 20 min followed by filtration through a 0.22 µm filter.
Enrichment plating
The enrichment method was used to isolate phages from the soil sample. The soil filtrate was mixed 1:1 with LB broth and 20 uL of overnight P. putida S12, incubated overnight, shaking at 250 rpm at 30 °C. The overnight culture was centrifuged at 845×g for 3 min and the supernatant was filtered through a 0.22 µm filter. Enrichment filtrates were tested for evidence of phage using a traditional double agar overlay31. Briefly, 100 uL of the enrichment filtrate and 100 uL of P. putida S12 were incubated 10 min then 3 mL molten top agar was added, plated onto LB agar plates, and incubated at 30 °C overnight.
Phage purification and lysate creation
To isolate a single phage, an individual plaque was picked with a sterile pipette tip into SM phage buffer and plated using double agar overlays and incubated at 30 °C overnight. This process was repeated three times to ensure a pure phage population.
A lysate was created by flooding a webbed plate (near complete lysis) with 8 mL of SM phage buffer, incubating for 30 min at room temp and filtering through a 0.22 µm syringe filter.
Host range testing
To determine the host range of MiCath, the phage lysate was titered using a tenfold serial dilution in SMPB and spot titered on several P. putida bacterial strains. Bacterial lawns for each tested bacteria were prepared from overnight cultures using the double agar overlap technique. 3 uL of each phage dilution was spotted onto the bacterial lawn and allowed to sit for 15 min at room temperature followed by overnight incubation at 30 °C. After incubation, plaques were counted on the lowest countable dilution, the phage titer was calculated followed by calculation of efficiency of plating.
Transmission electron microscopy
Phages were imaged by transmission electron microscopy (TEM) on a Themis Z transmission electron microscope operated in scanning transmission electron microscopy (STEM) mode. Fresh phage lysates > 109 pfu/mL were applied to a carbon grid (Ted Pella, catalog no. 1813) and incubated at room temperature for 10 min and wicked off with Whatman filter paper. The grids were washed twice with 10uL of diH2O for 2 min wicking off in between. The grid was then stained with 10 uL of uranyl acetate alternative stain (Ted Pella, catalog no. 19485, 10% gadolinium acetate tetrahydrate) for 10 min and wicked dry. Grids were allowed to completely dry in a chemical fume hood for 1 h and stored until imaging.
TEM images were visualized and analyzed in ImageJ software32 using the TIA plugin. Five images were selected for measurement analysis. Three independent measurements of the width and height of each capsid and the tail from capsid to end of tail spike were measured. The scale bar was also measured independently three times, averaged, and corrected to 50 nm (if needed). Each capsid or tail average was corrected based on the scale bar correction. The capsid and tail measurements were averaged, and standard deviation was calculated for all five images overall.
Burst size analysis
Burst size analysis was completed as previously described33. Briefly, 500 uL of an overnight P. putida S12 was inoculated into 9.5 mL of LB broth and incubated shaking at 30 °C until the OD600 reached 0.6. 1 mL of MiCath (corresponding to 106 phages, MOI = 10) was added and incubated at 30 °C without shaking for 5 min (Culture 1). After 5 min 100 uL of culture 1 was transferred to a new flask with 10 mL of fresh LB broth (Culture 2). Culture 2 was incubated for 30 seconds and 1 mL was transferred to a microcentrifuge tube for analysis. Immediately 20 uL was titered to determine the non-adsorbed titer (titer 1). Then chloroform was added to the original microcentrifuge tube and supernatant was removed and titered (titer 2). Culture 2 was incubated at 30 °C for 12 min and 1 mL was removed and chloroform treated, and supernatant was removed to obtain the free phage titer (titer 3). Phage titers at each step were performed by serial dilution of the phage lysate and double agar overlays on P. putida S12. The titer was calculated as previously described, followed by burst size calculation shown below.
Lysis assay
Overnight P. putida S12 was diluted to OD600 = 0.1 and split into six culture flasks. Three cultures were infected with MiCath at MOI = 10, the remaining three were left uninfected. The flasks were incubated shaking at 30 °C for 4 h, measuring the OD600 every 30 min.
Genome sequencing and analysis
Genomic DNA was isolated from the phage lysates through DNA extraction using Promega wizard Kit (Promega, catalog no. 0000457646) following the manufacturer protocol. A library was prepared using the Nextera DNA prep Kit (Illumina, catalog no. FC-121-1031) following the manufacturers recommended protocol and sequenced on a NextSeq 500/550 using 150-bp cycles high output kit (Illumina, catalog no. 20024907). The sequencing run produced 3,855,168 reads with a Q30 of 96.50%.
