
Abstract
Retinoic acid (RA) activates RA receptors (RAR), resulting in RA response element (RARE)-dependent gene expression in renal collecting duct (CD). Emerging evidence supports a protective role for this activity in acute kidney injury (AKI) and chronic kidney disease (CKD). Herein, we examined this activity in RARE-LacZ transgenic mice and by RARE-Luciferase reporter assays in CD cells, and investigated how this activity responds to neurotransmitters and mediators of kidney injury. In RARE-LacZ mice, Adriamycin-induced heavy albuminuria was associated with reduced RA/RAR activity in CD cells. In cultured CD cells, RA/RAR activity was repressed by acetylcholine, albumin, aldosterone, angiotensin II, high glucose, cisplatin and lipopolysaccharide, but was induced by aristolochic acid I, calcitonin gene-related peptide, endothelin-1, gentamicin, norepinephrine and vasopressin. Compared with age-matched normal human CD cells, CD-derived renal cystic epithelial cells from patients with autosomal recessive polycystic kidney disease (ARPKD) had significantly lower RA/RAR activity. Synthetic RAR agonist RA-568 was more potent than RA in rescuing RA/RAR activity repressed by albumin, high glucose, angiotensin II, aldosterone, cisplatin and lipopolysaccharide. Hence, RA/RAR in CD cells is a convergence point of regulation by neurotransmitters and mediators of kidney injury, and may be a novel therapeutic target.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI) is an abrupt renal insult followed by sudden decline of kidney functions1,2; chronic kidney disease (CKD) is a long-term condition where the kidneys do not work effectively, resulting in chronic loss of renal functions3,4. AKI occurs in 10–15% hospitalised patients and approximately 50% patients in intensive care, and is associated with high mortality2; affecting 8–16% of the world’s population3,4, CKD was the 12th most common cause of mortality and caused 4.6% of deaths globally in 20175. If this rising prevalence of CKD continues, it is predicted that CKD will become a top-5 cause of mortality worldwide by 20406.
AKI and CKD are not distinct disease entities but rather are closely interconnected syndromes7. Despite some shared aetiologies, risk factors and mechanisms, the pathophysiology of AKI and CKD can be quite different. Although AKI may be caused by systemic, vascular, glomerular and tubular diseases, the core pathophysiology of AKI is often predominated by tubulointerstitial injury2. On the other hand, although a wide range of aetiologies may all cause CKD, the latter is most often a glomerular disease4. However, regardless of the primary aetiologies of CKD, tubulointerstitial injury plays a crucial role in CKD progression to end-stage kidney disease8. Thus, it is critical to investigate the mechanism of tubulointerstitial injury, which is as yet poorly understood. What we do know, however, is that tubulointerstitial injury in AKI and CKD is associated with a wide range of overlapping aetiologies, risk factors and mediators. For example, infection, nephrotoxins and renal ischaemia–reperfusion contribute to tubulointerstitial injury in both AKI and CKD; while persistent albuminuria, diabetes and hypertension primarily cause CKD progression, although they may also contribute to increased risk of AKI1,2,3,4,7.
Endeavouring to better understand the mechanisms of tubulointerstitial injury, we have recently put forward a novel hypothesis that the canonical vitamin A signalling mediated by retinoic acids (RA) and retinoic acid receptors (RAR) in the renal collecting duct (CD) is a critical protector against renal injury and that mediators of kidney injury might contribute to tubulointerstitial injury by differentially regulating the RA/RAR activity in CD cells9. We have chosen to study the CD because emerging evidence suggests that the CD might play important roles in defence against infection10 and tubulointerstitial injury11, and might be involved in gentamicin- and ischaemia–reperfusion-induced AKI12,13 and obstructive CKD14. Furthermore, the CD meets many criteria for a key defence role in the kidney: it is in the right location to play a principal defence role—the CD is the only tubular structure that spans much of the kidney, ideally placed to protect the tubulointerstitial compartment; and it is equipped with defending cells and molecules: mesenchymal stem cells, antifibrotic miRNAs and the RA/RAR signalling9.
The activity of RA, especially all-trans RA (atRA), is mainly mediated through binding and activating heterodimers of RAR and retinoid X receptors (RXRs), thereby modulating gene transcription15,16. RARs and RXRs each have three isotypes (α, β and γ) with overlapping functions. Target genes of atRA are often characterised by the presence of one or more retinoic acid response elements (RAREs) in their gene regulatory regions, which serve as anchorage points for the RAR-RXR heterodimers17,18. The RAR-RXR heterodimers can be readily activated by RAR agonists, principally atRA, but cannot be activated by RXR agonists alone19. Thus, for convenience, the atRA-dependent RAR-RXR transcriptional activity is also widely referred to as the RA/RAR activity.
RARs and atRA are indispensable for embryonic kidney development, and even mild gestational vitamin A deficiency leads to a deficit in nephron number20. Using young and adult RARE-LacZ mice as a model to report endogenous RA/RAR activity, we have previously shown that the endogenous renal RA/RAR activity is physiologically confined to the CD21. Moreover, in embryonic kidneys after E11, the RA/RAR activity is confined to precursors of the CD known as ureteric bud (UB)22, so the UB/CD cell lineage appears to be the main site of RA/RAR activation in healthy kidneys.
To catalogue RA/RAR-dependent genes in CD cells in vitro, we first looked at mIMCD-3 cells, a well-characterised CD cell line derived from the mouse inner medulla23,24, which has been widely used in studying ion channel signalling25,26, signalling during osmotic and hypertonic stress27, urea signalling28 and branching morphogenesis29 of CD cells. We found that, similar to the CD, mIMCD-3 cells had significant endogenous RA/RAR activity and this activity was required for expression of a panel of RA/RAR-dependent genes, many of which are implicated in protection against bacterial infection, inflammation and fibrosis (Ppbp, Lcn2, Bmp7)23. Further supporting a renoprotective role for RA/RAR, vitamin A deficiency has been associated with increased susceptibility to pyelonephritis, urolithiasis, kidney inflammation and fibrosis19,30,31,32,33. On the other hand, vitamin A and RA treatment have well-documented therapeutic effects in infectious and non-infectious kidney diseases19,30,34,35,36,37,38 and the endogenous RA/RAR signalling in the kidney has been reported to be protective in ischaemia–reperfusion models of AKI in mice39, although the exact change and role for the RA/RAR in the CD remain un-answered.
To further examine our hypothesis about the role for the RA/RAR in the CD in tubulointerstitial injury in AKI and CKD, this study was designed to address whether and how neurotransmitters and mediators of kidney injury affect RA/RAR activity in CD cells. RA/RAR activity in CD cells was examined in RARE-LacZ mice40 and was quantified by RARE-Luciferase reporter assays in CD-derived cell cultures exposed to neurotransmitters and mediators of kidney injury, with or without atRA or RA-568, a selective agonist of RARα41, the most abundant RAR isotype in the kidney (Supplementary Figs. 1 and 2). Our results show that the RA/RAR activity in CD cells is a shared point of regulation by neurotransmitters and mediators of kidney injury and may be a potential target for clinical intervention by interacting drugs such as RA-568.
