
Abstract
The stink bug Arma custos (Hemiptera: Pentatomidae) is a predatory enemy successfully used for biocontrol of lepidopteran and coleopteran pests in notorious invasive species. In this study, a high-quality chromosome-scale genome assembly of A. custos was achieved through a combination of Illumina sequencing, PacBio HiFi sequencing, and Hi-C scaffolding techniques. The final assembled genome was 969.02 Mb in size, with 935.94 Mb anchored to seven chromosomes, and a scaffold N50 length of 135.75 Mb. This genome comprised 52.78% repetitive elements. The detected complete BUSCO score was 99.34%, indicating its completeness. A total of 13,708 protein-coding genes were predicted in the genome, and 13219 of them were annotated. This genome provides an invaluable resource for further research on various aspects of predatory bugs, such as biology, genetics, and functional genomics.
Similar content being viewed by others


Background & Summary
The stink bug Arma custos (Fabricius, 1794) (Hemiptera: Pentatomidae) is synonymous with Arma chinensis (Fallou, 1881), which has been recorded in China, Mongolia and Korea, as well as central and southern Europe (except the British Islands) and the neighboring parts of the Middle East1,2. Both nymphs and adults of this zoophytophagous bug can predate many agricultural and forestry pests belonging to the orders of Coleoptera, Lepidoptera, Hemiptera and Hymenoptera by utilizing a venomous cocktail produced by the salivary gland to capture and digest preys3,4. It can be easily mass-reared using artificial diet in a factory and exhibits strong adaptability to diverse ecological niches, enabling its successful use as a commercialized biocontrol agent3,5. Notably, it has shown effective management of notorious invasive pests such as the fall webworm Hyphantria cunea, the Colorado potato beetle Leptinotarsa decemlineata, and the fall armyworm Spodoptera frugiperda through the augmentative release6,7,8. However, limited attention has been given to the investigation of the biological characteristics9,10,11, artificial rearing methods3,5,12, chemoecology13, response to temperature and drought stresses8,14,15,16,17, and developmental regulation by miRNA18 of this predatory bug. In terms of its genetic information, only the mitochondrial genome and several transcriptomic datasets are available as the current genetic resources13,15,16,18,19. Obtaining high-quality genome for providing a whole set of gene resources of A. custos will greatly facilitate a wide range of biological researches and allow further investigations, such as population genetic diversity, venomics, adaptive evolution, and comparative genomics.
In this study, we have assembled a chromosome-level genome of A. custos by combining PacBio HiFi sequencing and High-throughput chromosome conformation capture (Hi-C) technologies. The genome assembly allowed us to identify repeat sequences and protein-coding genes. Predicted genes were annotated. The generated genomic resources will facilitate to the investigation of this predatory bug.
Methods
Sample collection and rearing
The population of A. custos used in this study originated from a colony collected in the suburb of Kunming, Yunnan Province, China. These bugs have been maintained in our laboratory for more than 20 generations. They were fed with larvae of the yellow mealworm Tenebrio molitor, the greater wax moth Galleria mellonella, and the fall armyworm S. frugiperda. Cages measuring 40 cm × 40 cm × 40 cm, constructed with Nylon netting (44 × 32 mesh) on all sides, were used to rear the bugs. Each cage housed approximately 100 bugs. Soybean plants were also provided in the cage for feeding and perching. The bugs were reared at a constant temperature of 25 ± 1 °C, 70 ± 5% relative humidity, and a photoperiod of 14 L:10D.
Sequencing
Genomic DNA was extracted from one newly emerged male adult using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Total RNA was isolated from various adult tissues including different glands of the salivary venom apparatus (anterior main gland, posterior main gland, and accessary gland), gut and residual body (adult deprived of salivary venom apparatus and gut). The integrity and contamination of the DNA and RNA were assessed on a 1% agarose gel. The purity of the DNA and RNA was measured with a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The DNA and RNA concentration of was determined using the Qubit DNA Assay Kit in Qubit 3.0 Flurometer (Invitrogen, Carlsbad, CA, USA).