Sequencing reads were processed with BBDuk34 to remove low quality reads using the following parameters: ktrim = r, k = 21, mink = 11, hdist = 1. After filtering there were 3,854,915 total reads with a Q30 of 96.51%. FastQC35 was used to evaluate the quality of reads before and after filtering with standard parameters. Filtered reads were assembled with SPAdes (v3.9.0)36. Any scaffold with less than 10% of the top scaffold coverage and smaller than 1000bp was removed from the assembly. The average coverage was 8667 × across the entire genome. Bowtie237 was used to align 95.74% (3,690,663 reads) of the raw filtered reads back to the final MiCath genome.
Coding sequences (CDS) were predicted by GeneMarkS38 and command-line multiPhATE v0.5 39 with Glimmer v3.0240, Prodigal v2.6.341, and Phanotate v0.13.042 gene callers turned on. The open reading frames (ORFs) predicted were functionally annotated by searching amino acid sequences against BLASTP13,14 standard databases and HHpred (using PDB_mmCIF70 and Pfam-A_v35 databases)15,16.
Phylogenetic analysis
Phylogenetic analysis was completed using proteins with high percent amino acid identity to the major capsid protein. The protein sequences that had any blastP high percent amino acid identity with the predicted major capsid protein of MiCath were collected from NCBI. The sequences were aligned with MUSCLE v3.8.3143 using standard options. A phylogenetic tree was made with FastTreeDBL v2.1.1022 using the Le-Gascuel 2008 model44 using standard options. Final trees were visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
We obtained the following complete and partial genome sequences from NCBI to compute intergenomic relatedness for the phages with major capsid proteins in the same clade as MiCath: Pseudomonas phage AF (NC_019923.1), Pseudomonas phage Iggy (NC_070970.1), Pseudomonas phage JG012 (KX898399.1), Pseudomonas phage JG054 (NC_072498.1), Pseudomonas phage NP1 (KX129925.1), Pseudomonas phage PaMx25 (NC_041953.1), Pseudomonas phage pPA-3099-2aT.3 (OP784576.1), Pseudomonas phage Quinobequin-P09 (NC_072501.1), Pseudomonas phage vB_PaeS_PAJD-1 (NC_072500.1), Escherichia phage vB_Eco_SLUR75 (LR025197.1), Escherichia phage vB_EcoS_SA80RD (OL960575.1), Escherichia phage SeppeHuegi (MZ501104.1), Pantoea phage vB_PagS_Vid5 (NC_042120.1), Sphingomonas phage vB_StuS_MMDA13 (NC_072503.1), Xanthomonas phage vB_Xar_IVIA-DoCa3 (NC_072499.1), Caudoviricetes sp. isolate ctFso4 (BK039317.1), Caudoviricetes sp. isolate ctgt12 (BK047545.1), Siphoviridae sp. ctJ7 × 27 (BK032517.1), Caudoviricetes sp. isolate ctX1H6 (BK020071.1). We used VIRIDIC23 web version with standard options to calculate whole genome nucleotide identity using a single multiFASTA of these phage genome sequences.
Restriction digestion
To isolate high quality MiCath DNA, a MiCath phage lysates was incubated overnight at 4 °C with a 2:1 lysate to PEG (1M NaCl, 20% PEG8000) ratio. The overnight PEG precipitation was centrifuged at 10,000×g for 30 min at 4 °C and the supernatant was discarded. The resulting pellet was resuspended in 1 mL of dH20. DNA was extracted using the Norgen Biotek Corp Kit (Norgen, catalog no. 46800) following manufacturer’s protocol including DNase I step followed by DNase inactivation and eluted in diH20.
MiCath and Lambda (NEB, catalog no. N3011S) DNAs were digested with EcoRI-HF (NEB, catalog no. R3101S), BamHI-HF (NEB, catalog no. R3136S), BstXI (NEB, catalog no. R0113S), EcoRV-HF (NEB, catalog no. R3195S), HaeIII (NEB, catalog no. R0108T), NdeI (NEB, catalog no. R0111S), SwaI (NEB, catalog no. R0604S), RsaI (NEB, catalog no. R0167S). Restriction enzymes digestions were prepared by combining 10 × NEB reaction buffer, 500 ng of MiCath DNA, sterile H2O, and 1 uL of restriction enzyme. The tube was briefly flicked to mixed and centrifuged before incubating at 37 °C for 1 h. The reaction was briefly spun and loaded and run on a 0.8% agarose gel.
