
Abstract
Although structural nuclear pore proteins (nucleoporins) are seemingly required in every cell type to assemble a functional nuclear transport machinery, mutations or deregulation of a subset of them have been associated with specific human hereditary diseases. In particular, previous genetic studies of patients with nephrotic syndrome identified mutations in Nup107 that impaired the expression or the localization of its direct partner at nuclear pores, Nup133. In the present study, we characterized the zebrafish nup133 orthologous gene and its expression pattern during larval development. Using a morpholino-mediated gene knockdown, we show that partial depletion of Nup133 in zebrafish larvae leads to the formation of kidney cysts, a phenotype that can be rescued by co-injection of wild type mRNA. Analysis of different markers for tubular and glomerular development shows that the overall kidney development is not affected by nup133 knockdown. Likewise, no gross defect in nuclear pore complex assembly was observed in these nup133 morphants. On the other hand, nup133 downregulation results in proteinuria and moderate foot process effacement, mimicking some of the abnormalities typically featured by patients with nephrotic syndrome. These data indicate that nup133 is a new gene required for proper glomerular structure and function in zebrafish.
Similar content being viewed by others

Introduction
Efficient and regulated bidirectional transport between the cytoplasm and the nucleus is an essential process in all eukaryotic cells. This function is achieved by nuclear pore complexes (NPCs), huge assemblies anchored within the nuclear envelope and composed of about 30 different proteins, termed nucleoporins (Nups) (reviewed in1). Despite the universal role of NPCs in all nucleated cells, some Nups are linked to human hereditary diseases affecting specific cell types or organs (reviewed in2,3,4).
In particular, genetic studies have implicated a restricted number of structural nucleoporins in specific kidney diseases termed nephrotic syndromes (NS). NS arise from defects or damages that impair the selectivity of the glomerular filtration barrier and lead to massive proteinuria and hypoalbuminemia, which in turn cause edema and dyslipidemia. The glomerular filtration barrier (GFB) surrounds the glomerular capillaries and comprises three layers: (i) a fenestrated endothelium, (ii) a basement membrane, and (iii) the podocytes. The latter are highly specialized epithelial cells characterized by long and thin cytoplasmic projections, termed foot processes (FPs), that interdigitate and are connected by specialized cell-cell junctions, the slit diaphragms (reviewed in5). While most patients with childhood-onset NS respond well to steroid treatments, 10–20% of the affected children do not achieve remission upon corticosteroid therapy. Steroid-resistant NS (SRNS) is associated with a high risk of progression to end-stage renal disease (ESRD)6. It frequently manifests histologically as focal segmental glomerulosclerosis (FSGS), characterized by scattered scarring of some glomeruli and is often associated with retractions (“effacement”) of podocytes foot processes (reviewed in7).
Although nonhereditary forms of SRNS seem to be prevalent, studies over the last years have identified over 50 dominant or recessive single-gene mutations in a significant percentage (30%) of patients with early-onset SRNS and FSGS (reviewed or discussed in6,8,9,10,11). While some of these genes have podocyte-specific or -restricted functions, these studies also unveiled the implication of multiple cellular processes in the establishment or maintenance of the glomerular filtration barrier7,12,13. In particular, these genetic studies have identified mutations in Nup93 and Nup205, two constituents of the inner ring of the NPC14 and in Nup107, a constituent of the Y-complex (Nup107/160-complex) that builds up the cytoplasmic and nuclear rings of the NPC15,16,17. Mutations within Nup107 were also identified in patients with a rare co-occurrence of microcephaly with nephrotic syndrome, similar to Galloway-Mowat syndrome (GAMOS)18. While patients with GAMOS-like presentation had a strong reduction in Nup107 protein level accompanied by decreased levels of Nup133, its direct partners within the Y-complex18, another SRNS-linked mutation affecting Nup107 was shown to impair its interaction with Nup13316. These data thus pointed towards a possible implication of Nup133 in NS.
In mice, a previous characterization of a Nup133 null mutant (mermaid, or merm) revealed that mouse embryos lacking a functional Nup133 allele developed through midgestation but die at e9.5–e10.519. While this indicates that Nup133 is not an obligate NPC component, merm mutants displayed abnormalities in a number of tissues, indicating that cell differentiation towards several epiblast-derived lineages likely requires Nup13319. However, the possible contribution of Nup133 to kidney development or function has never been assessed. To address this question, we used morpholino-mediated nup133 inactivation in zebrafish (Danio rerio, Dr), a well-established vertebrate model to study kidney development and renal diseases20,21,22,23. We report here that limited knockdown of zebrafish nup133, while not impairing early stages of kidney development, leads to glomerular abnormality that mimic nephrotic syndrome.
Results
Zebrafish nup133 ortholog is broadly expressed at early stages and becomes restricted to specific tissues at later stages
Query of the latest version of the genome databases identified a unique nup133 orthologue gene in zebrafish, Dr nup133 (ZFIN:ZDB-GENE-040426-2941; Ensembl:ENSDARG00000010078) containing 26 exons located on the forward strand of chromosome 1. The open reading frame is predicted to encode a protein of 1136 amino acids (aa) that shares 62.3% overall amino acid identity with human Nup133 (Supplementary Fig. S1).
To determine the spatio-temporal localization of nup133 transcripts in zebrafish embryonic tissues, we performed whole-mount in situ hybridization (ISH) at different developmental stages (Fig. 1a–f). The nup133 sense RNA probe was used as negative control (Supplementary Fig. S2). Ubiquitous expression of nup133 was observed at sphere stage (4 hours post fertilization, hpf, Fig. 1a and Supplementary Fig. S2). By 24 hpf, nup133 mRNA was detected in the central nervous system, with higher levels of expression in the retina, the tectum and the cerebellum (Fig. 1b). At 3 days post fertilization (dpf), in addition to a diffuse staining notably in the brain, we found evidence of nup133 mRNA enrichment in the liver, in the intestine and in neuromasts of the lateral line organ (Fig. 1c,d). At 5 dpf, the overall expression of nup133 is weaker, but the mRNA is still detectable in the brain, and enriched in the liver, the intestine and the swim bladder (Fig. 1e,f). Cross sections at 4 and 5 dpf confirmed the enrichment of nup133 mRNA in the liver and revealed the presence of nup133 mRNA also in the pronephric proximal tubules and the glomerulus, albeit in low amounts (Fig. 1g, and Supplemental Fig. S2). Consistent with these ISH data and with a previous genome wide RNA-seq dataset24 quantitative RT-PCR performed at 1, 2 and 5 dpf revealed a progressive decrease of nup133 mRNA level during development as compared to beta2 actin mRNA (actb2, previously reported to be extremely stable at these stages of zebrafish development (Supplementary Fig. S2f)24,25.

