
Abstract
Emergence of SARS-CoV-2 coronavirus has led to millions of deaths globally. We present three high-resolution crystal structures of the SARS-CoV-2 nsp14 N7-methyltransferase core bound to S-adenosylmethionine (1.62 Å), S-adenosylhomocysteine (1.55 Å) and sinefungin (1.41 Å). We identify features of the methyltransferase core that are crucial for the development of antivirals and show SAH as the best scaffold for the design of antivirals against SARS-CoV-2 and other pathogenic coronaviruses.
Similar content being viewed by others

Main
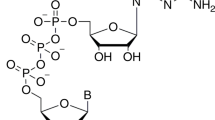
The SARS-CoV-2 mRNA is capped at the 5′-end by methyltransferases (MTases) nsp14 and nsp16 (ref. 1). Nsp14 methylates the N7 atom of guanosine to generate the N7MeGpppA2’OH-RNA structure, which is then methylated at the 2′O atom of the initiating nucleotide by nsp16 to make the N7MeGpppN2’OMe-RNA structure1. Both nsp14 and nsp16 use S-adenosylmethionine (SAM) as the methyl donor and generate S-adenosylhomocysteine (SAH) as the reaction byproduct. Nsp14 harbors an exoribonuclease domain (ExoN) at the N-terminus and the N7-MTase domain at the C-terminus (Fig. 1a)2,3,4. The nsp14 N7-MTase is an attractive target for the development of antivirals, but most structure-guided efforts thus far have depended on crystal structures of nsp14/nsp10 from SARS-CoV5,6, solved to 3.2–3.4 Å resolution2. Although the SARS-CoV-2 nsp14/nsp10 has been imaged by cryo-EM, the resolution of these structures is limited to 2.5–3.9 Å and they do not capture interactions with SAM, SAH or sinefungin (SFG)7,8. We employ here fusion protein-assisted crystallization9,10 and report high-resolution crystal structures of the nsp14 N7-MTase-TELSAM fusion (TEL-MTase; Fig. 1b) in complex with SAM, SAH and SFG (Supplementary Table 1).

a, Domain organization of SARS-CoV-2 nsp14 and nsp10. b, The overall structure of TELSAM-MTase fusion in complex with SAM shown in a ribbon representation. The nsp14 N7-MTase domain and TELSAM are colored in cyan and yellow, respectively. The secondary structure elements for the N7-MTase domain are labeled. The residues not modeled in the structure are shown by dashed lines. A zinc ion (Zn) is shown as a sphere and colored gray. c, Cα trace superposition of nsp14 N7-MTaseSAM, nsp14 N7-MTaseSAH and nsp14 N7-MTaseSFG.
The MTase core in the three structures is nearly identical, superimposing with root-mean-square deviations (RMSDs) between 0.085 and 0.09 Å for 187 Cα atoms, showing it to be essentially invariant when bound to SAM, SAH or SFG (Fig. 1c). The MTase core consists of an atypical Rossmann fold, composed of a central five stranded β-sheet (β1′, β2′, β3′, β4′ and β8′) instead of the seven stranded β-sheet (β1–β7) typically associated with class I MTases11, including those from most viruses. Helices α1′, α2′, α3′ and αC, β-strands βA and βB, and a Zn2+ coordinated substructure are located on one side of the β-sheet, and two short helices αA and αB on the other (Fig. 1b). SAM, SAH and SFG are located at the C-terminal ends of strands β1′, β2′ and β3′, and are cradled by loops between β1′ and β2′, β2′ and αA, and β3′ and β4′ (Fig. 2a–c).

a, Structure of nsp14 MTase domain bound to SAM (left), with a detailed view of the interactions between them (right). The Fo – Fc difference electron density for SAM is shown in a pink mesh and contoured at 3σ level. Hydrogen bonds between the MTase domain and SAM are depicted as dashed lines and the water molecules are shown as red spheres. b, Structure of nsp14 MTase bound to SAH (left), with a detailed view of the interactions between them (right). c, Structure of nsp14 MTase domain bound to SFG (left), with a detailed view of the interactions between them (right).
In the full-length nsp14 structures2,7,8, a characteristic of the MTase fold is a ‘hinge’ region composed of a three-stranded β-sheet (β5′, β6′ and β7′; residues 402–433) and an interdomain loop (residues 288–299) that precedes the MTase core (Extended Data Fig. 1a–c). The β-sheet extends from the MTase core and interacts with the ExoN domain, and flexibility of the hinge has been suggested to allow for the movement between the MTase core and the ExoN domain12. Intriguingly, this β-sheet is disordered in our three structures, suggesting that its interactions with the ExoN domain are required for its folding and stability (Extended Data Fig. 1a). Excluding the hinge region, the SARS-CoV-2 and SARS-CoV nsp14 N7-MTase cores superimpose with a RMSD of 0.67 Å for 183 Cα atoms. The most notable difference is in residues 467–482, which fold into helix αC and β-strand βB in SARS-CoV-2 nsp14 (Extended Data Fig. 1d).