Results
RA/RAR activity in CD cells was repressed by albumin
Persistent albuminuria is a leading risk factor for CKD progression and reducing albuminuria has been associated with renoprotection in CKD3,4,42. We have previously reported that renal RA/RAR activity is physiologically confined to CD cells in RARE-LacZ mice21. We further found that, in Adriamycin-induced nephropathy, RA/RAR activity is pathologically switched on in glomerular progenitor cells to drive podocyte regeneration; however, when albuminuria becomes heavy, albumin reabsorbed by glomerular progenitor cells sequesters intracellular RA and dose-dependently represses RA/RAR activity, thus blocking podocyte regeneration40. Recognising that CD cells are known to re-absorb albumin43,44 and the latter is known to specifically bind atRA45, we examined whether albuminuria may lead to repressed RA/RAR activity in the CD. We found that, in RARE-LacZ mice, Adriamycin treatment induced heterogenous phenotypes in terms of severity of albuminuria. In those manifesting trace albuminuria, there was an insignificant trend towards increase of RA/RAR activity in the CD; in contrast, those manifesting heavy albuminuria were associated with significantly repressed RA/RAR activity in CD cells (Fig. 1a,b). We propose that RA/RAR in the CD may be repressed by albuminuria, but oppositely regulated by other mediators of injury and repair, leading to biphasic changes. To determine the exact relation between albumin and endogenous RA/RAR activity in CD cells and CD-derived cells, we examined the effects of albumin on this activity, with the following RA/RAR-repressing controls: (i) 4-(diethylamino)-benzaldehyde (DEAB), an inhibitor of retinaldehyde dehydrogenases (Raldhs), which inhibits endogenous atRA biosynthesis21,46; (ii) 4-[2-[5,6-Dihydro-5,5-dimethyl-8-(4-methylphenyl)-2-naphthalenyl]ethynyl]-benzoic acid (AGN-193109), a pan-RAR antagonist23,47. As we reported previously, endogenous RA/RAR activity was defined as RARE dual luciferase reporter activity repressed by both 25 μM DEAB and 1 μM AGN19310923 (Fig. 2a). In the mIMCD-3 mouse inner medullary CD cell line23 (Fig. 2b) and the M-1 mouse cortical CD cell line48 (Fig. 2c), CD-derived Hoxb7 B2 mesenchymal cells (MSCs)49 (Fig. 2d), as well as a human cortical CD cell line50 (Fig. 2e), clear endogenous RA/RAR activity repressed by both DEAB and AGN193109 was detected. In all these CD-derived cell cultures, 10 mg/ml albumin significantly repressed RA/RAR activity. In mIMCD-3 cells, we found that 10 mg/ml albumin, but not IgG and transferrin, repressed RA/RAR activity, indicating albumin specifically repressed RA/RAR activity (Fig. 3a). The effect of albumin was also dose-dependent at clinically relevant concentrations ranging from 0.3 mg/ml (equivalent to microalbuminuria) to 10 mg/ml (equivalent to heavy albuminuria) (Fig. 3b). To further confirm the specificity of albumin in repressing RA/RAR in CD cells and to shed light into the underlying mechanism, we found that overexpression of wild-type ALB, but not the gene truncated of the sequence encoding its RA-binding domain40, significantly repressed the endogenous RA/RAR activity (Fig. 3c). These data indicate that albumin excreted into urine could be reabsorbed by CD cells, leading to sequestration of RA and repression of RA/RAR activity in the cells, similar to what we reported previously in glomerular progenitor cells40.

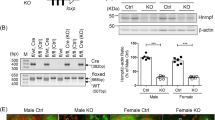
In Adriamycin nephropathy of RARE-LacZ mice, heavy albuminuria was associated with repressed RA/RAR activity. (a) RA/RAR activity in CD cells as a function of RARE-LacZ expression was reported as the ratio between number of β-gal-expressing CD cells and total number of DBA positive CD cells. Heavy albuminuria was associated with a 2.6-fold reduction of RA/RAR activity in CD cells; (b) Representative images of kidney tissues stained with an anti–β-galactosidase antibody (green), DBA-lectin (red, CD cells) and Topro-3 (blue, nuclei). In healthy control RARE-LacZ mice, RA/RAR activity (green signal) was physiologically confined to CD cells. In Adriamycin nephropathy mice, heavy albuminuria was associated with reduced RA/RAR activity in CD cells. *, ***: p < 0.05, p < 0.001, respectively.

Mouse CD cells, CD-derived MSCs and human CD cells had endogenous RA/RAR activity, which was repressed by AGN193109, DEAB and albumin. (a) RA/RAR activity was defined as RARE-LacZ or RARE-Luciferase reporter activity. In cells, all-trans retinol (atRol), i.e., vitamin A, is oxidised reversibly to all-trans retinaldehyde (atRal), catalysed by the enzymes retinol dehydrogenases (Rdhs) or alcohol dehydrogenases (Adhs). atRal is then oxidised irreversibly to all-trans RA (atRA), catalysed by retinaldehyde dehydrogenases (Raldhs). atRA translocates into cell nucleus to bind and activate RARs, modulating gene transcription. AGN193109 (purple) was used to antagonise RAR. DEAB (orange) was used to inhibit Raldh-mediated atRA synthesis. In mIMCD-3 cells (b), M-1 cells (c) Hoxb7 B2 mouse CD-derived MSCs (d) and HCD cells (e), 1 μM AGN193109, 25 μM DEAB or 10 mg/ml albumin treatment for 24 h resulted in significant repression of RA/RAR activity. ***: p < 0.001 vs Vehicle control group.

Albumin specifically and dose-dependently repressed RA/RAR activity. In mIMCD-3 cells, 24 h exposure to 10 mg/ml albumin, but not IgG or transferrin, repressed RA/RAR activity (a); Albumin repression of RA/RAR activity was dose-dependent (b); overexpression of ALB, but not ALB truncated of its RA-binding domain, repressed RA/RAR activity (c). *, **, ***: p < 0.05–0.001 vs Control, Vehicle-treated group. Tet-On, transgene induced by tetracycline; Vehicle-off, transgene silent in the presence of vehicle only.
Although no RA/RAR activity was detected in any non-CD cells in the healthy kidney in RARE-LacZ mice21,22, we do not rule out the possibility that non-CD renal cells have functional RA/RAR machinery that is physiologically below the detection limit of X-gal assay for reporting RA/RAR-dependent expression of LacZ gene21. Using in vitro cellular models and quantitative RARE dual luciferase reporter assays, we found that cultured mouse and rat proximal tubular cell lines (mPTEC and NRK-52E) and rat renal fibroblasts (NRK-49F) had endogenous RA/RAR activity, equivalent to 1/3–1/4 of the activity of mIMCD-3 cells. As in mIMCD-3 cells, the RA/RAR activity in these non-CD cell lines was repressed by DEAB, AGN193109, as well as albumin (Supplementary Figs. 3 & 4). Thus, although this project focuses on CD cells, some of our findings may also have implications for other relevant renal cell types. For example, physiologically, proximal tubules and the CD reabsorb 71% and 3% of albumin filtrated from glomeruli, respectively51. This means that albumin may exert stronger physiological repression on RA/RAR activity in proximal tubules, likely contributing to lack of visible physiological RA/RAR activity in these tubules in RARE-LacZ mice21.
RA/RAR activity in CD cells was repressed by high glucose
Diabetes characterised by high blood and urinary glucose levels is a leading cause of CKD52. To examine whether high glucose may regulate RA/RAR activity, we compared the effects of 24 h exposure to 25 mM glucose (high glucose) and 5.5 mM glucose (normal medium control) on RA/RAR activity in mIMCD-3 cells, with 25 mM mannitol as an osmotic control for high-glucose treatment. We found that high glucose, but not equal molar concentrations of mannitol, significantly repressed RA/RAR activity (Fig. 4a).