Expression of nup133 in the developing zebrafish detected by in situ hybridization (ISH). Whole mount ISH with nup133 antisense probe of embryos at: (a) sphere stage (4 hpf; embryo shown with animal pole to the top); (b) 24 hfp (lateral view); (c–f) 3 and 5 dpf (left panels: dorsal view; right panels: lateral view). Arrows point to tissues with enriched expression of nup133. Abbreviations: E: eyes; T: tectum; C: cerebellum; L: liver; I: intestine; N: neuromasts; SB: swim bladder. Scale bars, 200 µm. (g) Transverse section of a 5 dpf embryo at the level of the pectoral fins (as shown in the dotted line in e) confirms nup133 expression in the liver, and show in addition a diffuse staining in the proximal tubules (PT) and a faint signal in the glomerulus (G). Scale bar, 50 µm.
Partial loss of nup133 causes glomerular cysts in zebrafish
In order to characterize the in vivo function of nup133 in zebrafish larvae, we generated knockdown larvae using splice-blocking antisense morpholino oligos (MO) targeting the exon-intron boundary (splice donor E3I3) of exon 3 and the intron-exon boundary of exon 4 (splice acceptor I3E4) of the nup133 gene (Fig. 2a). Reverse transcription-PCR (RT-PCR) demonstrated that the morpholinos interfere with the splicing of exon 3, as revealed by the sequencing of the additional RT-PCR product detected in MO-treated compared to control embryos (Fig. 2a,b and Supplemental Fig. S3). Retention of intron 3 in nup133 transcripts is predicted to produce a truncated protein of 124 aa. Quantitative analyses revealed that the levels of properly spliced nup133 mRNA (red and orange bars in Fig. 2c) drop to about 30% and 40% in 24 and 48 hpf embryos respectively upon nup133 MO injection as compared to control embryos (uninjected or injected with control MO). These MO-injected embryos only contained 20–30% of intron 3-containing nup133 mRNAs (difference between the red/oranges and blue bars in Fig. 2c), indicating that most of the intron 3-containing mRNAs, which encode premature stop codons, are degraded via non-sense mediated mRNA decay. Western blot analysis confirmed these data by revealing a clear albeit moderate decrease of Nup133 protein level in 24 hpf embryos (Supplemental Fig. S3).

Splice Morpholinos (MO) targeting nup133 lead to a partial degradation of nup133 mRNAs. (a) Exon structure of Danio rerio (Dr) nup133 around the binding sites of the E3I3 and I3E4 splice morpholinos. Blue arrowheads above the scheme indicate the position of the RT-PCR primers used in (b) and blue/red/orange arrows below indicate the position of primers and RT-qPCR products used in (c). The size of intron 3 is indicated. (b) RT-PCR from total RNAs of 48 hpf uninjected embryos (control) and of embryos coinjected with the two splicing morpholinos (nup133 MO) reveal an additional band caused by retention of intron 3. (c) nup133 mRNA levels relative to actb2 expression was determined by RT-qPCR on 24 and 48 hpf embryos, uninjected, injected with control MO or nup133 MO, or sequentially injected with 3xHA-mCherry-Dr nup133 mRNA (wt mRNA) and nup133 MO. The E3-E4/5 (red) and E3/4-E4/5′ (orange) primer pairs only amplify nup133 mRNA with properly spliced intron 3 (i.e. wt nup133 mRNA not targeted by the morpholinos) while the E1/2-E3 and E7/8-E9 primer pairs (blue bars) recognize both the spliced and unspliced mRNAs.
Following nup133 MOs injections, zebrafish larvae frequently developed pericardial edema and exhibited an expansion of the glomerulus detectable at 3 dpf by the formation of pronephric cysts (Fig. 3a and Supplemental Fig. S3). Using the Tg(wt1b:eGFP) transgenic line26, in which podocytes and proximal pronephric tubules express EGFP under the wt1b promoter, the glomerular expansion could be directly observed under a fluorescence microscope (Fig. 3b, asterisks). Analysis of semi-thin transverse sections of the glomerulus and proximal tubules at 5 dpf confirmed that the Bowman’s space of the glomerulus was dilated in nup133 morphants compared with the control larvae (Supplemental Fig. S3). To establish the specificity of MOs effects, we determined whether nup133 MO phenotypes could be rescued by co-injection of a synthetic zebrafish nup133 (Dr nup133) mRNA. The Dr nup133 mRNA was fused to a triple-hemagglutinin (HA) epitope and mCherry that enabled us to confirm the expression of the resulting 3xHA-mCherry-Dr Nup133 protein by western blot (Supplementary Fig. S4). Co-injection of the splice MOs with the 3xHA-mCherry-Dr nup133 mRNAs reduced significantly the percentage of larvae with glomerular cysts (Fig. 3c and Supplementary Fig. S4). This demonstrates that the glomerular phenotype observed in nup133 morphants is due to a specific effect of nup133 knockdown.

Partial knockdown of nup133 causes glomerular expansion in zebrafish. (a) Gross morphology of 3 dpf control and nup133 MO embryos (left panels: lateral view; middle panel: dorso-lateral view from two distinct embryos). Scale bar, 500 µm. Two-fold magnification of the indicated area is shown in the rightmost panels. Arrows indicate the pronephric cysts detected in the nup133 MO embryos. (b) Dorsal view of 3dpf Tg(wt1b:EGFP) embryos uninjected (control, top panels), injected with nup133 MO (middle panels), or sequentially injected with 3xHA-mCherry-Dr nup133 mRNA (wt mRNA) and nup133 MO (bottom panels). Overlays of transmission (gray) and GFP-signal (green) images reveal the glomerulus, proximal tubules, and exocrine pancreas. Scale bar, 500 µm. Two-fold magnification of the indicated area and of the same area from a distinct larvae are shown in the right and rightmost panels, respectively. The glomerular structure is indicated in brackets. Asterisks point to cystic dilations of the pronephros in nup133 MO (middle panels). Note that the nup133 MO + wt mRNA embryo displays a left-sided exocrine pancreas (arrowhead) (see also Supplemental Fig. S5). (c) Relative proportion of embryos showing or not kidney cysts at 3 dpf. For each condition, the total number of embryos analyzed is indicated (n=, quantified in 2 distinct experiments for control MO injections and 5 experiments for nup133 MO injections. See also Supplementary Fig. S3). Unlike the embryos injected with control MO, those injected with nup133 MO frequently feature kidney cysts. On the other hand nup133 MO + wt mRNA showed significantly fewer cysts than nup133 MO alone. ***P < 0.0001 using a Fisher exact probability test.
In the course of this study, we also noticed that the exocrine pancreas, an organ normally positioned on the right side of the zebrafish embryo and visualized by the wt1b:GFP transgene in 3 dpf embryos, was misplaced in about 15–20% of the nup133 morphants (Supplemental Fig. S5). Analysis of heart looping in 2 dpf embryos confirmed this Left-Right patterning defect27 (Supplemental Fig. S5). Importantly however, and unlike the frequency of kidney cysts, these Left-Right patterning defects that are classically linked to ciliopathy were not rescued by injection of the 3xHA-mCherry-Dr nup133 mRNA (Arrowhead in Fig. 3b and Supplemental Fig. S5). This indicates that the appearance of kidney cysts upon partial nup133 knockdown is not correlated with a Left-Right patterning defect and may therefore not result from a primary defect in cilia assembly or function (see discussion).