The adenine base of SAM, SAH and SFG is ensconced in a cavity formed by the Ala353, Phe367, Tyr368, Cys387 and Val389 side chains, while the N1 and N6 atoms make hydrogen bonds with the backbone amide and carboxyl groups of Tyr368, respectively, and the N3 atom makes a hydrogen bond with the amide group of Ala353 (Fig. 2a–c). The ribose sugar makes direct hydrogen bonds with the Asp352 side chain, as well as water-mediated interactions with both the Gln354 side chain and main chain. Asp352 is conserved in coronaviruses and its mutation to alanine in SARS-CoV has been shown to abrogate N7-MTase activity2,3,13. The tail portion is fixed by numerous interactions, including direct hydrogen bonds with the Arg310 side chain and the Gly333 and Trp385 main chain atoms, as well as intricate water-mediated interactions with Gln313, Asp331 and Asn386 side chains and the Ile332 and Trp385 main chains (Fig. 2a–c). In addition, the Pro335 ring is involved in van der Waals contacts with the nonpolar portion (atoms Cβ and Cγ) of SAM/SAH/SFG. Arg310, Asp331 and Asn386 are conserved in coronaviruses, and their mutation to alanine in SARS-CoV has been shown to abolish N7-MTase activity2,3,13. Thus, although Asp331 is not involved in a direct hydrogen bond with the ligand, its interaction via a water molecule makes it crucial for N7-MTase activity2,3. Indeed, the entire nsp14 MTase-ligand interface is defined by an unusually large number of well-ordered water molecules that mediate hydrogen bonds between the ligand and the protein (Fig. 2a–c). Many of these are ‘good waters’ in that they bridge the MTase and SAM/SAH/SFG and can be considered as extensions of the MTase amino acids in the SAM/SAH/SFG binding pocket. Substitution of these water molecules will be an important feature to take into account in the design of SAM competitive inhibitors of the SARS-CoV-2 N7-MTase.
All the amino acids at the interface are conserved in the SARS-CoV nsp14 N7-MTase. The crystal structure of SARS-CoV nsp14/nsp10 with SAM captured a subset of the interactions (Extended Data Fig. 2), but some key interactions, such as between Arg310, Gln313, Asn386 and the terminal carboxylate group of SAM were not observed, possibly because of the moderate resolution of the structure. Also, the configuration of the bound SAM is different, wherein the donor methyl group points in the opposite direction to what we observe here (Extended Data Fig. 2). Most importantly, the limited resolution of the SARS-CoV structure did not allow for the observation of water molecules, which form a crucial part of the N7-MTase-SAM interface (Extended Data Fig. 2).
Interestingly, because of the interconnection between the MTase domain and the ExoN domain (Extended Data Fig. 3), the MTase activity of the nsp14 is influenced by noncatalytic mutations in the ExoN domain13,14,15. To explore this further, we expressed and purified just the SARS-CoV-2 MTase domain (residues 289–527). We find that the MTase activity of the isolated MTase domain and the TEL-MTase fusion is nearly identical, showing that addition of TELSAM to the MTase domain does not impact its activity (Extended Data Fig. 4). But, consistent with the previous mutational and deletional analysis of SARS-CoV nsp14 (ref. 13,14,15), the activity of MTase domain and the TEL-MTase is reduced in the absence of the ExoN domain (Extended Data Fig. 4). This is probably due to a stabilizing allosteric effect of one domain on the other, as mutations in the MTase domain have also been found to reciprocally effect the ExoN activity3.
From isothermal titration calorimetry (ITC) analysis, SARS-CoV-2 nsp14/nsp10 complex binds SAM and SFG with similar affinities (Kd of 5.7 μM versus 4.4 μM), but binds SAH substantially better (Kd of 0.3 μM) (Supplementary Table 2 and Extended Data Fig. 5). The MTase domain and the TEL-MTase fusion bind SAM/SAH/SFG in a similar pattern, though the absolute affinities (Kd of around 22–25 µM for SAM/SFG and Kd of 5 µM for SAH) are lower than those observed with full-length nsp14/10 (Extended Data Fig. 6). This further reinforces the notion that the ExoN domain has a stabilizing allosteric effect on the MTase domain and helps to increase its affinity for SAM/SAH/SFG. Importantly, the residues that interact between the two domains are distant from the SAM/SAH/SFG binding site (Extended Data Fig. 3).