Neurotransmitters and mediators of kidney injury differentially regulated RA/RAR activity in mIMCD-3 cells. High-glucose treatment (but not 25 mM mannitol) for 24 h significantly reduced RA/RAR activity (a). In addition, 24 h exposure to angiotensin II (b), aldosterone (c), acetylcholine (h), cisplatin (k) and LPS (l) dose-dependently repressed, while endothelin-1 (d), vasopressin (e), norepinephrine (f), CRGP (g), aristolochic acid I (i) and gentamicin (j) dose-dependently induced RA/RAR activity. *, **, ***: p < 0.05, p < 0.01, p < 0.001 vs 5.5 mM D-Glucose or vehicle control groups.
RA/RAR activity in CD cells was differentially regulated by mediators of hypertension and neurotransmitters
Hypertension is another major risk factor for CKD53. To examine whether mediators of hypertension may regulate RA/RAR activity in CD cells, we compared the effects of four well-established mediators of hypertension on RA/RAR activity in mIMCD-3 cells. We found that angiotensin II (0.01–1 μM) and aldosterone (0.01–1 μM) dose-dependently repressed, while endothelin-1 (1–50 nM) and vasopressin (100 nM) dose-dependently induced RA/RAR activity (Fig. 4b–e). More recently, renal nerves have been shown to contribute to hypertension54,55, AKI and its transition to CKD56,57,58,59. We then examined whether neurotransmitters norepinephrine, calcitonin gene-related peptide (CGRP) and acetylcholine may also regulate RA/RAR activity in CD cells. As shown in Fig. 4f,g, norepinephrine (10 nM) and CGRP (1–100 nM) activated, while acetylcholine (0.1–10 µM) repressed RA/RAR activity in CD cells (Fig. 4h). These results suggest that mediators of hypertension and neurotransmitters may have opposite effects on the RA/RAR activity in CD cells, apart from their systemic effects on blood pressure and organ perfusion. In view that angiotensin II and aldosterone are well-established mediators of CKD progression3,4,53, while vasopressin and norepinephrine are vasopressors often used to boost organ perfusion and prevent AKI in shock patients60, it is possible that differential regulation of RA/RAR activity in the CD may be a novel determinant in their differing roles in AKI and CKD, in addition to their systemic effects on blood pressure.
Of note, although we tested 0.1–10 nM norepinephrine and 1–100 nM vasopressin, only at the highest concentrations they induced RA/RAR activity. 10 nM norepinephrine is 4–20-fold higher than the plasma concentrations of healthy subjects61 and comparable with plasma concentrations of patients with phaeochromocytoma61 or those receiving norepinephrine as a vasopressor62. Vasopressin, however, primarily targets the renal CD. Thus, although its circulating concentrations in healthy subjects and patients are at picomolar levels63,64, it might be enriched at the CD and need higher local concentrations to regulate CD functions. In keeping with this notion, in mIMCD-3 cells, it took 10 nM vasopressin to activate the expression of the H+-K+-ATPase α2-subunit (HKα2) gene cells65 and 100 nM vasopressin was required to activate RA/RAR activity. Thus, both pathophysiological and pharmacological implications of our findings deserve further investigation.
RA/RAR activity in CD cells was differentially regulated by nephrotoxic agents and other inducers of kidney injury
Depending on dosing regimens and duration of exposure, nephrotoxic agents aristolochic acid I and gentamicin, the chemotherapeutic drug cisplatin and endotoxin lipopolysaccharide (LPS) can induce AKI and/or CKD1,66,67,68,69. Although they are all broadly considered nephrotoxic, they are known to cause renal damage by different mechanisms66. For example, instead of direct cytotoxicity, cisplatin and LPS both induce AKI in a Toll-like receptor-4 (TLR4)-dependent manner67,68. As shown in Fig. 4i,j, aristolochic acid I (20–40 μM) and gentamicin (0.25–1 mg/ml) dose-dependently induced, while cisplatin (10–40 μM) and LPS (1–10 μg/ml) dose-dependently repressed RA/RAR activity in mIMCD-3 cells (Fig. 4k,l). Given the emerging evidence of a critical protective role for the RA/RAR activity in CD cells9, our results suggest that, LPS and cisplatin may disarm RA/RAR-mediated defensive mechanisms, leading to more progressive renal kidney injury, while activation of RA/RAR activity by aristolochic acid I and gentamicin may mount defence and mitigate injury.
RA/RAR activity in autosomal recessive polycystic kidney disease (ARPKD) renal cystic cells was significantly lower than in age-matched normal CD cells
In ARPKD, all renal cysts derive from the CD70. To examine whether RA/RAR activity in CD-derived ARPKD renal cystic cells is abnormal, we compared the endogenous RA/RAR activity in primary cultures of embryonic (foetal 24 weeks), perinatal (newborn) and paediatric (18 months old) ARPKD renal cystic cells and their age-matched normal human CD cells. Endogenous RA/RAR activity was calculated in two forms: (i) Endogenous RA/RAR activity repressed by 1 μM RAR antagonist AGN193109 (EndoActAGN) = RARE-Luc activity (Vehicle) − RARE-Luc activity (AGN193109); (ii) Endogenous RA/RAR activity repressed by 25 μM of the RA biosynthesis inhibitor DEAB (EndoActDEAB) = RARE-Luc activity (Vehicle) − RARE-Luc activity (DEAB). As shown in Fig. 5a–c, CD-derived ARPKD renal cystic cells consistently had significantly lower EndoActAGN and EndoActDEAB than age-matched normal CD cells. These data suggest that RA/RAR activity in ARPKD renal cystic cells may be reduced and the biological importance of this change in ARPKD deserves further investigation.

Endogenous RA/RAR activity in ARPKD renal cystic cells was significantly lower than age-matched normal CD cells. EndoActAGN: Endogenous RA/RAR activity repressible by AGN193109; EndoActDEAB: Endogenous RA/RAR activity repressible by DEAB. (a) embryonic; (b) perinatal; and (c) paediatric. **, ***: p < 0.01, 0.001, respectively.
RA-568 was more potent than atRA in rescuing RA/RAR activity repressed by albumin, aldosterone, angiotensin II, cisplatin, high glucose and LPS
Given the hypothesised protective role for the RA/RAR activity in the CD9, to prevent CKD and its progression, it may be desirable to prevent RA/RAR repression by albumin, aldosterone, angiotensin II, cisplatin, high glucose and LPS in these cells. To this end, we propose that the RARα agonist RA-56841 may be more suited than natural RAR pan-agonist atRA, because (i) it is structurally distinct from atRA (Supplementary Fig. 3), RA-568 may not be sequestered by albumin as much as atRA40,45; (ii) RA-568 activates RARα, but not RARβ and RARγ, nor RXR41, thus it will selectively activate RARα, the most abundant RAR isotype in CD cells71 and the kidney (Supplementary Fig. 1), while minimising RARγ-mediated irritating skin syndrome19 and RXR-mediated pro-fibrotic effects of atRA metabolites72. Indeed, in both mouse (mIMCD-3) and human (HCD) CD cell lines, we found that RA-568 was at least 100-fold more efficacious than atRA in restoring RA/RAR activity repressed by 10 mg/ml albumin. Though not totally unexpected, it was striking to find that atRA at concentrations as high as 100 ng/ml had no effect on the RA/RAR activity repressed by 10 mg/ml albumin. This was likely because high-concentration albumin sequesters both endogenous and exogenous atRA. In contrast, in both mIMCD-3 mouse CD cells and HCD human CD cells, RARα agonist RA-568 efficaciously restored RA/RAR activity repressed by 10 mg/ml albumin. At 1–10 nM, it restored RA/RAR loss of activity; and at 100 nM the RA/RAR activity was significantly higher than the vehicle control group (Fig. 6a,b). Further, as shown in Supplementary Fig. 5, RA/RAR activity induced by 100 nM RA-568 in both mIMCD-3 and HCD cells was not repressed by increasing concentrations of albumin (0.3–10 mg/ml), in contrast to 100 nM atRA, which neither boosted RA/RAR activity nor prevented RA/RAR activity from being repressed by albumin in a dose-dependent manner (1–10 mg/ml). These data suggest that selectively activating RARα by RA-568 is likely a better approach than pharmaceutical atRA to restoring and boosting RA/RAR activity in CD cells.