Finally, because mutations of Nup107 that affect the stability of Nup133, also leads to microcephaly in human patients18, we measured the size of the heads of control, nup133 MO and nup133 MO + wt nup133 mRNA embryos at 3 dpf. While a very mild decrease of head width and length was observed in nup133 morphants as compared to control embryos, this defect was not rescued by injection of nup133 mRNA (Supplemental Fig. S5). This slight delay in development may thus reflect unspecific effects of the morpholinos28 rather than a specific effect of nup133 downregulation.
nup133 morphants do not feature major NPC assembly defects and properly express molecular markers of kidney development
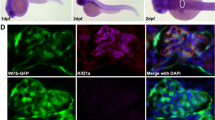
Because Nup133 is a structural nucleoporin, we next analyzed the consequence of its partial depletion on NPC assembly, using as readout the mAb414 antibody that recognizes a subset of FG-containing nucleoporins29. We therefore stained cryosections of 3 dpf Tg(wt1b:EGFP) embryos either treated with control MO or treated with nup133 MO and featuring detectable kidney cysts. While this analysis uncovered a large variability of staining intensity in-between tissues in the control embryos, it did not reveal major alterations of the NPC density in the nup133 MO compared to control MO-treated embryos (Fig. 4). Although specific alteration of a given nucleoporin cannot be excluded, these data suggest that unlike previously reported for Nup107 depletion in zebrafish30 the partial depletion of Nup133 does not affect the overall assembly of NPCs.

Nup133 depletion does not result in major nuclear pore complex assembly defects. (a) Representative transverse sections of the glomerulus of 3 dpf Tg(wt1b:EGFP) embryos treated with either control or nup133 morpholinos (MO) and stained with mAb414 (not shown in these panels) and DAPI. In these sections, GFP-positive cells mark the glomerulus and the neck region of the proximal tubules. Scale bars, 100 μm. (b) Fivefold magnification of the areas indicated in (a) encompassing the glomerulus (G, indicated by the dashed lines) and the liver (L). mAb414 antibody, that recognizes a subset of FG-nucleoporins, shows specific staining around the nucleus of all cells (visualized with DAPI), with however cell-type-dependent variations in intensity. Note for instance the more prominent staining of liver as compared to glomerular cells. In contrast, no major difference can be seen between control and nup133 MO-treated embryos. Insets show a fourfold magnification of representative liver nuclei, revealing the punctate staining typical of NPC staining (mAb414). Scale bars, 20 μm.
We next wanted to determine whether the glomerular expansion in nup133 morphants is caused by a developmental defect of the glomerulus and/or of the pronephros. For this purpose, we used whole-mount ISH to screen established markers of pronephros and glomerulus development. We first assayed expression of the intermediate mesoderm marker pax2a (paired box gene 2a). At 24 hpf, pax2a is expressed during early somitogenesis in the developing pronephric tubules and is important in establishing the boundary between podocytes and the neck segment of the nephron31,32. In nup133 morphants, expression of pax2a at 24 hpf was similar to that of wild type embryos, suggesting that the tubular development is not compromised in the morphants (Fig. 5a). We further examined the pronephric tubules and ducts in nup133 knockdown embryos by checking the expression of the kidney specific marker cadherin 17 (cdh17) at 24 hpf and 72 hpf. Again, no obvious difference in expression of cdh17 was observed between control and nup133 morphant larvae at 24 or 72 hpf (Fig. 5b). Based on these markers, we conclude that tubular development is not impaired upon nup133 knockdown.

Normal development of the pronephric tubules and glomerulus in nup133 morphants. (a) Lateral view of pax2a mRNA expression in pronephric tubules at 24 hpf in uninjected (control) and nup133 MO-injected embryos reveal that the developmental expression of pax2a is not altered in nup133 morphants. Two-fold magnification of the pronephric tubules is also shown. (b) Pronephros marker cdh17 mRNA expression in nup133 MO is comparable to that of uninjected controls at 24 and 72 hpf (dorsal view). (c) Glomerular development in control and nup133 MO embryos visualized using the podocyte differentiation marker wt1a (dorsal view). At 30 hpf (upper panels), wt1a marks future podocytes with two distinct domains in both control and nup133 MO. At 48 hpf (middle panels), the glomerular primordia merge to the midline to form a single glomerulus. At these stages, mRNA expression does not differ between control and nup133 MO (middle panels). At 58 hpf (lower panels), after the onset of glomerular filtration, the increased area labeled by the wt1a probe (arrow) reflects the glomerular expansion observed in 7 out of 13 nup133 morphants analyzed. Scale bars, 500 µm.
We next used an mRNA probe for Wilms tumor suppressor 1a (wt1a) that is predominantly expressed in podocytes throughout pronephric development33. At 24 hpf, wt1a is still expressed in two distinct domains, the glomerular primordials, that fuse to form a single glomerulus by 40 hpf34,35. In nup133 morphants, expression of wt1a was comparable at 30 and 48 hpf to that of wild type embryos (Fig. 5c). wt1a expression sometimes revealed some glomerular enlargement already at 48hpf, a phenotype that became more evident at 58 hpf, after the onset of the glomerular filtration (Fig. 5c, bottom panels). Nevertheless, wt1a transcripts were still expressed in nup133 morphants podocytes. These results suggest that knockdown of nup133 does not affect the gross development of zebrafish glomerulus.
nup133 deficiency affects the normal function of the glomerular filtration barrier
Because the glomerular enlargement upon nup133 knockdown becomes striking at the onset of glomerular filtration, we next analyzed the functionality of the pronephros in the morphant larvae. The glomerular filtration barrier (GFB) of the kidney allows the free filtration of water and small solutes, while restricting the flow of large plasma proteins such as albumin36. However, if the GFB is damaged, albumin and other large proteins pass the barrier to a great extent. Part of the albumin is endocytosed by the epithelial cells of the proximal tubules, while the rest gets then excreted via the final urine leading to proteinuria. In zebrafish larvae, it is possible to inject fluorescent compounds of specific size into the general circulation and then to monitor the appearance of fluorescent endosomes in the apical cytoplasm of pronephric tubule cells37,38. To assay for kidney function, we therefore injected fluorescently labeled albumin from Bovine Serum (BSA, Alexa Fluor™ 647 conjugate) into the common cardinal vein of wild type and nup133 morphant larvae at 4 dpf and then examined the appearance of fluorescent endosomes in the cytoplasm of pronephric tubule cells. Larvae were fixed 20 minutes after BSA injections and sections of the pronephric proximal tubules were imaged. In the control larvae (n = 6, arising from two distinct experiments), we did not observe fluorescent signal from the injected BSA in the apical endosomes of the proximal tubules, indicating that BSA was not able to pass through the intact filtration barrier (Fig. 6, left panels). In most (5/6 imaged larvae) nup133 morphants, however, fluorescently labeled endosomes were detectable in the proximal tubules (Fig. 6, right panels). This suggests that the filtration barrier of the glomerulus is impaired upon nup133 knockdown, allowing the passage of macromolecules with a size exceeding the size selectivity of an intact GFB, a condition defined in patients as proteinuria. A statistically relevant analysis of phenotypic rescue would have required BSA injection and imaging of more than 100 embryos and was thus not performed. However, the BSA filtration defects observed in the nup133 morphants are consistent with the presence of kidney cysts (a phenotype that we could successfully rescue with injections of mRNA). This suggests that the altered functionality of the GFB is most likely a specific consequence of nup133 knockdown.