For short-read genomic and transcriptome sequencing, the library with an insert size of 350 bp was constructed using the NEBNext Ultra DNA Library Prep Kit (Illumina, San Diego, CA, USA) following manufacturer’s recommendations. This library was then sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). The genomic short-read data yielded from the Illumina NovaSeq 6000 platform amounted to 74.86 Gb with a Q20 value of 96.56% and a Q30 value of 90.84% (Table 1). A total of 72.86 Gb transcriptomic data were generated, which have Q20 values over 96.56% and Q30 values more than 90.84%.
For PacBio HiFi long-read sequencing, the SMRTbell library was prepared with the SMRTbell Express template preparation kit 2.0 (Pacific Biosciences, Menlo Park, CA) and subsequently sequenced using the Sequel II Sequencing Kit 2.0 with SMRT Cell 8 M Tray on a PacBio sequel II instrument (Pacific Biosciences, Menlo Park, CA). In total, 34.41 Gb high-quality HiFi reads (34.85 × coverage) were obtained with an average length of 14.96 kb and an N50 length of 15.18 kb (Table 1).
The Hi-C library was generated using the restriction endonuclease Mbol following the standard protocol described previously20, which was sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) using a 150-bp paired-end strategy. A total of 163.62 Gb (165.72 × coverage) of raw data was generated.
Genome survey
To ensure data quality, adapter sequences and low-quality reads were removed with fastp v0.21.021. The resulting clean reads were used to generate a histogram of the 17-mer distribution with Jellyfish v2.2.7 with parameters ‘count -g generators -G 4 -s 5 G -m 17 -C -t 10’22 (Fig. 1), followed by calculation of genome heterozygosity. Based on these analyses, the estimated genome size was determined to be 987.35 Mb, with a heterozygosity of 0.80%.

The 17-mer analysis of the genome of Arma custos. The X-axis represents the k-mer depth. The Y-axis indicates the k-mer frequency for a given depth.
Genome assembly
The PacBio HiFi reads were utilized to assemble the genome into contigs using hifiasm v0.16.123. The assembled draft genome was polished by employing the genomic short-reads generated by Illumina NovaSeq 6000 sequencer with the NextPolish v1.4.024. To identify and remove potential contaminant sequences, Kraken2 was employed against a custom database25. A total of 137 contigs were identified as bacteria and subsequently eliminated. The resulting draft genome was 969.02 Mb with a contig N50 of 2.11 Mb, and the GC content of 33.18% (Table 2).
Hi-C scaffolding
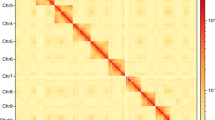
The raw HiC data were processed using Hi-C-Pro v2.8.026, followed by quality control with fastp v0.21.021. The resulting data were aligned to the draft genome assembly utilizing bowtie 2 v2.2.327 to obtain the uniquely mapped paired-end reads. Among the 8,661,026 reads, 4,330,513 reads were paired, with a total paired ratio of 38.70%. And a total of 1,470,719 reads were uniquely mapped to the genome, with an effect rate of 33.96%, representing valid interaction pairs. These valid interaction pairs were used to anchor the assembled contigs to near-chromosomal level using the Allhic v0.9.828. Then, juicebox v1.11.0829 was employed for manual correction based on chromosome interaction strength, ultimately resulting a chromosome-level genome. After curation, a total of 935.94 Mb of contigs, accounting for 96.58% of the assembled draft genome, were anchored into seven chromosomes, ranging from 77.33 Mb to 234.11 Mb (Table 3). The number of anchored chromosomes matched the result of chromosome karyotype analysis following the previously reported method30 (Fig. 2). The final genome exhibited an N50 of 135.75 Mb. A genome-wide chromatin interaction HiC heatmap was constructed using the ggplot2 software in the R package. According to the heatmap, all chromosomes were clearly distinguishable from each other (Fig. 3). The Advanced Circos tool implanted in TBtools v1.09876531 was used to visualize the landscape of the chromosomes (Fig. 4).