RA-568 was more potent than atRA in preventing RA/RAR repression by mediators of kidney injury. RA-568 (1–100 nM), but not atRA (1–100 nM) dose-dependently prevented RA/RAR repression by 24 h exposure to 10 mg/ml albumin (a,b), 50 μM cisplatin (c), 10 μg/ ml LPS (c), 1 μM angiotensin II (d), 1 μM aldosterone (e) and 25 mM glucose (f) in mIMCD-3 (a,c–f) and HCD cells (b). *, **, ***: p < 0.05, p < 0.01, p < 0.001, respectively.
We next examined the effects of atRA and RA-568 in preventing RA/RAR repression by LPS and cisplatin in mIMCD-3 cells. As shown in Fig. 6c, RA/RAR activity repression by either LPS (10 μg/ml) or cisplatin (50 μM) was not affected by 1–100 nM atRA but was significantly and dose-dependently prevented by RA-568, at 10–100 nM and 1–100 nM, respectively. Similarly, we compared effects of atRA and RA-568 in preventing RA/RAR repression by angiotensin II, aldosterone and high glucose. As shown in Fig. 6d–f, RA/RAR repression by either angiotensin II, aldosterone or high glucose in mIMCD-3 cells was prevented by 100 nM RA-568; 10 nM RA-568 also effectively prevented RA/RAR activity repression by aldosterone and high glucose. In contrast, 1–100 nM atRA did not prevent angiotensin II, aldosterone and high glucose from repressing RA/RAR activity. The ineffectiveness of atRA in all these settings was in stark contrast to the excellent efficacy of 1–100 nM atRA in preventing RA/RAR repression by 25 μM DEAB, an inhibitor of RA synthesis (Figs. 2a, 6d)21.
Discussion
AKI and CKD are multifactorial disorders2,3,4,7. This study highlights the RA/RAR activity in CD cells as a convergence point of regulation by the multiple factors involved in AKI and CKD. Intriguingly, it responds in an opposite fashion to mediators of kidney injury (induced by gentamicin and aristolochic acid I; repressed by cisplatin and LPS), mediators of hypertension (induced by endothelin-1, vasopressin and norepinephrine; repressed by aldosterone and angiotensin II) and neurotransmitters (induced by CGRP and norepinephrine; repressed by acetylcholine). Further investigations into the mechanisms and biological consequences of RA/RAR activity changes induced by these factors may facilitate deeper understanding of the heterogeneity and multifactorial nature of AKI and CKD and guide refined therapies.
In particular, this report establishes that RA/RAR activity responds differentially to a wide range of neurotransmitters and mediators of kidney injury and thus is unlikely a bystander in AKI and CKD. This rationalise the next step of studies to test the exact roles for the RA/RAR in the CD in different AKI and CKD models, e.g. by selectively up- and down-regulating RA/RAR activity and its downstream genes in the CD cell lineage using Hoxb7 or AQP2 promoters and then examining whether this changes the outcomes of AKI and CKD models in mice9.
The main limitation of this work is that evidence we have presented so far is mainly from in vitro models. Although our in vitro and in vivo data both support the capacity of heavy albuminuria to repress RA/RAR activity in CD cells (Fig. 1, 2, 3), we are acutely aware that regulation of the RA/RAR pathway in the CD must be more complex in vivo than in cultured CD cells, and eventually, in vitro, in vivo and clinical studies must be analysed together to fully understand the regulation of the RA/RAR pathway in the CD. For example, high glucose has been reported to repress RAR expression in mIMCD-3 cells71. Thus, reduced RAR expression may, at least in part, contribute to high-glucose repression of RA/RAR activity in mIMCD-3 cells. In diabetic mouse kidneys, atRA biosynthesis and atRA-dependent gene expression are compromised73, at least in part, due to o-GlcNAcylation of proteins crucial for retinoid binding, metabolism and signalling74. Furthermore, a biphasic regulation of renal RA/RAR signalling has been suggested by transcriptomic analysis of renal biopsy tissues of early- and late-stage diabetic nephropathy, respectively75.
Similar to our findings in mIMCD-3 cells, LPS has been previously reported to repress RA/RAR signalling in hepatic stellate cells76 and gentamicin has been reported to induce RA/RAR activity in renal tubules in zebra fish39. Given the potential importance of RA/RAR signalling in the CD9 and the potential differences in vitro and in vivo, it is desirable to clarify how renal RA/RAR activity is regulated in different types of AKI and CKD in RARE-reporter animals21,22,39,40 and in related clinical settings. To know exactly how this pathway is regulated will pave the way for further mechanistic studies, which will guide the development of intervening strategies, if indicated.
Taking ARPKD, a disease of the focal adhesion complex and the primary cilia due to mutations of fibrocystin (encoded by PKHD1)70, for example, we found that ARPKD renal cystic cells had significantly lower endogenous RA/RAR activity than age-matched control CD cells. Although we have three age-matched pairs leading to exactly the same findings, larger sample size may be needed to further confirm our findings. To examine a possible causal relation, it may be necessary to examine whether genetic mutations of PKHD1 in normal CD cells repress RA/RAR signalling and, if so, its role in ARPKD pathogenesis. Based on RA/RAR-dependent gene profiling in CD cells21, we hypothesise that decreased RA/RAR activity in CD cells may reduce expression of focal adhesion complex components tensin (Tns1) and α2-integrin (Itga2), and the cilium-specific transcription factor Foxj1 (Foxj1), impair signalling through focal adhesion complexes and cilia, leading to disease progression. If this is true, restoring the RA/RAR activity in the CD cell lineage may curb progression of this devastating disease and becomes a cost-effective strategy to improve ARPKD prognosis.
The inefficacy of atRA in preventing RA/RAR repression by albumin, high glucose, cisplatin, LPS, aldosterone and angiotensin II was an important finding. Since in parallel studies atRA was always highly efficacious in preventing RA/RAR repression by the atRA biosynthesis inhibitor DEAB, RA/RAR repression by the aforementioned mediators of kidney injury may not be simply due to lack of the bioactive ligand atRA. Thus, further mechanistic studies are clearly needed to guide development of both retinoid- and non-retinoid-based rescuing strategies. The good news is that, in contrast to the poor efficacy of atRA, the synthetic RARα agonist RA-568 has shown excellent effect in preventing RA/RAR repression. As this compound has an established safety profile in preclinical studies in mice and rats41, it could be an ideal drug lead for AKI and/or CKD, by boosting or restoring RA/RAR activity in kidneys, particularly in the CD. Thus, our in vitro data justify further translational studies comparing the efficacy and safety of RA-568 and atRA as potential renal therapeutics.