How to explain the higher affinity of SAH compared with SAM or SFG? In the nsp14 MTaseSAM structure, the donor methyl group of SAM (attached to its Sδ atom) abuts the Asn386 main chain carbonyl and seems to displace a water molecule that would normally be coordinated to the main chain carbonyl (Fig. 2a). Indeed, in the nsp14 MTaseSAH structure, we observe a well-ordered water molecule coordinated to the Asn386 main chain carbonyl at a position that would be incompatible with the methyl group of SAM (Fig. 2b). The entry of this water molecule may provide a partial explanation for the higher affinity of SAH relative to SAM, particularly the more favorable enthalpic contribution to binding (Supplementary Table 2 and Extended Data Fig. 5). It is less clear, however, why SAH would bind better than SFG. The amino group of SFG (attached to its Cδ) makes a direct hydrogen bond with the Asn386 main chain carbonyl and would seem to compensate for the loss of a water molecule (Fig. 2c). Whether this hydrogen bond is less favorable enthalpically than a coordinated water molecule to the Asn386 main chain carbonyl is uncertain at present.
An attractive feature of SARS-CoV-2 N7-MTase as a drug target is its high conservation of sequence across other coronaviruses and nearly total conservation of sequence across all the strains of SARS-CoV-2 (Extended Data Fig. 7a,b). Interestingly, we find that the affinity of SAH for nsp14 is substantively better than for SAM or SFG; positing SAH as the scaffold of choice for the design of more potent SAM competitors. Indeed, when Devkota et al. added a nitrile group to position 7 of the adenine base of SAH, it further improved its potency and binding (Kd of 0.05 µM)6 and a bulky aromatic substituent at the same place led to single-digit nanomolar inhibitors16. Notably, the N7-MTase-SAM/SAH/SFG interface also contains a conserved cysteine (Cys387) at 3.9 Å and 4.6 Å from the N7 and N6 atoms of the adenine base, respectively (Fig. 2), allowing for a suitable ‘warhead’ on the adenine base to make a covalent bond with the conserved cysteine. Such covalent inhibitors have been designed previously for other MTases17, including one that forms a covalent bond with Cys449 in the active site of protein arginine methyltransferase 5 (PRMT5)18.
Overall, the high-resolution structures of SARS-CoV-2 nsp14 N7-MTase presented here will aid in the development of new antivirals against SARS-CoV-2 and other pathogenic coronaviruses.
Methods
Protein expression and purification
Full-length nsp14/10 complex
For ITC binding studies, a single pRSFDuet-1 plasmid bearing both C-terminal 6×His-tagged full-length nsp14 (NdeI and XhoI) and nsp10 (NcoI and NotI) was transformed into E. coli BL21Gold(DE3) cells (Agilent). The cells were grown at 37 °C until the culture reached an optical density OD600 of around 0.5, after which the temperature was reduced to 30 °C and ZnCl2 added at a final concentration of 20 µM. At an OD600 of around 0.8, the temperature was reduced to 15 °C and expression of the complex was induced by addition of 0.5 mM IPTG. The culture was incubated for 18 h at 180 r.p.m. The cells were harvested by centrifugation and resuspended in binding buffer (25 mM Tris pH 7.5, 250 mM NaCl, 10% glycerol, 0.01% IGEPAL, 25 mM imidazole, 10 µM ZnCl2 and 10 mM 2-mercaptoethanol). The cells were lysed by sonication in the presence of EDTA-free Pierce Protease Inhibitor tablets (Thermo Fisher) and 1 mM PMSF, and the cell debris were clarified by centrifugation. The filtered supernatant was loaded onto a HisTrap HP affinity column (GE Healthcare). The column was washed with the binding buffer to remove the nonspecific proteins bound to the column and the desired complex was eluted using the binding buffer with 500 mM Imidazole. The fractions containing the nsp14/nsp10 complex were concentrated and further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 (GE Healthcare) column, pre-equilibrated with 100 mM KH2PO4/K2HPO4 buffer pH 8.0, 100 mM KCl, 0.01% IGEPAL, 5 mM 2-mercaptoethanol and 10% glycerol. The fractions containing pure nsp14/nsp10 complex were concentrated and used for ITC without freezing.