Karyotype analysis of Arma custos reveals a chromosome count of seven. The chromosomes from two nuclei are shown.

Heatmap of the Hi-C assembly of Arma custos. The interaction intensity of Hi-C links represents by colors shown in the left bar, ranging from yellow (low) to red (high).

Overview of the genome characteristics of Arma custos in a circos plot. (a), length of chromosomes at the Mb scale; (b), gene density per Mb; (c), CG content per Mb.
Genome annotation
A combined strategy of homology alignment and de novo search was applied to identify repetitive elements in the genome. Tandem repeats were detected using Tandem Repeats Finder (TRF) v4.0932. Repetitive elements homologous to those available in the Repbase28.0633 were identified with RepeatMasker v4.1.0 and RepeatProteinMask v4.1.034. In addition, a de novo repetitive elements database was generated using LTR_FINDER v1.0.635, RepeatScout v1.0.536, and RepeatModeler v2.0.137. The resulting repeat sequences with lengths greater than 100 bp and gap ‘N’ less than 5%, obtained from both two strategies, were combined to construct the raw transposable element library. This library was then processed by UCLUST algorithm38 to yield a non-redundant library, followed by DNA-level repeat identification using RepeatMasker v2.0.137. The results indicated that the genome contained 52.78% repetitive elements, most of which were long terminal repeat (LTR) retrotransposons, representing 40.05% of the genome (Table 4).
For non-coding RNA (ncRNAs) annotation, the transfer RNAs (tRNAs) were predicted using tRNAscan-SE v1.439. As ribosomal RNAs (rRNAs) are highly conserved, they were predicted by searching against selected rRNA sequences from closely related species as references using the BLAST v2.2.2640. Other ncRNAs, including micro RNAs (miRNAs) and small nuclear RNAs (snRNAs), were identified by searching against the Rfam database v14.141 using the Infernal v1.1.242. Overall, 20,337 tRNAs, 1,556 rRNAs, 2,790 miRNAs and 596 snRNAs were predicted, resulting in a total of 25,279 ncRNAs (Table 5).
A combined three-pronged strategy, involving de novo prediction, homology-based gene prediction, and transcriptome-based prediction, was employed to annotate the genes in the genome. De novo gene models were generated by multiple programs, namely Augustus v3.3.343, GlimmerHMM v3.0.444, SNAP v2013.11.2945, Geneid v1.446, and Genscan v1.047. For homology-based prediction, protein sets from five bugs including Halyomorpha halys48, Nesidiocoris tenui49, Oncopeltus fasciatus50, Rhodnius prolixus51 and Apolygus lucorum52 were downloaded from Insectbase 2.053 on January 3, 2022. These protein sets were aligned to the assembled genome using TblastN v2.2.2640 with an E-value threshold of ≤1e−5. The matching proteins from these bugs were used to predict the gene structure of the assembled genome with GeneWise v2.4.154. For transcriptome-based prediction, raw reads from five transcriptomic libraries were subjected to quality control with fastp v0.21.021. After eliminating adapter sequences and low-quality reads with Trimmomatic v1.455, clean data were assembled into transcripts using Trinity v2.11.056 and StringTie2 v2.1.657. The candidate coding regions in these transcripts were predicted using TransDecoder v5.5.056, which is implemented in the Trinity software. The resulting protein sequences were used to predict the gene structures following the procedure as described for homology-based prediction. In addition, the clean transcriptomic data were aligned to the assembled genome using HISAT2 v2.2.158 to identify the exons and splice sites, and these were used to extract the gene structures using PASA v2.4.159. A non-redundant reference gene set was generated by merging genes predicted by the three strategies with EVidenceModeler (EVM) v1.1.160. The gene models were further updated with PASA v2.4.159 to identify untranslated regions. Finally, the final comprehensive gene set was generated, resulting in a total of 13,708 protein-coding genes (Table 6). These genes had an average gene length of 25,698.42 bp. The average lengths of their coding sequence (CDS), exon, and intron length were 1,400.68 bp, 192.17 bp, and 3,863.69 bp, respectively. On average, each gene contained 7.29 exons (Table 7).