Despite the focus on CD cells of the present study, we acknowledge the possibility that RA/RAR activity in other renal cell types could be activated in pathological conditions. If so, how renal RA/RAR activity pathologically re-distributes in different AKI and CKD, whether such pathologically activated signalling in other renal cells, such as proximal tubular cells and infiltrating cells in ischaemia–reperfusion AKI39 and renal progenitor cell in Adriamycin-induced nephropathy40, has the same roles as the RA/RAR in the CD, and how all these RA/RAR signalling interacts are important questions to answer. Eventually, all such knowledge should need be integrated to fully understand the pathological roles for the RA/RAR signalling in AKI and CKD and to devise intervention strategies.
In conclusion, we have found that RA/RAR activity in cultured CD cells responds to a wide range of neurotransmitters and mediators of kidney injury, either being induced or repressed. When repressed, the activity is poorly rescued by exogenous atRA, but is effectively reversed by RARα agonist RA-568. Further studies revolving around this newly found convergent point of regulation in AKI and CKD may help us better understand these interconnected syndromes and lead to novel therapeutics.
Methods
Studies on animal tissues
Immunofluorescence confocal microscopy
No new animal study was performed in this study. Instead, we performed confocal microscopy (LSM510 META, Carl Zeiss, Jena, Germany) on 10 μm frozen sections of renal tissues of RARE-LacZ mice (CD1 background, 6- to 10-week-old, female) collected in our previously published project, including nine with trace albuminuria at dip-stick tests (approximately 0.1–0.2 mg/ml albumin or albumin:creatinine ratio < 0.3) and six with high levels of albuminuria, with albumin:creatinine ratios of > 7 (17.7 ± 2.99), induced by two retro-orbital injections of 14 mg/kg Adriamycin in phosphate-buffered saline (PBS), as well as five healthy controls treated by two successive injections of PBS40. Animal experiments were approved by the University of Florence Animal Studies Review Board, performed in accordance with institutional, regional, and state guidelines, and adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Dolichos biflorus agglutinin (DBA)-lectin (Vector Laboratories, Peterborough, UK) was used to stain CD cells. Chicken anti–β-galactosidase polyclonal antibody (Abcam, Cambridge, UK) and AlexaFluor 488-labelled donkey anti-chicken antibody (Jackson ImmunoResearch, West Grove, PA) were used to stain LacZ-expressing cells and Topro-3 (Molecular Probes, Invitrogen, Carlsbad, CA) was used for nuclear staining. Sections were also stained with secondary antibodies only to serve as negative control, as we previously reported40,77. To quantify the number of LacZ-expressing cells in the CD, β-gal- and DBA-lectin-positive cells were counted in at least 4 renal cortex sections of each mouse and percentage of dual positive cells was calculated. Five fields were counted on each section.
Cell biology studies
Reagents
Acetylcholine, AGN193109, aldosterone, aristolochic acid I, cisplatin, CGRP, DEAB, dexamethasone, D-glucose, endothelin-1, gentamicin, human albumin, human angiotensin II, human IgG, human transferrin, LPS, mannitol, norepinephrine, tetracycline, atRA and vasopressin were purchased from Sigma-Aldrich Company Ltd, Gillingham, UK. RA-568 was synthesised by Sygnature Discovery, Nottingham, UK. All reagents dissolved in ethanol and/or dimethylsulphoxide (DMSO) were first diluted to 1000 × stock solution and then diluted 1000 × with culture medium to the working concentrations. Control groups were treated with 0.1% ethanol and/or DMSO.
Cell cultures
mIMCD-3 and M-1 cells (LGC Standards, Middlesex, UK) and mPTEC cells (kind gift by Dr Mark Dockrell, South West Thames Institute for Renal Research, Surrey, UK) originally derived from the inner medullary CD, cortical CD and the S3 segment of the proximal tubule of a Brinster transgenic mouse [Tg(SV40E)Bri7], respectively24,48,78. They were routinely grown in DMEM-F12 medium containing 100 μg/ml penicillin and 100 μg/ml streptomycin (thereafter known as antibiotics) (PAA Laboratories Ltd, Somerset, UK), supplemented with 5% foetal calf serum (FCS; Life Technologies Ltd, Paisley, UK). Both mIMCD-3 and M-1 cells were 100% positive for aquaporin-2 (AQP2), a CD principal cell marker, and mPTEC cells were 100% positive for proximal tubular cell marker aquaporin-1 (AQP1). NRK-52E and NRK-49F cells (LGC Standards) are epithelial and fibroblast-like cell clones from kidneys of an adult non-inbred Osborne-Mendel rat79. They were routinely grown in 5% FCS DMEM with antibiotics (PAA). NRK-49F cells stained positive for vimentin and negative for cytokeratin and factor VIII. RNAs extracted from M-1, mIMCD-3 and mPTEC were selectively amplified using mouse specific primers, while RNA extracted from NRK-49F cells was selectively amplified using rat specific primers only. HCD cells are a well-characterised human cortical CD cell line50 provided by Dr Remi Piedagnel (INSERM, Paris, France). They were cultured in 2% FCS DMEM-F12 with antibiotics and insulin-transferrin-selenium supplement (ITS) (PAA). Mouse CD-derived MSCs (Hoxb7 B2) were provided by Dr Joan Li (University of Queensland, Queensland, Australia) and fully characterised as reported before49. They were cultured in 20% FCS α-MEM medium with antibiotics (PAA). Primary human CD cells and their age-matched embryonic (24 weeks), newborn and paediatric (18-month old) ARPKD renal cystic cells were fully characterised by Professor Patricia Wilson80,81,82 and were obtained from the Polycystic Kidney Disease Charity Biobank. Both foetal and paediatric ARPKD renal CD cells and their age-matched normal CD cells were routinely grown in 3% FCS 50% Click’s medium: 50% RPMI 1640 media (Gibco, Life Technologies) supplemented with antibiotics and 5 µg/ml human transferrin, 5 × 10–8 M dexamethasone, 2 mM GlutaMax (Gibco). Cells were plated in 6-well plates coated with 4.1 mg/ml collagen type-I (Corning, NY) and left to attach overnight before further studies. All cell cultures were maintained at 37 °C and 5% CO2.
Transient transfection and RARE dual luciferase assays
Three tetracycline-inducible gene expression plasmids were procured from VectorBuilder, Santa Clara, CA for transient transfection studies: (i) ALB-EGFP, a plasmid with DNA inserts of enhanced green fluorescence protein (EGFP) and the wild-type ALB gene (Accession NM_000477.5); (ii) mutant ALB-EGFP, a plasmid with DNA inserts of EGFP and mutated ALB genes, encoding human albumin deleted of amino acids 139–189, including 11 of 12 amino acids critical for binding atRA45; and (iii) a control vector, with the EGFP DNA insert only. The maps of these plasmids are shown in Supplementary Fig. 6. Lipofectamine LTX, Plus reagents and Opti-MEM I Reduced Serum Medium (Gibco) were used in transient transfection of plasmid DNAs according to the manufacturer’s instructions.