Virtual DNA digests and virtual gels were created using Benchling39. The FASTA sequence for MiCath was input into the software and enzymes were selected and a virtual gel was created.
Data availability
MiCath whole genome sequence has been deposited at NCI Genbank under the accession number OP882271.
References
Planchamp, C., Glauser, G. & Mauch-Mani, B. Root inoculation with Pseudomonas putida KT2440 induces transcriptional and metabolic changes and systemic resistance in maize plants. Front. Plant. Sci. 5, 719. https://doi.org/10.3389/fpls.2014.00719 (2014).
Loeschcke, A. & Thies, S. Pseudomonas putida-a versatile host for the production of natural products. Appl. Microbiol. Biotechnol. 99, 6197–6214. https://doi.org/10.1007/s00253-015-6745-4 (2015).
Cook, R. et al. INfrastructure for a PHAge REference database: Identification of large-scale biases in the current collection of cultured phage genomes. Phage (New Rochelle) 2, 214–223. https://doi.org/10.1089/phage.2021.0007 (2021).
Magill, D. J. et al. Pf16 and phiPMW: Expanding the realm of Pseudomonas putida bacteriophages. PLoS ONE 12, e0184307. https://doi.org/10.1371/journal.pone.0184307 (2017).
Valero-Rello, A. Diversity, specificity and molecular evolution of the lytic arsenal of Pseudomonas phages: In silico perspective. Environ. Microbiol. 21, 4136–4150. https://doi.org/10.1111/1462-2920.14767 (2019).
Campbell, J. I., Albrechtsen, M. & Sørensen, J. Large Pseudomonas phages isolated from barley rhizosphere. FEMS Microbiol. Ecol. 18, 63–74 (1995).
Shaburova, O. et al. Search for destruction factors of bacterial biofilms: Comparison of phage properties in a group of Pseudomonas putida bacteriophages and specificity of their halo-formation products. Russ. J. Genet. 45, 161–170 (2009).
Ackermann, H. W. Frequency of morphological phage descriptions in the year 2000. Brief review. Arch. Virol. 146, 843–857. https://doi.org/10.1007/s007050170120 (2001).
Ellis, E. L. & Delbrück, M. The growth of bacteriophage. J. Gen. Physiol. 22, 365–384. https://doi.org/10.1085/jgp.22.3.365 (1939).
Kirzner, S., Barak, E. & Lindell, D. Variability in progeny production and virulence of cyanophages determined at the single-cell level. Environ. Microbiol. Rep. 8, 605–613. https://doi.org/10.1111/1758-2229.12409 (2016).
Parks, D. H. et al. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785-d794. https://doi.org/10.1093/nar/gkab776 (2022).
Martínez-García, E., Nikel, P. I., Aparicio, T. & de Lorenzo, V. Pseudomonas 2.0: Genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb. Cell Fact. 13, 159. https://doi.org/10.1186/s12934-014-0159-3 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
Gish, W. & States, D. J. Identification of protein coding regions by database similarity search. Nat. Genet. 3, 266–272. https://doi.org/10.1038/ng0393-266 (1993).
Gabler, F. et al. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinform. 72, e108. https://doi.org/10.1002/cpbi.108 (2020).
Zimmermann, L. et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243. https://doi.org/10.1016/j.jmb.2017.12.007 (2018).
Hutinet, G. et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 10, 5442. https://doi.org/10.1038/s41467-019-13384-y (2019).
Jakobsson, E., Schwarzer, D., Jokilammi, A. & Finne, J. Endosialidases: Versatile tools for the study of polysialic acid. Top. Curr. Chem. 367, 29–73. https://doi.org/10.1007/128_2012_349 (2015).
Hudaiberdiev, S. et al. Phylogenomics of Cas4 family nucleases. BMC Evol. Biol. 17, 232. https://doi.org/10.1186/s12862-017-1081-1 (2017).
Ongenae, V. et al. Genome sequence and characterization of Streptomyces phage Pablito, representing a new species within the genus Janusvirus. Sci. Rep. 12, 17785. https://doi.org/10.1038/s41598-022-22784-y (2022).
Al-Shayeb, B. et al. Clades of huge phages from across Earth’s ecosystems. Nature 578, 425–431. https://doi.org/10.1038/s41586-020-2007-4 (2020).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
Moraru, C., Varsani, A. & Kropinski, A. M. VIRIDIC-A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses https://doi.org/10.3390/v12111268 (2020).