The annotation of the protein-coding genes was performed using BLAST v2.2.2640 against SwissProt and National Center for Biotechnology Information (NCBI) non-redundant (Nr) database with DIAMOND v2.2.2261, parameters used ‘-ultra-sensitive -max-target-seqs. 1 -evalue 1e−5’ with a threshold of E-value ≤ 1e−5. The motifs and domains present in the predicted proteins encoding by these genes were annotated using InterProScan v86.0 with parameters ‘-disable-precalc, -goterms, -pathways’ and Pfam62. Additionally, these genes were classified into functional categories based on KEGG63 and GO64 with a threshold of E-value ≤ 1e−5. Overall, 13,219 predicted genes were annotated using the databases of Nr, SwissProt, InterProScan, Pfam, KEGG and GO, representing 96.43% of the total gene set (Table 8).
Data Records
The raw data of Illumina short reads, PacBio HiFi long reads and Hi-C reads for assembling the genome of A. custos, as well as the transcriptome Illumina sequencing data for genomic annotation, have been deposited in the NCBI SRA (Sequence Read Archive) database under BioProject number PRJNA1001510. Illumina sequencing data for genome survey can be accessed and downloaded with accession number SRR2549817865. PacBio sequel II sequencing data for genome assembly can be accessed and downloaded with accession number SRR2550303466. Hi-C sequencing data can be accessed and downloaded with accession number SRR2551832167. Transcriptome sequencing data for genome annotation can be accessed and downloaded from NCBI SRA database (https://identifiers.org/ncbi/insdc.sra:SRP453032)68. The genome sequence has been deposited in Genbank under the accession number JBBAGI000000000 (https://www.ncbi.nlm.nih.gov/nuccore/JBBAGI000000000)69. The final chromosome assembly, genome structure annotation, amino acid sequences and CDS sequences data are available at the Figshare database (https://doi.org/10.6084/m9.figshare.25284943)70.
Technical Validation
The accuracy of the assembled genome was assessed using two methods. Firstly, the clean Illumina genomic short reads were aligned back to the genome by Burrows–Wheeler Aligner (BWA) v.0.7.12-r103971. Approximately 97.81% of the short reads were successfully aligned to the genome, providing a genome coverage of 99.95%. The heterozygous and homozygous nucleotide polymorphisms (SNPs) in the genome were 0.407191% and 0.00011%, respectively. The results indicate a high accuracy of the genome assembly. Secondly, the accuracy of the assembled genome was evaluated using Merqury v1.472. A quality value of 46.78 was obtained, affirming the base-level accuracy genome assembly. The completeness of the assembled genome was evaluated using three methods. Firstly, Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.4.7 (-l insecta_odb10 -m genome)73 was employed. The results showed that the complete and fragment scores were 99.34% and 0.22%, respectively. Among the retrieved complete single-copy genes, only 2.3% of them are duplicated. Secondly, Core Eukaryotic Genes Mapping Approach (CEGMA, v2.5)74 was employed. Among the 248 most highly conserved core eukaryotic genes (CEGs) within CEGMA, 230 CEGs were successfully assembled, accounting for 92.74%, and 222 CEGs were complete, accounting for 89.52%. Thirdly, LTR Assembly Index (LAI) was assessed using LTR_retriever v. 2.9.075, resulting in a value of 8.44. These results indicated a high level of completeness in the genome assembly.
Code availability
In this study, no custom scripts or command lines were utilized. All software employed for data processing and analysis are publicly available. The specific versions and parameters of each software are detailed in the Methods section. If no specific parameters were mentioned for a particular software, default parameters were used. The software was applied following the manuals and protocols provided by the respective bioinformatic tools.
References
Rider, D. A. & Zheng, L. Y. Checklist and nomenclatural notes on the Chinese Pentatomidae (Heteroptera) I, Asopinae. Entomotaxonomia 24, 107–115 (2002).