Glomerular filtration is impaired in nup133 morphants. Representative images of cross sections of 4 dpf control (left panels) and nup133 morphants larvae (right panels) fixed 20 minutes after injection of Alexa Fluor™ 647 conjugated-BSA into the common cardinal vein. Lower panels represent a higher magnification view of the proximal tubule region (dotted lines). Note the uptake of fluorescent BSA in the apical endosomes of the proximal tubules of the Nup133-depleted larva (arrows). Scale bars 50 µm.
nup133 knockdown leads to ultrastructural abnormalities of the glomerular filtration barrier
In order to determine whether the glomerular filtration impairment observed in nup133 morphants was a consequence of a defect in the glomerular filtration barrier (GFB), we conducted electron microscopy studies to compare the ultrastructure of the glomerulus in nup133 morphants and wild type larvae at 5 dpf, a time point when the larval glomerulus development is considered nearly complete34,39 (Fig. 7 and Supplementary Fig. S6). In wild type larvae, the glomerulus showed the typical organization with podocytes harboring well-developed interdigitated foot processes on the outer side, and fenestrated endothelial cells on the inner side of the glomerular basement membrane (GBM) (Fig. 7a–c). In nup133 knockdown larvae with kidney cysts, however, irregularly-shaped processes covering the GMB were frequently observed, although typical foot processes were still present in some regions (Fig. 7d–f). In addition to this moderate foot process effacement, more extreme alterations were observed around the expanded bowman space (Fig. 7h, and Supplementary Fig. S6). In the latter case, podocyte cell bodies in direct contact with the GBM were frequently observed (Supplementary Fig. S6). Quantification of foot processes density along the basement membrane confirmed the disorganization of podocyte foot process architecture in the nup133 morphants (Fig. 7g). Observation of proximal tubules (PTs) revealed variable phenotypes in the morphants: a normal organization, with well-organized microvilli and ciliated cells was observed in morphants with a mild phenotype (Supplementary Fig. S7); in contrast larvae presenting a more severe phenotype featured also an expanded lumen and a less organized brush border (Supplementary Fig. S7). Multiple cross sections of cilia were nevertheless observed inside these expanded PT lumens (Supplementary Fig. S7). These ultrastructural data, combined with the observation of leakage and recapture of fluorescently labeled albumin, suggest that zebrafish nup133 is mainly required for the structural integrity of the GFB and that the observed defects in proximal tubules are secondary to an altered GFB.

nup133 knockdown induces moderate foot processes effacement. (a,d) Transmission electron micrographs showing the glomerular capillary wall of 5 dpf uninjected control (a) and nup133 MO injected larvae (d). (b,e) Two-fold magnification of the areas indicated in (a,d), and (c,f,h) representative images from distinct larvae. Images were pseudocolored to better highlight the glomerular filtration barrier components (podocyte foot processes in green, fenestrated endothelium in blue). Arrows point to irregular-shaped foot processes that are more frequently found in nup133 MO larvae. (g) Quantification of foot processes (FP) density along the basement membrane, presented as average number of FP/µm, was performed as described under materials and methods. The distinct labels reflect values measured on the 3 wt or morphant embryos. Statistical analyses were performed using Wilcoxon-Mann-Whitney Rank Sum Test. **P ≤ 0.01 (p = 0.002486). Image (h) corresponds to a basement membrane surrounding a large cyst (presented in Supplemental Fig. S6e). Note that statistical analyses performed without the latter nup133 morphant larva (values indicated by black squares on the graph) still revealed a significant difference between the control and the nup133 morphants (p = 0.01855). Abbreviations: BS: bowman space; C: capillary lumen; EC: endothelial cells; FP: foot processes. Scale bars 1 µm.
Discussion
In this study we used zebrafish as model organism to evaluate the contribution of Nup133 to vertebrate kidney function. Using splice-blocking MO-based depletion of Nup133 followed by rescue experiments with nup133 mRNA, we have identified nup133 as a new regulator of glomerular structure and functional integrity.
Our study of glomerulus and proximal tubules expression markers shows that the decreased level of Nup133 does not affect the initial stages of nephron development. In addition, TEM analysis of the glomerulus of Nup133-depleted zebrafish reveals that all the cell types constituting the GFB, including the podocytes, are in place. However, the podocytes of Nup133-depleted zebrafish present some degrees of effacement that correlate with a defective GFB. This result therefore suggests that Nup133 plays a fundamental role in the maintenance rather than in the establishment of the GFB.
While we describe here a kidney-specific alteration caused by a moderate depletion of Nup133, Fujita et al.40 very recently reported a translation-blocking morpholino leading to a more efficient depletion of Nup133 protein level in zebrafish and causing both glomerular defects and microcephaly. On the same line, more severe developmental phenotypes were previously observed upon Nup133 inactivation in mouse19. Similarly, while MO-induced depletion of zebrafish Nup107 mainly causes glomerular defects16, transposon insertion in nup107tsu068Gt transgenic embryos affects multiple tissues including the pharyngeal skeleton, the intestine, the swim bladder and the eyes30,41, and a CRISPR/Cas9-generated homozygous truncating “null” mutation of nup107 showed early lethality at 5 dpf associated with developmental malformations42. Together, these data indicate that the extent of depletion of structural nucleoporins can lead to distinct graded phenotypes in zebrafish, and that the kidney appears to be the organ with the highest sensitivity to moderate Nup133 or Nup107 deficiencies.
As we submitted our manuscript, the only Y-complex nucleoporin gene found to be mutated in SRNS was Nup10715,16,17,18. In addition, a study had revealed that normal expression level of another Y-complex component, Nup160, is critical for the proliferation and viability of podocytes in vitro43. Because our data indicated that Nup133, the direct partner of Nup107 within the Y-complex, is also required for proper glomerular function in zebrafish, we proposed that in addition to Nup107 at least these two other Y-Nups, or possibly all the nine Y-complex subunits (Nup107, Nup133, Nup160, Nup96, Nup85/75, Nup43, Nup37, Seh1, and Sec13) were candidate genes that would be worth testing in SRNS patients. While our manuscript was under revision, two publications came out that confirmed our hypothesis: specific hypomorphic mutations in 4 Y-complex components, namely Nup107, Nup133, Nup160 and Nup85 were reported to cause SRNS42 whereas a homozygous splicing mutation in NUP133 was identified in a GAMOS family with SRNS combined to brain atrophy40. Noteworthy, the mutation found in this GAMOS family caused reduced Nup133 protein level40, as also previously described for the consanguineous families with homozygous mutation in NUP107 and a GAMOS-like presentation18. Although Nup133 protein levels were not investigated in patients solely featuring SRNS, the data obtained in zebrafish (our study and40) suggest that more severe alteration leading to decreased expression or stability of Nup133 would cause neuronal in addition to renal defects.