Turner, D. et al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch. Virol. 168, 74. https://doi.org/10.1007/s00705-022-05694-2 (2023).
Simmonds, P. et al. Four principles to establish a universal virus taxonomy. PLoS Biol. 21, e3001922. https://doi.org/10.1371/journal.pbio.3001922 (2023).
Carstens, A. B., Kot, W. & Hansen, L. H. Complete genome sequences of four novel Escherichia coli bacteriophages belonging to new phage groups. Genome Announc. https://doi.org/10.1128/genomeA.00741-15 (2015).
Kot, W. et al. Complete genome sequence of Streptococcus pneumoniae virulent phage MS1. Genome Announc. 5, 23. https://doi.org/10.1128/genomeA.00333-17 (2017).
Sazinas, P. et al. Comparative genomics of bacteriophage of the genus Seuratvirus. Genome Biol. Evol. 10, 72–76. https://doi.org/10.1093/gbe/evx275 (2018).
Tsai, R., Corrêa, I. R., Xu, M. Y. & Xu, S. Y. Restriction and modification of deoxyarchaeosine (dG(+))-containing phage 9 g DNA. Sci. Rep. 7, 8348. https://doi.org/10.1038/s41598-017-08864-4 (2017).
Software, B. B. https://benchling.com (2023).
Adams, M. H. Bacteriophages (Interscience Publishers Inc., 1956).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. https://doi.org/10.1038/nmeth.2089 (2012).
Hernandez-Morales, A. C. et al. Genomic and biochemical characterization of Acinetobacter podophage petty reveals a novel lysis mechanism and tail-associated depolymerase activity. J. Virol. 92, 23. https://doi.org/10.1128/jvi.01064-17 (2018).
Bushnell, B. BBTools software packag. e (2014).
FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Besemer, J. & Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451-454. https://doi.org/10.1093/nar/gki487 (2005).
Ecale Zhou, C. L. et al. multiPhATE: Bioinformatics pipeline for functional annotation of phage isolates. Bioinformatics (Oxford, England) 35, 4402–4404. https://doi.org/10.1093/bioinformatics/btz258 (2019).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics (Oxford, England) 23, 673–679. https://doi.org/10.1093/bioinformatics/btm009 (2007).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119. https://doi.org/10.1186/1471-2105-11-119 (2010).
McNair, K., Zhou, C., Dinsdale, E. A., Souza, B. & Edwards, R. A. PHANOTATE: a novel approach to gene identification in phage genomes. Bioinformatics (Oxford, England) 35, 4537–4542. https://doi.org/10.1093/bioinformatics/btz265 (2019).
Edgar, R. C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 113. https://doi.org/10.1186/1471-2105-5-113 (2004).
Le, S. Q. & Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. https://doi.org/10.1093/molbev/msn067 (2008).
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under the Secure Biosystems Design Initiative. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. This article has been authored by an employee of National Technology & Engineering Solutions of Sandia, LLC under Contract No. DE-NA0003525 with the U.S. Department of Energy (DOE). The employee owns all right, title and interest in and to the article and is solely responsible for its contents. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this article or allow others to do so, for United States Government purposes. The DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan https://www.energy.gov/downloads/doe-public-access-plan. We thank Doug Medlin for TEM imaging of MiCath. We thank Anupama Sinha for guidance during sequencing. We thank Grant Rybnicky (Northwestern University) for providing P. putida strains S12, DOT-T1E, and F1, Huseyin Tas (Harvard University) for P. putida EM383, Samuel Buck (Baylor College of Medicine) for P. putida JUb85, and Carey-Ann Burnham (Washington University School of Medicine in St. Louis) for P. putida p106. We thank Kelly Williams for help in obtaining strains JUB85 and p106.
Author information
Authors and Affiliations
Contributions
The experimental plan was designed by C.M.M., J.D.J., and J.S.S.. Experiments were performed by J.D.J. and C.M.M. The phage genome was analyzed by C.M.M and J.D.J. The manuscript was written, and figures prepared by C.M.M. and J.D.J. The manuscript was edited and reviewed by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jaryenneh, J., Schoeniger, J.S. & Mageeney, C.M. Genome sequence and characterization of a novel Pseudomonas putida phage, MiCath. Sci Rep 13, 21834 (2023). https://doi.org/10.1038/s41598-023-48634-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48634-z
This article is cited by
-
Genome analysis and classification of Xanthomonas bacteriophage AhaSv, a new member of the genus Salvovirus
Archives of Virology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