Zhao, Q., Wei, J., Bu, W., Liu, G. & Zhang, H. Synonymize Arma chinensis as Arma custos based on morphological, molecular and geographical data. Zootaxa 4455, 161–176 (2018).
Zou, D. Y. et al. Taxonomic and bionomic notes on Arma chinensis (Fallou) (Hemiptera: Pentatomidae: Asopinae). Zootaxa 3382, 41–52 (2012).
Pan, M., Zhang, H., Zhang, L. & Chen, H. Effects of starvation and prey availability on predation and dispersal of an omnivorous predator Arma chinensis Fallou. J. Insect Behav. 32, 134–144 (2019).
Zou, D. Y. et al. Performance and cost comparisons for continuous rearing of Arma chinensis (Hemiptera: Pentatomidae: Asopinae) on a zoophytogenous artificial diet and a secondary prey. J. Econ. Entomol. 108, 454–461 (2015).
Wang, W. L. et al. Preliminary observation of preyed ability of Arma chinensis (Fallou), a new natural enemy of Hyphantria cunea (Drury). Shandong For. Sci. Technol. 1, 11–14 (2012).
Tang, Y. T. et al. Predation and behaviour of Arma chinensis to Spodoptera frugiperda. Plant Protection 45, 65–68 (2019).
Liu, J., Liao, J. & Li, C. Bottom-up effects of drought on the growth and development of potato, Leptinotarsa decemlineata Say and Arma chinensis Fallou. Pest Manag. Sci. 78, 4353–4360 (2022).
Li, J. J. et al. Effects of three prey species on development and fecundity of the predaceous stinkbug Arma chinensis (Hemiptera: Pentatomidae). Chin. J. Biol. Control. 32, 552–561 (2016).
Wang, J. et al. Population growth performance of Arma custos (Faricius) (Hemiptera: Pentatomidae) at different temperatures. J. Insect Sci. 22, 12 (2022).
Liu, J., Liu, X., Liao, L. & Li, C., Biological performance of Arma chinensis on three preys Antheraea pernyi, Plodia interpunctella and Leptinotarsa decemlineata, Int. J. Pest Manag. https://doi.org/10.1080/09670874.2023.2216173, 1-8 (2023).
Guo, Y., Liu, C. X., Zhang, L. S., Wang, M. Q. & Chen, H. Y. Sterol content in the artificial diet of Mythimna separata affects the metabolomics of Arma chinensis (Fallou) as determined by proton nuclear magnetic resonance spectroscopy. Arch. Insect Biochem. Physiol. 96, e21426 (2017).
Wu, S. et al. Analysis of chemosensory genes in full and hungry adults of Arma chinensis (Pentatomidae) through antennal transcriptome. Front. Physiol. 11, 588291 (2020).
Zou, D. Y. et al. A meridic diet for continuous rearing of Arma chinensis (Hemiptera: Pentatomidae: Asopinae). Biol. Control 67, 491–497 (2013).
Zou, D. Y. et al. Performance of Arma chinensis reared on an artificial diet formulated using transcriptomic methods. Bull. Entomol. Res. 109, 24–33 (2019).
Zou, D. et al. Differential proteomics analysis unraveled mechanisms of Arma chinensis responding to improved artificial diet. Insects 13, 605 (2022).
Meng, J. Y., Yang, C. L., Wang, H. C., Cao, Y. & Zhang, C. Y. Molecular characterization of six heat shock protein 70 genes from Arma chinensis and their expression patterns in response to temperature stress. Cell Stress Chaperones 27, 659–671 (2022).
Yin, Y., Zhu, Y., Mao, J., Gundersen-Rindal, D. E. & Liu, C. Identification and characterization of microRNAs in the immature stage of the beneficial predatory bug Arma chinensis Fallou (Hemiptera: Pentatomidae). Arch. Insect Biochem. Physiol. 107, e21796 (2021).