Several mechanisms could explain the implication of Nup133, and more generally, of Y-complex Nups, in glomerular function and thus NS. While it was recently reported that alterations of several Y-Nups including NUP133 causes dysregulation of Cdc42 activity in human podocytes, the authors suggested that this was likely an indirect effect, possibly linked to altered nucleocytoplasmic transport42. Our immunofluorescence data using mAb414 did not reveal any major alteration of overall NPC assembly in nup133 morphants. However, Nup133 deficiency may alter the nuclear transport of specific molecules, as for instance reported for Nup93 that belongs to another structural domain of the NPC14. Indeed, human podocytes and HEK293 cells expressing Nup93 mutations identified in SRNS patients exhibited impaired nuclear import of SMAD4 upon stimulation with bone morphogenetic protein 7 (BMP7), a secreted molecule involved in kidney development and response to renal injury. This result was consistent with a previous study in which depletion of Drosophila Nup93 led to a defective import of the SMAD4 orthologue44. Noteworthy, the latter study also revealed the implication of two Drosophila Y-Nups, Nup85/75 and Sec13, but not of Nup133, in this specific import pathway44. While we cannot formally exclude Nup133 contribution to the nuclear import of SMAD4 in vertebrates, Nup133 depletion may also affect other signaling cascades required for the maintenance of an intact GFB. Indeed, two Y-complex subunits, Nup107 and Nup37, were reported to be regulators of the ERK and YAP pathways, respectively45,46.
Nup133 knockdown may also alter nuclear mechanotransduction, for instance by interfering with the ‘linker of nucleoskeleton and cytoskeleton’ (LINC) complex that establish a stable and cross-linked network between the cytoskeleton and the lamina underneath the NE (reviewed in47). Indeed, while Nup133 is required for the proper assembly of the NPC basket48, intimate links between the NPC basket, the LINC subunit Sun1 and Lamin-C have been reported49,50. As previously discussed16, deregulation of mechanotransduction signaling pathways may in turn affect podocytes that are subjected to mechanical stretching caused by capillary pressure. As reported for a subset of nucleoporins (reviewed in51), Nup133 may also be involved in gene regulation and thus modulate the expression of specific genes required for proper function of the GFB, notably in podocytes.
Finally, one may also keep in mind that several nucleoporins localize at the base of the cilia52,53. In particular, another member of the Y-complex, Nup85, was recently reported to be required for proper cilia localization of Nup98, a nucleoporin that regulates diffusion of soluble molecules through the ciliary base54. Moreover, mutations affecting the kinetochore protein Cenp-F, an established partner of Nup13355, were identified in patients affected by severe ciliopathy and microcephaly56. Because the appearance of kidney cysts in zebrafish can also be caused by inactivation of several ciliopathy genes (reviewed in57,58), the observation of a mild Left-Right patterning defect in nup133 morphants was noteworthy. However, our data revealed that unlike the glomerular cysts phenotype, the Left-Right patterning defect is not rescued by the 3xHA-mCherry-Dr nup133 transgene, and is therefore not correlated with the glomerular defect observed in these larvae. As previously proposed for other mutants37,59,60), the pronephric cyst phenotype observed in the nup133 morphants might thus be primarily caused by alterations of the glomerular filtration barrier and the subsequent inability to osmoregulate, rather than the indirect consequence of cilia defect. While this non-rescued situs inversus phenotype may reflect an off-target effect of the morpholinos (as previously reported in another study)61, we do not formally rule out a possible impact of Nup133 depletion on ciliary function. The lack of rescue of the situs inversus by our nup133 construct would then reflect an improper expression of the transgene in the Kupffer’s vesicle (the ciliated organ that initiates left-right development in the zebrafish embryo)62. Analysis of a potential cell-type-dependent ciliary function of Nup133 will deserve a full and independent study.
In conclusion, our study on zebrafish Nup133, together with an increasing number of recent studies correlating single-gene mutations in Nups with cell type-specific defects, strengthens the emerging concept of specific “nucleoporopathies”1, an exciting development in the nuclear pore complex field.
Materials and Methods
Fish maintenance and breeding
Zebrafish (Danio rerio) were kept at 26 °C under a 14-h/10-h light/dark cycle and bred as previously described63. Larval stages were raised at 28 °C in E3 embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) containing 0.01% methylene blue. 0.003% PTU (1-phenyl-2-thiourea; Sigma Aldrich) was added in embryo medium to inhibit melanin synthesis during larval development and facilitate fluorescent microscopy. Tg(wt1b:EGFP) line was a kind gift from Dr. Christoph Englert (Leibniz Age Research, Jena, Germany). All experiments were performed in accordance with internationally recognized and with Swiss legal ethical guidelines for the use of fish in biomedical research and experiments were approved by the local authorities (Veterinäramt Zürich Tierhaltungsnummer 150 and TV4206).
Whole-mount in situ hybridization
Sequences were identified and annotated using combined information from expressed sequence tags and genome databases (GeneBank, http://www.ncbi.nlm.nih.gov; Ensembl, http://www.ensembl.org/index.html). The primers used for probe preparation are listed in supplementary Table S1. nup133, pax2a, wt1a and cdh17 cDNAs were all isolated by RT-PCR from total RNA from 24–48 hpf embryos and cloned into TOPO pCRII vector (TA Cloning Kit Dual Promoter, Invitrogen) as previously described64. The resulting plasmids were linearized for SP6 and T7 in vitro transcription and purified with phenol-chloroform. Digoxigenin (DIG)-labeled antisense (or sense, as negative control for nup133 expression) RNA probes were generated using DIG-RNA-labeling kit (Roche Diagnostic). Zebrafish embryos were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) at 4 °C overnight and whole mount in situ hybridization was performed as previously described65. Briefly, on day 1 the larvae were treated with proteinase K and then fixed with 4% paraformaldehyde (PFA) before prehybridization at 64 °C. Hybridization of RNA probes was done at 64 °C overnight. On day 2, after several stringency washes at 64 °C, probes were blocked in 1 × Roche blocking solution in Tris/NaCl/Tween. Anti-DIG AP antibody was applied overnight at 4 °C. On day 3, after several washing steps, signal was detected by incubation in staining buffer. Stained embryos were postfixed with PFA overnight and imaged in glycerol with an Olympus BX61 light microscope or with a stereomicroscope (Olympus MVX10). Following PFA postfixation, 4 and 5 dpf embryos were cryoprotected in 30% sucrose overnight and embedded in tissue freezing medium (Richard-Allan Scientific Neg-50 Frozen Section Medium Thermo Fisher Scientific). 14–16 μm transverse sections were cut on a Microm HM 550 cryostat and imaged with a Olympus BX61 wide-field microscope (Volketswil, Switzerland). Images were processed and assembled using Adobe Photoshop and Adobe Illustrator CS6.