For RARE dual luciferase assay, pGL3-RARE-luciferase (https://www.addgene.org/13458/)83 and PRLSV40 (https://www.addgene.org/vector-database/3949/) plasmids (Promega, Southampton, UK) were used to co-transfect cells at a ratio of 5:1 to report RARE-dependent expression of Firefly luciferase and constitutive expression of Renilla luciferase. Cells were lysed with Reporter Lysis Buffer (Promega) and luminescence signal was detected with Luciferase Assay System and Beta-Glo Assay System (Promega). “Background signal” was determined as the average readout from wells treated with transfection medium only without any plasmid DNA and this was subtracted from readouts of other wells. The reporter assay results were expressed as relative unit (RLU, ratios of readouts of the two reporters). Alternatively, RLU of treatment group was normalised to vehicle control group and expressed as fold-change. Because pGL3-RARE-Luc and the control PRLSV40 plasmid both have the SV40 minimal promoter, the RLU (ratios of readouts of the two reporters) always comprises two parts, one dependent on RA/RAR and RARE (RA/RAR activity) and the rest being “baseline signal” mediated by the minimal promoter in the pGL3-RARE-Luc plasmid. The expected effects of DEAB and AGN193109 on the reporter assay are as follows: (i) DEAB represses RA/RAR activity induced by endogenously synthesised RA but does not repress the effects of the trace amount of RA in 1% FCS in the culture medium and any exogenous RAR agonists, if applicable, nor does it repress any RA-independent transcription activity of RAR mediated by the AF-2 domain of the nuclear receptor84; (ii) AGN193109 is not only a potent pan-RAR antagonist (antagonising effects of RAR agonists and inhibiting RA/RAR-mediated AF1-mediated gene expression) but also a RAR inverse agonist (actively recruiting corepressors to RAR and thus repressing RAR agonist-independent AF2-mediated transcriptional activity of RAR)84. At the concentrations used in this study, both DEAB and AGN193109 almost completely switched off RA/RAR-dependent Dhrs3, Sprr1a and Ppbp mRNA expression, but AGN193109 was consistently more potent in pressing these gene expression (− 99%, − 95% and − 93%) than DEAB (− 96%, − 85% and − 86%)23. Thus, DEAB incomplete repression of RAR activation by endogenous RA, DEAB inability to repress effects of RA in 1% FCS cell culture medium and AGN193109 incomplete repression of RA/RAR activity may not play any major role in the RLU values unrepressed by DEAB and AGN193109 in the RARE dual luciferase results in this project, although some minor roles cannot be completely ruled out. Nonetheless, this study particularly focuses on endogenous RA/RAR activity, which is defined as net RLU values repressed by both DEAB and AGN193109.
Experimental protocols
For dual luciferase reporter assays, mIMCD-3, HCD, M-1, Hoxb7 B2 MSCs, mPTEC, NRK-49F and NRK-52E cells were seeded into 24-well plates at 4 × 104 cells/well in 500 μl of the corresponding complete medium with FCS but without antibiotics. After culturing for 16 h, cells were co-transfected with pGL3-RARE-luciferase and PRLSV40 plasmids and cultured for another 6 h. Medium was then changed to 1% FCS fresh medium. After an overnight incubation, medium was changed to 1% FCS fresh medium containing 1 μM AGN193109, 25 μM DEAB, 10 mg/ml albumin and cultured for 24 h. mIMCD-3 cells were also treated for 24 h with albumin ranging from 0.3 to 10 mg/ml, 10 mg/ml IgG, 10 mg/ml transferrin, angiotensin II (0.01–1 μM), aldosterone (0.01–1 μM), 5.5 mM and 25 mM D-glucose, 25 mM mannitol, LPS (1–10 μg/ml), cisplatin (10–40 μM), vasopressin (1–100 nM), CGRP (1–100 nM), endothelin-1 (1–50 nM), gentamicin (0.25–1 mg/ml), aristolochic acid I (10–40 μM), norepinephrine (0.1–10 nM) and acetylcholine (0.1–10 μM).
In some studies, RARE dual luciferase reporter assays were conducted in mIMCD-3 and HCD cells cultured in 96-well plates. The cells (5 × 103/well) were seeded in 200 μl of culture medium without antibiotics. Following medium changes and transient transfection same as above, effects of albumin (0.3–10 mg/ml), with and without atRA (100 nM) or RA-568 (100 nM), and of albumin (10 mg/ml), DEAB (25 μM), high glucose (25 mM), angiotensin II (1 μM), aldosterone (1 μM), LPS (10 μg/ml) or cisplatin (50 μM), with or without 1–100 nM atRA or RA-568 for 24 h, on RA/RAR activity was analysed.
For ALB overexpression studies, mIMCD-3 cells were seeded into a 24-well plate at 4 × 104 cells/well in 500 μl 5% FCS-DMEM-F12 without antibiotics. After culturing for 16 h, cells were co-transfected with tetracycline-inducible ALB-EGFP, mutant ALB-EGFP or the EGFP-only control plasmid, together with pGL3-RARE-luciferase and PRLSV40 plasmids for 6 h. Medium was then changed to 1% FCS DMEM-F12. After an overnight incubation, medium was changed to 1% FCS DMEM-F12 supplemented with 2 μg/ml tetracycline for 24 h to switch on transgenes.
For reporter assays involving primary human CD cells and age-matched ARPKD renal cystic cells, cells were seeded into 96-well plates, 3.5 × 104/well, in 500 μl 3% FCS 50% Click’s medium: 50% RPMI 1640 without antibiotics. After culturing to about 70% confluence, cells were co-transfected with pGL3-RARE-luciferase and PRLSV40 plasmids and cultured for another 6 h. Medium was then changed to fresh 1% FCS medium. After an overnight incubation, medium was changed to 1% FCS medium with 1 μM AGN193109, 25 μM DEAB, 10 mg/ml albumin or vehicle and cultured for 24 h.
Data analysis
Immunofluorescence confocal microscopy data are expressed as percentage of LacZ-expressing DBA-positive cells over total DBA-positive cells per renal cortex section. All cell biology experiments were performed in quadruplicate wells for each group, with reproducible results in at least two independent experiments. Results are presented as mean ± SEM. For comparison of two and more than two groups, t-tests (non-paired, two-tail; with Welsh’s correction if any data of comparison failed Shapiro–Wilk normality tests) and one-way ANOVA analysis (including Tukey’s multiple comparisons) were conducted, respectively. For comparison of fold-changes, data were log-transformed before statistical analysis. Statistical significance was determined as p < 0.05. Analyses were performed using GraphPad Prism, Version 8.0 (GraphPad Software Inc., CA).
References
Bunel, V., Qu, F., Duez, P. & Xu, Q. Herbal medicines for acute kidney injury: Evidence, gaps and frontiers. World J. Trad. Chin. Med. 1, 47–66 (2015).
Ronco, C., Bellomo, R. & Kellum, J. A. Acute kidney injury. Lancet 394, 1949–1964 (2019).
Jha, V. et al. Chronic kidney disease: global dimension and perspectives. Lancet 382, 260–272 (2013).
Webster, A. C., Nagler, E. V., Morton, R. L. & Masson, P. Chronic kidney disease. Lancet 389, 1238–1252 (2017).
GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020).
Foreman, K. J. et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 392, 2052–2090 (2018).
Chawla, L. S., Eggers, P. W., Star, R. A. & Kimmel, P. L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 371, 58–66 (2014).
Schnaper, H. W. The tubulointerstitial pathophysiology of progressive kidney disease. Adv. Chronic Kidney Dis. 24, 107–116 (2017).