MTase domain
pGEX-6p-1 plasmid containing N-terminal GST tagged MTase domain (residues 289–527) was transformed into the E. coli C41(DE3) cells. The cells were grown at 37 °C until the OD600 reached around 0.8 and then the temperature was reduced to 18 °C and 0.5 mM IPTG and 20 µM ZnCl2 added. The cells were harvested 18 h postinduction and resuspended in a GST-binding buffer (25 mM Tris pH 7.5, 500 mM NaCl, 10% glycerol, 0.01% IGEPAL, 10 µM ZnCl2 and 2 mM DTT). After sonication, the supernatant was incubated with Glutathione Sepharose 4B beads (GE Healthcare) and washed with the GST-binding buffer to remove the nonspecific proteins. PreScission Protease was then added to the column and incubated overnight at 4 °C. The released MTase domain was collected as flowthrough. The fractions containing pure MTase domain were combined and the protein was further subjected to size exclusion chromatography.
TEL-MTase
Our efforts to crystallize various constructs and mutants of the nsp14/nsp10 complex and the N7-MTase domain alone (with and without various expressions tags and protein fusions such as green fluorescent protein (GFP)) were unsuccessful. Reports on the fusion of TELSAM with target proteins to improve their crystallization9,10 motivated us to fuse the nsp14 MTase domain (residues 300–527) with TELSAM (residues 47–124) with different linkers (A, PA and PAA) and we carried out expression and protein purification as follows. The pRSFDuet-1-smt3 plasmids containing N-terminal 6×His-SUMO-TELSAM-MTase (TEL-MTase) were transformed into E. coli BL21Gold (DE3) cells. The cells were grown at 37 °C until OD600 reached 0.8. The temperature was reduced to 15 °C and IPTG and ZnCl2 added to final concentrations of 0.5 mM and 20 µM, respectively. The cells were harvested 18 h postinduction and resuspended in the binding buffer (25 mM Tris pH 7.5, 500 mM NaCl, 10% glycerol, 0.05% IGEPAL, 30 mM imidazole, 10 µM ZnCl2 and 10 mM 2-mercaptoethanol) in the presence of EDTA-free Pierce Protease Inhibitor tablets (Thermo Fisher) and 1 mM PMSF. The cells were lysed by sonication and the filtered supernatant was loaded onto a HisTrap HP affinity column (GE Healthcare). The column was washed with the binding buffer containing 1 M NaCl to remove nonspecific proteins bound to the column. The column was then re-equilibrated with binding buffer and Ulp1-Protease was added to the column to cleave the 6×His-SUMO tag. The cleaved protein was eluted and the fractions containing the 1TEL-MTase fusion protein were diluted to a final concentration of 50 mM NaCl and loaded onto a 5 ml HiTrap Q HP anion-exchange column (GE Healthcare). The protein was eluted in the unbound fractions and was further purified by size exclusion chromatography on a HiLoad 16/600 Superdex 200 (GE Healthcare) column using 25 mM Tris pH 8.3, 200 mM KCl and 2 mM TCEP. All of the purified proteins were concentrated and stored in −80 °C. For ITC studies, the size exclusion buffer was 100 mM KH2PO4/K2HPO4 buffer pH 8.0, 100 mM KCl, 0.01% IGEPAL, 5 mM 2-mercaptoethanol and 10% glycerol.
MTase activity assays
The MTase activity was measured using the MTase-Glo Methyltransferase bioluminescence assay (Promega)19 following the manufacturer’s instructions. The reaction mix containing 20 mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 0.1 mg ml–1 BSA, 1 mM DTT, 20 µM protein (nsp14/10, MTase domain or TEL-MTase), 20 µM SAM and 0.15 mM G(5′)ppp(5′)A RNA cap analog (NEB, S1406S) were incubated for 1 h at room temperature. The detection solution from the kit was then added, and the mixture was further incubated for 30 min at room temperature, before the addition of the developing solution. Luminescence was measured by using a TECAN infinite 200Pro microplate reader. The averages and the s.d. of three measurements were plotted as a histogram using Origin v.7.0.
Isothermal titration calorimetry
The titrations were performed on a Microcal ITC200 instrument at 25 °C with the standard 10 µcals s–1 reference power and at 600 r.p.m. The ligand SAM/SAH/SFG was loaded in the syringe (400 µM) and titrated into 40 µM of nsp14/nsp10 complex in the cell. For the MTase domain and TEL-MTase, concentrations of the protein and the ligands were 60 µM and 600 µM, respectively. Care was taken to ensure buffer match for the ligand and proteins to eliminate heat production due to the buffer mismatch. The titrations consisted of 15 injections of 2.5 µl ligand solution at a rate of 0.5 µl s–1 at 180 s time intervals. An initial injection of 0.4 µl was made and discarded during data analysis. The data were fit to a single binding site model using the Origin v.7.0 software, supplied by MicroCal. All the experiments were repeated twice and average value reported.