Morpholino and mRNA injections
The nup133 gene was targeted with specific antisense splice-blocking Morpholinos (MOs) (GeneTools, Philamath, OR) designed to target the exon-intron boundary of exon 3 (splice donor, nup133_E3I3) and the intron-exon boundary of exon 4, respectively (splice acceptor, nup133_I3E4) (MO sequences are provided in Supplemental Table S1). The morpholinos were diluted in RNase-free water with 0.1% phenol red as an injection tracer and ~1 nl was injected into fertilized embryos at 1–2 cell stage. Uninjected sibling embryos and embryos injected with standard control morpholinos were used as controls. Evaluation of morphological changes was performed at 3 dpf. A range of concentration was first used to determine the optimal MO amount required to induce a specific phenotype without inducing toxicity (Supplemental Fig. S3). The two MOs were then always injected at 1.75 ng each.
For the rescue experiments, a pBluescript KSM vector containing 3xHA-mCherry-Dr nup133 was used. The plasmid was generated using PCR amplification using proofreading DNA polymerases (Phusion HF, NEB) and NEBuilder HiFi DNA Assembly Cloning Kits. Briefly, the sequence encoding three copies of the influenza virus hemagglutinin (HA) epitope was amplified from pFA6a-3HA-kanMX666, the mCherry was amplified from plasmid #193748, full-length zebrafish nup133 was amplified from a Dharmacon cDNA vector (Clone ID: 2600558) and the three PCR products were inserted by recombination in a pBluescript KSM vector (Stratagene). PCR-amplified fragments and junctions were checked by sequencing. Plasmid map is available upon request. For in vitro mRNA production the 3xHA-mCherry-Dr nup133 coding fragment was linearized by digestion with the restriction enzyme NotI and purified with phenol-chloroform extraction. Capped and tailed RNA was in vitro transcribed using the mMessage mMachine T3 kit (Life Technologies, Zug, Switzerland) according to the manufacturer’s instruction. A polyA tail was added with polyA tailing kit (Invitrogen by Thermo Fischer Scientific) followed by purification with MEGAclear Transcription Clean-Up Kit (Ambion). The mRNA (300 nM) was injected into the embryos at one cell stage before MO injection.
Real-time PCR and real time quantitative PCR
To examine splicing defects caused by nup133_E3I3 and nup133_I3E4 injection, total RNAs were extracted from 10–40 control or MO-injected embryos at 1 dpf by using the ReliaPrep RNA Tissue Miniprep System (Promega), and RNAs were reverse transcribed and amplified using SuperScript III First-Strand Synthesis SuperMix with random hexamers (Invitrogen). The resulting cDNAs were then characterized by RT-PCR using primers designed from flanking exon-coding sequence (Fig. 2a,b and Supplementary Table S1). The RT-PCR products were purified from gel, re-amplified with nested primers and altered splicing was confirmed by sequencing.
Real-time quantitative PCR was performed on independent batches of cDNAs from 1, 2 or 5 dpf embryos with a LightCycler480 instrument (Roche Life Sciences) by using SYBR Green incorporation (SYBR Green PCR-Master Mix; Applied Biosystems) and specific primer pairs (listed in Supplementary Table S1). The relative amounts of cDNAs in the samples were quantified according to the manufacturer’s instructions and normalized by reference to the actin beta 2 (actb2) cDNAs.
Whole mount live-animal imaging
To evaluate morphological changes, larvae were anesthetized with 200 mg/ml 3-aminobenzoic acid methyl ester (MESAB, Sigma-Aldrich), mounted in 1.5% low melting temperature agarose in E3 medium and imaged using a stereomicroscope (Olympus MVX10) equipped with a color camera (ColorViewIII, Soft imaging System, Olympus).
Western blotting
For western blot analyses, groups of 20–40 embryos at 24 hpf were homogenized by sonication in 50–100 µl of Laemmli buffer. The lysates were separated either on 4–20% Mini-Protean® TGX Stain-Free™ gel or on NuPAGETM 4–12% Bis-Tris gels, using Tris-Glycine and MOPS as running buffer, respectively, and transferred to nitrocellulose membranes. The resulting blots were stained using Ponceau, saturated with TBS, 0.1% Tween, and 5% dried milk and probed overnight at 4 °C with Nup133 antibody (Rabbit monoclonal [EPR10809] to NUP133, ab181355; Abcam, 1:500), rabbit polyclonal anti-ß-Actin (Cell Signaling Technology, #4967; 1:1000), or mouse monoclonal antibody HA.11 (clone 16B12; Eurogentec #MMS-101R; 1:2,000). Incubations of the membrane with primary and HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were done in TBS buffer (0.1% Tween, 5% dried milk), and signals were detected by enhanced chemiluminescence (SuperSignal® West Femto or Pico PLUS; Thermo Scientific).
NPC labeling on cryosections
Because the glomerular phenotype is not fully penetrant (observed, depending on the experiment, in 40–90% of the injected embryos, Supplemental Fig. S4), only nup133 MO embryos displaying kidney cysts (visualized using the wt1b:GFP transgene) were analyzed. For mAb414 staining, 3dpf zebrafish larvae were fixed for 30 min in 3% PFA, cryoprotected in 30% sucrose and embedded in tissue freezing medium (OCT - Leica 14020108926). Following freezing, 14 μm transverse sections were cut on a Leica CM3050S cryostat and placed on SuperFrost Ultra Plus™ adhesion slides (Thermo Scientific). The slides were dried for 30 min at 37 °C, washed for 5 min in PBS, saturated for 30 min with PBS containing 0.1% Triton X-100 and 1% BSA (PBS-T-BSA) and incubated overnight at 4 °C with mAb414 mouse monoclonal antibody (Covance, MMS-120P) diluted at 1/5000 in the same buffer. Following rapid washes in PBS-T, slides were then incubated for 2 hours at room temperature with CY3-conjugated anti-mouse antibody (Jackson ImmuoResearch, # 715-165-151) diluted 1/500 in PBS-T-BSA containing 0.1 µg/ml DAPI. The slides were then washed in PBS-T and mounted in Moviol medium.
Spinning disk images were acquired on an inverted microscope (DMI8; Leica) with a CSU-W1 spinning disk head (Yokogawa) and a sCMOS ORCA-Flash 4 V2+ camera (Hamamatsu) using 100×/1.4 oil objective or a 20×/0.75 oil objective. Images were acquired without camera binning using MetaMorph (Universal Imaging Corp.), processed using ImageJ/Fiji software and assembled using Adobe Photoshop CS. A unique plane is presented.
Glomerular filtration assay
Zebrafish larvae at 4 dpf were anesthetized with 200 mg/ml 3-aminobenzoic acid methyl ester (MESAB, Sigma-Aldrich). 1 nl of Albumin from bovine serum (BSA), Alexa Fluor 647 conjugate (Thermo Fischer scientific) was injected into the common cardinal vein according to38. The larvae were transferred to E3 medium for recovery. 20 mins after injections, the larvae were fixed in 4% PFA overnight and processed for cryosections and imaging.