Xu, Q. The renal collecting duct rises to the defence. Nephron 143, 148–152 (2019).
Chassin, C. et al. Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J. Immunol. 177, 4773–4784 (2006).
Huang, M. et al. Integrin-linked kinase deficiency in collecting duct principal cell promotes necroptosis of principal cell and contributes to kidney inflammation and fibrosis. J. Am. Soc. Nephrol. 30, 2073–2090 (2019).
Huang, H. et al. Gentamicin-induced acute kidney injury in an animal model involves programmed necrosis of the collecting duct. J. Am. Soc. Nephrol. https://doi.org/10.1681/ASN.2019020204 (2020).
Miyazaki, T. et al. Cell-specific image-guided transcriptomics identifies complex injuries caused by ischemic acute kidney injury in mice. Commun. Biol. 2, 326 (2019).
Fujiu, K., Manabe, I. & Nagai, R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J. Clin. Invest. 121, 3425–3441 (2011).
Napoli, J. L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta. 1821, 152–167 (2012).
Rochette-Egly, C. & Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 7, e005 (2009).
Balmer, J. E. & Blomhoff, R. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J. Steroid. Biochem. Mol. Biol. 96, 347–354 (2005).
Lalevee, S. et al. Genome-wide in silico identification of new conserved and functional retinoic acid receptors response elements (directs repeats separated by 5 bp). J. Biol. Chem. 286, 33322–33334 (2011).
Xu, Q. et al. Retinoids in nephrology: Promises and pitfalls. Kidney Int. 66, 2119–2131 (2004).
Lelievre-Pegorier, M. et al. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int. 54, 1455–1462 (1998).
Wong, Y. F. et al. Endogenous retinoic acid activity in principal cells and intercalated cells of mouse collecting duct system. PLoS ONE 6, e16770 (2011).
Rosselot, C. et al. Non-cell-autonomous retinoid signaling is crucial for renal development. Development 137, 283–292 (2010).
Wong, Y. F. et al. Retinoic acid receptor-dependent, cell autonomous, endogenous retinoic acid signaling and its target genes in mouse collecting duct cells. PLoS ONE 7, e45725 (2012).
Rauchman, M. I., Nigam, S. K., Delpire, E. & Gullans, S. R. An osmotically tolerant inner medullary collecting duct cell line from an SV40 transgenic mouse. Am. J. Physiol. 265, F416–F424 (1993).
Goel, M. & Schilling, W. P. Role of TRPC3 channels in ATP-induced Ca2+ signaling in principal cells of the inner medullary collecting duct. Am. J. Physiol. Renal. Physiol. 299, F225–F233 (2010).
Sproul, A. et al. N-methyl-D-aspartate receptor subunit NR3a expression and function in principal cells of the collecting duct. Am. J. Physiol. Renal. Physiol. 301, F44–F54 (2011).
Cai, Q. et al. Pax2 expression occurs in renal medullary epithelial cells in vivo and in cell culture, is osmoregulated, and promotes osmotic tolerance. Proc. Natl. Acad. Sci. USA 102, 503–508 (2005).
Cohen, D. M., Gullans, S. R. & Chin, W. W. Urea signaling in cultured murine inner medullary collecting duct (mIMCD3) cells involves protein kinase C, inositol 1,4,5-trisphosphate (IP3), and a putative receptor tyrosine kinase. J. Clin. Invest. 97, 1884–1889 (1996).
Lee, D. C., Chan, K. W. & Chan, S. Y. RET receptor tyrosine kinase isoforms in kidney function and disease. Oncogene 21, 5582–5592 (2002).
Kavukcu, S. et al. The role of vitamin A in preventing renal scarring secondary to pyelonephritis. BJU Int. 83, 1055–1059 (1999).
Kancha, R. K. & Anasuya, A. Contribution of vitamin A deficiency to calculogenic risk factors of urine: Studies in children. Biochem. Med. Metab. Biol. 47, 1–9 (1992).
van Leersum, E. C. Vitamin A deficiency and urolithiasis. Br. Med. J. 2, 873–874 (1927).
Zile, M., DeLuca, H. F. & Ahrens, H. Vitamin A deficiency and urinary calcium excretion in rats. J. Nutr. 102, 1255–1258 (1972).
Zhang, G. Q., Chen, J. L. & Zhao, Y. The effect of vitamin A on renal damage following acute pyelonephritis in children: A meta-analysis of randomized controlled trials. Pediatr. Nephrol. 31, 373–379 (2016).
Neveus, T. Can postpyelonephritic renal scarring be prevented?. Pediatr. Nephrol. 28, 187–190 (2013).
Sobouti, B., Hooman, N. & Movahed, M. The effect of vitamin E or vitamin A on the prevention of renal scarring in children with acute pyelonephritis. Pediatr. Nephrol. 28, 277–283 (2013).
Dalirani, R. et al. Role of vitamin A in preventing renal scarring after acute pyelonephritis. Iran J. Kidney. Dis. 5, 320–323 (2011).
Ayazi, P., Moshiri, S. A., Mahyar, A. & Moradi, M. The effect of vitamin A on renal damage following acute pyelonephritis in children. Eur. J. Pediatr. 170, 347–350 (2011).
Chiba, T. et al. Retinoic acid signaling coordinates macrophage-dependent injury and repair after AKI. J. Am. Soc. Nephrol. 27, 495–508 (2016).
Peired, A. et al. Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J. Am. Soc. Nephrol. 24, 1756–1768 (2013).
Clarke, E. et al. Design and synthesis of a potent, highly selective, orally bioavailable, retinoic acid receptor alpha agonist. Bioorg. Med. Chem. 26, 798–814 (2018).
Heerspink, H. J. L. et al. Drug-induced reduction in albuminuria is associated with subsequent renoprotection: A meta-analysis. J. Am. Soc. Nephrol. 26, 2055–2064 (2015).
Erkan, E. Proteinuria: It is time to look beyond the proximal tubule. Am. J. Physiol. Renal Physiol. 305, F1107–F1108 (2013).
Dizin, E. et al. Albuminuria induces a proinflammatory and profibrotic response in cortical collecting ducts via the 24p3 receptor. Am. J. Physiol. Renal Physiol. 305, F1053–F1063 (2013).
Belatik, A., Hotchandani, S., Bariyanga, J. & Tajmir-Riahi, H. A. Binding sites of retinol and retinoic acid with serum albumin. Eur. J. Med. Chem. 48, 114–123 (2012).
Russo, J. E., Hauguitz, D. & Hilton, J. Inhibition of mouse cytosolic aldehyde dehydrogenase by 4-(diethylamino)benzaldehyde. Biochem. Pharmacol. 37, 1639–1642 (1988).
Johnson, A. T. et al. Synthesis and characterization of a highly potent and effective antagonist of retinoic acid receptors. J. Med. Chem. 38, 4764–4767 (1995).
Stoos, B. A. et al. Characterization of a mouse cortical collecting duct cell line. Kidney Int. 39, 1168–1175 (1991).
Li, J. et al. Collecting duct-derived cells display mesenchymal stem cell properties and retain selective in vitro and in vivo epithelial capacity. J. Am. Soc. Nephrol. 26, 81–94 (2015).
Prié, D. et al. Role of adenosine on glucagon-induced cAMP in a human cortical collecting duct cell line. Kidney Int. 47, 1310–1318 (1995).