Zou, D. et al. Nutrigenomics in Arma chinensis: transcriptome analysis of Arma chinensis fed on artificial diet and Chinese oak silk moth Antheraea pernyi pupae. PLoS One 8, e60881 (2013).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Cheng, H., Concepcion, G., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 1–13 (2019).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Shi, J. et al. Chromosome conformation capture resolved near complete genome assembly of broomcorn millet. Nat. Commun. 10, 464 (2019).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Imai, H. T., Taylor, R. W., Crosland, M. W. & Crozier, R. H. Modes of spontaneous chromosomal mutation and karyotype evolution in ants with reference to the minimum interaction hypothesis. Jpn. J. Genet. 63, 159–185 (1988).
Chen, C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Bergman, C. M. & Quesneville, H. Discovering and detecting transposable elements in genome sequences. Brief Bioinform. 8, 382–392 (2007).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 48 (2019).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–402 (1997).
Griffiths-Jones, S. et al. Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinformatics 18, 4.3.1–4.3.28 (2007).
Aggarwal, G. & Ramaswamy, R. Ab initio gene identification: Prokaryote genome annotation with GeneScan and GLIMMER. J. Biosci. 27, 7–14 (2002).
Sparks, M. E. et al. Brown marmorated stink bug, Halyomorpha halys (Stål), genome: putative underpinnings of polyphagy, insecticide resistance potential and biology of a top worldwide pest. BMC Genomics 21, 227 (2020).
Shibata, T. et al. High-quality genome of the zoophytophagous stink bug, Nesidiocoris tenuis, informs their food habit adaptation. G3 14, jkad289 (2024).
Panfilio, K. A. et al. Molecular evolutionary trends and feeding ecology diversification in the Hemiptera, anchored by the milkweed bug genome. Genome Biol. 20, 64 (2019).
Mesquita, R. D. et al. Genome of Rhodnius prolixus, an insect vector of Chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc. Natl. Acad. Sci. USA 112, 14936–14941 (2015).
Liu, Y. et al. Apolygus lucorum genome provides insights into omnivorousness and mesophyll feeding. Mol. Ecol. Resour. 21, 287–300 (2021).
Mei, Y. et al. InsectBase 2.0: a comprehensive gene resource for insects. Nucleic Acids Res. 50, D1040–D1045 (2022).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Kim, D., Paggi, J., Park, C., Bennett, C. & Salzberg, S. Graph-based genome alignment and genotyping with HISAT2 and HISAT genotype. Nat. Biotechnol. 37, 907–915 (2019).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Buchfink, B., Reuter, K. & Drost, H. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR25498178 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR25503034 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR25518321 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP453032 (2024).
Wang, Y. Q. & Zhu, J. Y. Arma custos isolate FDSW210240299, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBBAGI000000000 (2024).
Wang, Y. Q. et al. Chromosome-level genome assembly of the predatory stink bug Arma custos (Hemiptera: Pentatomidae). Figshare. https://doi.org/10.6084/m9.figshare.25284943 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, e323 (2021).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Acknowledgements
This work was supported by the Joint Special Key Project of Agricultural Basic Research in Yunnan Province (202101BD070001-024), the National Natural Science Foundation of China (32360686); the Xing Dian Talents Support Program of Yunnan Province to Shujun Wei, the National Science and Technology Innovation Talent Program in Forestry and Grassland for Young Top-notch Talents (2019132615), and the Funding for the Construction of First-Class Discipline of Forestry in Yunnan Province.
Author information
Authors and Affiliations
Contributions
J.Z. and S.W. conceived this study. Y.W., Y.L., Y.G. and C.W. collected the samples, and prepared DNA and RNA for sequencing. Y.W., Y.L. and Y.G. performed the experiments and analysed the data. Y.W. drew the figures. S.L., W.L. and C.W. assisted in data analysis. J.Z. drafted the manuscript. J.Z. and S.W. revised the manuscript. All authors read the final manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Luo, Y., Ge, Y. et al. Chromosome-level genome assembly of the predatory stink bug Arma custos. Sci Data 11, 417 (2024). https://doi.org/10.1038/s41597-024-03270-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03270-8