Transmission electron microscopy and histology
Zebrafish larvae at 5 dpf were fixed overnight in 2.5% glutaraldehyde (along with 3% EM grade PFA for the larvae shown in Fig. 7h and in Supplementary Figs. S6, panels b, d and e, and S7, panel f) in 0.1 M cacodylate buffer, pH 7.2. As indicated above for the IF studies, nup133 MO embryos displaying kidney cysts were selected prior to EM studies. To achieve a better penetration of the fixative, the tail of each larva was cut off with a scalpel prior to fixation. The larvae were rinsed in 0.1 M cacodylate buffer before postfixation in 1% osmium tetroxide in cacodylate buffer for 1 hour at room temperature. The samples were rinsed in distilled water before en block staining in 1% aqueous uranyl acetate for 1 hour at room temperature. Following dehydration through a graded series of ethanol ranging from 50% to 100%, the larvae were infiltrated overnight in 66% Epon/Araldite in propylene oxide. Finally, the specimens were embedded in 100% Epon/Araldite and placed in a polymerizing oven at 60 °C for 26 h. Semi thin section (2 µm) were stained with toluidine blue and used for histological studies. Ultrathin sections (65 nm) of the glomerulus and the proximal tubules, obtained using a Leica EM FCS ultramicrotome were collected on formvar coated copper grids, stained in lead for 5 minutes to increase the contrast, and examined with a Philips CM-100 scope at 80 kV. Images were acquired using the Gatan Microscopy Software.
For quantification of FP density, images acquired from 3 control and 3 nup133-MO treated embryos were analyzed. For each embryo, the number of foot processes and the length of the basement membrane were measured from 3–5 distinct fields each. Images used for quantification, the selected ROI, and original Excel file used to generate the graph are available at Mendeley under https://doi.org/10.17632/j78ddshctz.1.
Statistical analyses were performed using Wilcoxon-Mann-Whitney Rank Sum Test provided by the KaleidaGraph software.
Data Availability
pBluescript KSM vector containing 3xHA-mCherry-Dr nup133 and its sequences are available upon request. Images used for EM quantification (Fig. 7g) and for the quantifications of kidney cysts appearance, LR asymmetry (based on pancreas positioning) and head sizes (Supplemental Fig. S5) are available at Mendeley under https://doi.org/10.17632/j78ddshctz.1.
Change history
19 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
15 January 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Beck, M. & Hurt, E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18, 73–89 (2017).
Nofrini, V., Di Giacomo, D. & Mecucci, C. Nucleoporin genes in human diseases. Eur J Hum Genet 24, 1388–1395 (2016).
Hezwani, M. & Fahrenkrog, B. The functional versatility of the nuclear pore complex proteins. Semin Cell Dev Biol 68, 2–9 (2017).
Sakuma, S. & D’Angelo, M. A. The roles of the nuclear pore complex in cellular dysfunction, aging and disease. Semin Cell Dev Biol 68, 72–84 (2017).
Patrakka, J. & Tryggvason, K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun 396, 164–169 (2010).
Trautmann, A. et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol 10, 592–600 (2015).
Machuca, E., Benoit, G. & Antignac, C. Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Hum Mol Genet 18, R185–194 (2009).
Lovric, S., Ashraf, S., Tan, W. & Hildebrandt, F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31, 1802–1813 (2016).
Preston, R., Stuart, H. M. & Lennon, R. Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol, https://doi.org/10.1007/s00467-017-3838-6 (2017).
Lepori, N., Zand, L., Sethi, S., Fernandez-Juarez, G. & Fervenza, F. C. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J 11, 179–190 (2018).
Jin, Y. Y., Feng, B. Y. & Mao, J. H. The status quo and challenges of genetic diagnosis in children with steroid-resistant nephrotic syndrome. World J Pediatr, s12519-018-0156-4 (2018).
Akchurin, O. & Reidy, K. J. Genetic causes of proteinuria and nephrotic syndrome: impact on podocyte pathobiology. Pediatr Nephrol 30, 221–233 (2015).
Ha, T. S. Genetics of hereditary nephrotic syndrome: a clinical review. Korean J Pediatr 60, 55–63 (2017).
Braun, D. A. et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet 48, 457–465 (2016).
Alazami, A. M. et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep 10, 148–161 (2015).
Miyake, N. et al. Biallelic Mutations in Nuclear Pore Complex Subunit NUP107 Cause Early-Childhood-Onset Steroid-Resistant Nephrotic Syndrome. Am J Hum Genet 97, 555–566 (2015).
Park, E. et al. NUP107 mutations in children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant 32, 1013–1017 (2017).
Rosti, R. O. et al. Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J Med Genet 54, 399–403 (2017).
Lupu, F., Alves, A., Anderson, K., Doye, V. & Lacy, E. Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev Cell 14, 831–842 (2008).
Morales, E. E. & Wingert, R. A. Zebrafish as a Model of Kidney Disease. Results Probl Cell Differ 60, 55–75 (2017).
Outtandy, P., Russell, C., Kleta, R. & Bockenhauer, D. Zebrafish as a model for kidney function and disease. Pediatr Nephrol, s00467-018-3921-7 (2018).
Jerman, S. & Sun, Z. Using Zebrafish to Study Kidney Development and Disease. Curr Top Dev Biol 124, 41–79 (2017).
Swanhart, L. M. et al. Zebrafish kidney development: basic science to translational research. Birth Defects Res C Embryo Today 93, 141–156 (2011).
White, R. J. et al. A high-resolution mRNA expression time course of embryonic development in zebrafish. Elife 6, https://doi.org/10.7554/eLife.30860 (2017).
Tang, R., Dodd, A., Lai, D., McNabb, W. C. & Love, D. R. Validation of zebrafish (Danio rerio) reference genes for quantitative real-time RT-PCR normalization. Acta Biochim Biophys Sin (Shanghai) 39, 384–390 (2007).
Perner, B., Englert, C. & Bollig, F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev Biol 309, 87–96 (2007).
Shu, X. et al. Na,K-ATPase alpha2 and Ncx4a regulate zebrafish left-right patterning. Development 134, 1921–1930 (2007).
Bedell, V. M., Westcot, S. E. & Ekker, S. C. Lessons from morpholino-based screening in zebrafish. Brief Funct Genomics 10, 181–188 (2011).
Davis, L. I. & Blobel, G. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci USA 84, 7552–7556 (1987).
Zheng, X. et al. Loss of zygotic NUP107 protein causes missing of pharyngeal skeleton and other tissue defects with impaired nuclear pore function in zebrafish embryos. J Biol Chem 287, 38254–38264 (2012).
Krauss, S. et al. Zebrafish pax[zf-a]: a paired box-containing gene expressed in the neural tube. EMBO J 10, 3609–3619 (1991).
Majumdar, A., Lun, K., Brand, M. & Drummond, I. A. Zebrafish no isthmus reveals a role for pax2.1 in tubule differentiation and patterning events in the pronephric primordia. Development 127, 2089–2098 (2000).
Kreidberg, J. A. et al. WT-1 is required for early kidney development. Cell 74, 679–691 (1993).
Ichimura, K. et al. A comparative analysis of glomerulus development in the pronephros of medaka and zebrafish. PLoS One 7, e45286 (2012).
Drummond, I. A. et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development 125, 4655–4667 (1998).
Arif, E. & Nihalani, D. Glomerular Filtration Barrier Assembly: An insight. Postdoc J 1, 33–45 (2013).