Tojo, A. & Kinugasa, S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int. J. Nephrol. 2012, 481520 (2012).
Alicic, R. Z., Rooney, M. T. & Tuttle, K. R. Diabetic kidney disease: challenges, progress and possibilities. Clin. J. Am. Soc. Nephrol. 12, 2032–2045 (2017).
Tedla, F. M., Brar, A., Browne, R. & Brown, C. Hypertension in chronic kidney disease: navigating the evidence. Int. J. Hypertens. 2011, 132405 (2011).
Böhm, M. et al. Efficacy of catheter-based renal denervation in the absence of antihypertensive medications (SPYRAL HTN-OFF MED Pivotal): a multicentre, randomised, sham-controlled trial. Lancet 395, 1444–1451 (2020).
Azizi, M. et al. Optimum and stepped care standardised antihypertensive treatment with or without renal denervation for resistant hypertension (DENERHTN): A multicentre, open-label, randomised controlled trial. Lancet 385, 1957–1965 (2015).
Kim, J. & Padanilam, B. J. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 87, 350–358 (2015).
Kim, J. & Padanilam, B. J. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J. Am. Soc. Nephrol. 24, 229–242 (2013).
Yao, Y. et al. Chronic bilateral renal denervation attenuates renal injury in a transgenic rat model of diabetic nephropathy. Am. J. Physiol Renal Physiol. 307, 251–262 (2014).
Cao, W. et al. A salt-induced reno-cerebral reflex activates renin-angiotensin systems and promotes CKD progression. J. Am. Soc. Nephrol. 26, 1619–1633 (2015).
Antal, O. et al. Hemodynamic predictors for sepsis-induced acute kidney injury: A preliminary study. J. Clin. Med. 9, E151 (2020).
Ross, G. A. et al. Plasma and 24 H-urinary catecholamine concentrations in normal and patient populations. Ann. Clin. Biochem. 30, 38–44 (1993).
Oualha, M. et al. Population pharmacokinetics and haemodynamic effects of norepinephrine in hypotensive critically ill children. Br J Clin Pharmacol. 78, 886–897 (2014).
Baylis, P. H. & Thompson, C. J. Osmoregulation of vasopressin secretion and thirst in health and disease. Clin. Endocrinol. 29, 549–576 (1988).
Russell, J. A. Bench-to-bedside review: Vasopressin in the management of septic shock. Crit. Care 15, 226 (2011).
Xu, X., Zhang, W. & Kone, B. C. CREB trans-activates the murine H(+)-K(+)-ATPase alpha(2)-subunit gene. Am. J. Physiol. Cell Physiol. 287, C903–C911 (2004).
Uber, A. M. & Sutherland, S. M. Nephrotoxins and nephrotoxic acute kidney injury. Pediatr. Nephrol. https://doi.org/10.1007/s00467-019-04397-2 (2019).
Nair, A. R., Masson, G. S., Ebenezer, P. J., Del Piero, F. & Francis, J. Role of TLR4 in lipopolysaccharide-induced acute kidney injury: Protection by blueberry. Free Radic. Biol. Med. 71, 16–25 (2014).
Andrade-Silva, M. et al. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. (Lond) 132, 1725–1739 (2018).
Sumida, K. & Kovesdy, C. P. The gut-kidney-heart axis in chronic kidney disease. Physiol Int. 106, 195–206 (2019).
Wilson, P. D. Polycystic kidney disease. N. Engl. J. Med. 350, 151–164 (2004).
Ogawa, D. et al. Nuclear hormone receptor expression in mouse kidney and renal cell lines. PLoS ONE 9, e85594 (2014).
Rankin, A. C., Hendry, B. M., Corcoran, J. P. & Xu, Q. An in vitro model for the pro-fibrotic effects of retinoids: mechanisms of action. Br. J. Pharmacol. 170, 1177–1189 (2013).
Starkey, J. M. et al. Altered retinoic acid metabolism in diabetic mouse kidney identified by O isotopic labeling and 2D mass spectrometry. PLoS ONE 5, e11095 (2010).
Chen, C. H. et al. O-GlcNAcylation disrupts STRA6-retinol signals in kidneys of diabetes. Biochim. Biophys. Acta Gen. Subj. 1863, 1059–1069 (2019).
Fan, Y. et al. Comparison of kidney transcriptomic profiles of early and advanced diabetic nephropathy reveals potential new mechanisms for disease progression. Diabetes 68, 2301–2314 (2019).
Chen, M., Liu, J., Yang, W. & Ling, W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 13, 1813–1827 (2017).
Angelotti, M. L. et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 30, 1714–1725 (2012).
Hosoyamada, M., Obinata, M., Suzuki, M. & Endou, H. Cisplatin-induced toxicity in immortalized renal cell lines established from transgenic mice harboring temperature sensitive SV40 large T-antigen gene. Arch. Toxicol. 70, 284–292 (1996).
de Larco, J. E. & Todaro, G. J. Epithelioid and fibroblastic rat kidney cell clones: epidermal growth factor (EGF) receptors and the effect of mouse sarcoma virus transformation. J. Cell. Physiol. 94, 335–342 (1978).
Israeli, S., Amsler, K., Zheleznova, N. & Wilson, P. D. Abnormalities in focal adhesion complex formation, regulation, and function in human autosomal recessive polycystic kidney disease epithelial cells. Am. J. Physiol. Cell. Physiol. 298, C831–C846 (2010).
Wilson, P. D., Dillingham, M. A., Breckon, R. & Anderson, R. J. Defined human renal tubular epithelia in culture: growth, characterization, and hormonal response. Am. J. Physiol. 248, F436–F443 (1985).
Wilson, P. D., Schrier, R. W., Breckon, R. D. & Gabow, P. A. A new method for studying human polycystic kidney disease epithelia in culture. Kidney Int. 30, 371–378 (1986).
Hoffman, L. M. et al. BMP action in skeletogenesis involves attenuation of retinoid signaling. J. Cell. Biol. 174, 101–113 (2006).
Klein, E. S. et al. Identification and functional separation of retinoic acid receptor neutral antagonists and inverse agonists. J. Biol. Chem. 271, 22692–22696 (1996).
Acknowledgements
This project was generously supported by Kidney Research UK (RP38/2014) and the Polycystic Kidney Disease Charity (ARPKD-19-02).
Author information
Authors and Affiliations
Contributions
Q.X. designed and supervised the whole project and developed the full manuscript based on a draft by A.P., who conducted all the in vitro studies, data analysis and initial writing. P.R., D.F. and B.H. are co-investigators of the Kidney Research UK Research Grant (RP38/2014) and contributed ideas and discussion throughout the project. In P.R.’s laboratory, M.L.A. generated data included in Fig. 1. M.N. provided technical training and support, as well as helpful discussions. J.C. provided the RA-568 and related discussions. P.W. is a co-investigator of the Polycystic Kidney Disease Charity grant (ARPKD-19–02). P.W. and K.R. cultured and characterised ARPKD renal cystic cells and their age-matched human CD cells and offered invaluable discussions. J.L. and R.P. characterised and provided the CD-derived MSCs and HCD cells, respectively. All co-authors were involved in discussion and approval of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Papadimitriou, A., Romagnani, P., Angelotti, M.L. et al. Collecting duct cells show differential retinoic acid responses to acute versus chronic kidney injury stimuli. Sci Rep 10, 16683 (2020). https://doi.org/10.1038/s41598-020-73099-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73099-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