Sugano, Y. et al. The Rho-GTPase binding protein IQGAP2 is required for the glomerular filtration barrier. Kidney Int 88, 1047–1056 (2015).
Drummond, I. A. & Davidson, A. J. Zebrafish kidney development. Methods Cell Biol 134, 391–429 (2016).
Zhu, X. et al. Ultrastructural characterization of the pronephric glomerulus development in zebrafish. J Morphol 277, 1104–1112 (2016).
Fujita, A. et al. Homozygous splicing mutation in NUP133 causes Galloway-Mowat syndrome. Ann Neurol 84, 814–828 (2018).
Niu, X. et al. The nuclear pore complex function of Sec13 protein is required for cell survival during retinal development. J Biol Chem 289, 11971–11985 (2014).
Braun, D. A. et al. Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J Clin Invest 128, 4313–4328 (2018).
Wang, P., Zhao, F., Nie, X., Liu, J. & Yu, Z. Knockdown of NUP160 inhibits cell proliferation, induces apoptosis, autophagy and cell migration, and alters the expression and localization of podocyte associated molecules in mouse podocytes. Gene 664, 12–21 (2018).
Chen, X. & Xu, L. Specific nucleoporin requirement for Smad nuclear translocation. Mol Cell Biol 30, 4022–4034 (2010).
Kim, S. Y., Kang, H. T., Choi, H. R. & Park, S. C. Reduction of Nup107 attenuates the growth factor signaling in the senescent cells. Biochem Biophys Res Commun 401, 131–136 (2010).
Luo, X. et al. NUP37, a positive regulator of YAP/TEAD signaling, promotes the progression of hepatocellular carcinoma. Oncotarget 8, 98004–98013 (2017).
Kirby, T. J. & Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat Cell Biol 20, 373–381 (2018).
Souquet, B. et al. Nup133 Is Required for Proper Nuclear Pore Basket Assembly and Dynamics in Embryonic Stem Cells. Cell Rep 23, 2443–2454 (2018).
Liu, Q. et al. Functional association of Sun1 with nuclear pore complexes. J Cell Biol 178, 785–798 (2007).
Xie, W. et al. A-type Lamins Form Distinct Filamentous Networks with Differential Nuclear Pore Complex Associations. Curr Biol 26, 2651–2658 (2016).
Raices, M. & D’Angelo, M. A. Nuclear pore complexes and regulation of gene expression. Curr Opin Cell Biol 46, 26–32 (2017).
Kee, H. L. et al. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol 14, 431–437 (2012).
Del Viso, F. et al. Congenital Heart Disease Genetics Uncovers Context-Dependent Organization and Function of Nucleoporins at Cilia. Dev Cell 38, 478–492 (2016).
Endicott, S. J. & Brueckner, M. NUP98 Sets the Size-Exclusion Diffusion Limit through the Ciliary Base. Curr Biol 28, 1643–1650 (2018).
Berto, A. et al. Disentangling the molecular determinants for Cenp-F localization to nuclear pores and kinetochores. EMBO Rep 19, https://doi.org/10.15252/embr.201744742 (2018).
Waters, A. M. et al. The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J Med Genet 52, 147–156 (2015).
Shi, Y., Su, Y., Lipschutz, J. H. & Lobo, G. P. Zebrafish as models to study ciliopathies of the eye and kidney. Clin Nephrol Res 1, 6–9 (2017).
Song, Z., Zhang, X., Jia, S., Yelick, P. C. & Zhao, C. Zebrafish as a Model for Human Ciliopathies. J Genet Genomics 43, 107–120 (2016).
Kramer-Zucker, A. G., Wiessner, S., Jensen, A. M. & Drummond, I. A. Organization of the pronephric filtration apparatus in zebrafish requires Nephrin, Podocin and the FERM domain protein Mosaic eyes. Dev Biol 285, 316–329 (2005).
Ebarasi, L. et al. A reverse genetic screen in the zebrafish identifies crb2b as a regulator of the glomerular filtration barrier. Dev Biol 334, 1–9 (2009).
Maurya, A. K. et al. Positive and negative regulation of Gli activity by Kif7 in the zebrafish embryo. PLoS Genet 9, e1003955, https://doi.org/10.1371/journal.pgen.1003955 (2013).
Essner, J. J., Amack, J. D., Nyholm, M. K., Harris, E. B. & Yost, H. J. Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 132, 1247–1260 (2005).
Westerfield, M. The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio). 5th edn, (Univ. of Oregon Press, 2007).
Gesemann, M., Lesslauer, A., Maurer, C. M., Schonthaler, H. B. & Neuhauss, S. C. Phylogenetic analysis of the vertebrate excitatory/neutral amino acid transporter (SLC1/EAAT) family reveals lineage specific subfamilies. BMC Evol Biol 10, 117, https://doi.org/10.1186/1471-2148-10-117 (2010).
Huang, Y. Y., Haug, M. F., Gesemann, M. & Neuhauss, S. C. Novel expression patterns of metabotropic glutamate receptor 6 in the zebrafish nervous system. PLoS One 7, e35256, https://doi.org/10.1371/journal.pone.0035256 PONE-D-11-21707 (2012).
Bahler, J. et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 (1998).
Acknowledgements
We are grateful to Dr. Christoph Englert (Leibniz Age Research, Jena, Germany) for sharing the Tg(wt1b:EGFP) line. We also thank Zhiyong Chen, Yuya Sugano, Ruxandra Bachmann-Gagescu and Matthias Gesemann and members of our labs for support and helpful advices and Benoit Palancade for critical reading of the manuscript. We acknowledge the assistance and support of the Center for Microscopy and Image Analysis, University of Zurich for support with electron microscopy experiments. We also acknowledge the ImagoSeine core facility of the Institut Jacques Monod, member of IBISA and of the France-Bioimaging (ANR-10-INBS-04) infrastructures. Work in the laboratory of VD is supported by the Centre National de la Recherche Scientifique (CNRS), the “Fondation pour la Recherche Médicale” (Foundation for Medical Research) under grant No DEQ. 20150734355, “Equipe FRM 2015” and the Labex Who Am I? (ANR-11-LABX-0071; Idex ANR-11-IDEX-0005-02). AB received PhD fellowships from the “Ministère de l’Enseignement Supérieur et de la Recherche” and the “Ligue Nationale contre le Cancer” and a “transition post-doc” grant from the Labex Who Am I? SCNF and JL are supported by research funds from the Swiss National Science Foundation (31003A_173083 and 310030_143929/1, respectively). CCC received a fellowship and research funds from the Italian Ri.MED foundation.
Author information
Authors and Affiliations
Contributions
C.C.C., A.B., J.L., S.N. and V.D. conceived and designed the experiments; C.C.C., A.B. S.P. and M.H. performed the experiments; C.C.C., A.B., S.P., J.L., S.N. and V.D. analyzed the data; C.C.C., A.B. and V.D. wrote the manuscript with contribution from all co-authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cianciolo Cosentino, C., Berto, A., Pelletier, S. et al. Moderate Nucleoporin 133 deficiency leads to glomerular damage in zebrafish. Sci Rep 9, 4750 (2019). https://doi.org/10.1038/s41598-019-41202-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41202-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