Crystallization and structure determination of TEL-MTase with ligands
Crystallization trials for all the constructs were carried out at 15 mg ml–1 with fivefold molar excess of the ligand (SAM, SAH and SFG). Initial screens were set up with Oryx Nano (Douglas Instruments) at 20 °C using commercially available screens in a sitting drop format with 0.3 µl of protein mixed with equal volume of reservoir solution. Among the three fusion constructs, only the fusion construct with a PAA linker produced initial hits. Initial crystals were observed in solutions containing 15% reagent alcohol, 0.2 M lithium sulfate and 0.1 M sodium citrate pH 5.5 in 2 days. The crystals were further optimized by varying both concentration of the reagent alcohol and also the pH of the buffer in hanging drop format using 1 µl protein with 1 µl reservoir. The crystals were cryoprotected in a stepwise manner with reservoir solutions containing 5–30% glycerol and flash-cooled in liquid nitrogen. X-ray diffraction data were collected at the NSLS-II 17-ID-1 and 17-ID-2 beamlines at the Brookhaven National Laboratory under cryogenic conditions.
The diffraction data were processed using DIALS and AIMLESS in the CCP4 suite20,21. The experimental data showed significant anisotropy and an anisotropic correction was performed using the STARANISO server (https://staraniso.globalphasing.org/cgi-bin/staraniso.cgi) with a surface threshold of I/σ(I) ≥ 1.2. The structure was solved by molecular replacement with Phaser-MR22 using the MTase domain of SARS-CoV (PDB-5C8T2) and TELSAM domain from PDB-7N1O10 as search models. Subsequent iterative manual building and refinement were performed with Coot and Phenix Refine, respectively23,24. Ligand restraint file for SFG was generated using eLBOW25 from the PHENIX suite. All molecular graphic figures were prepared using PyMOL (Schrödinger LLC).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Atomic coordinates and structure factors for TELSAM-MTase-SAM, TELSAM-MTase-SAH and TELSAM-MTase-SFG have been deposited in the Protein Data Bank under the accession codes of 7TW7, 7TW8 and 7TW9, respectively. Source data are provided with this paper.
References
V’Kovski, P., Kratzel, A., Steiner, S., Stalder, H. & Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170 (2021).
Ma, Y. et al. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl Acad. Sci. USA 112, 9436–9441 (2015).
Ogando, N. S. et al. Structure-function analysis of the nsp14 N7-guanine methyltransferase reveals an essential role in Betacoronavirus replication. Proc. Natl Acad. Sci. USA 118, e2108709118 (2021).
Lin, S. et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 49, 5382–5392 (2021).
& Gorgulla, C. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. iScience 24, 102021 (2021).
Devkota, K. et al. Probing the SAM binding site of SARS-CoV-2 Nsp14 in vitro using SAM competitive inhibitors guides developing selective bisubstrate inhibitors. SLAS Discov. 26, 1200–1211 (2021).
Liu, C. et al. Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science 373, 1142–1146 (2021).
Yan, L. et al. Coupling of N7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell 184, 3474–3485 e11 (2021).
Nauli, S. et al. Polymer-driven crystallization. Protein Sci. 16, 2542–2551 (2007).
Nawarathnage, S. et al. Crystals of TELSAM-target protein fusions that exhibit minimal crystal contacts and lack direct inter-TELSAM contacts. Open Biol. 12, 210271 (2022).
Schubert, H. L., Blumenthal, R. M. & Cheng, X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 (2003).
Ferron, F. et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl Acad. Sci. USA 115, E162–E171 (2018).
Chen, Y. et al. Structure-function analysis of severe acute respiratory syndrome coronavirus RNA cap guanine-N7-methyltransferase. J. Virol. 87, 6296–6305 (2013).
Jin, X. et al. Characterization of the guanine-N7 methyltransferase activity of coronavirus nsp14 on nucleotide GTP. Virus Res. 176, 45–52 (2013).
Chen, Y. et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl Acad. Sci. USA 106, 3484–3489 (2009).
Otava, T. et al. The structure-based design of SARS-CoV-2 nsp14 methyltransferase ligands yields nanomolar inhibitors. ACS Infect. Dis. 7, 2214–2220 (2021).
Ferreira de Freitas, R., Ivanochko, D. & Schapira, M. Methyltransferase inhibitors: competing with, or exploiting the bound cofactor. Molecules 24, 4492 (2019).
Lin, H. et al. Discovery of potent and selective covalent protein arginine methyltransferase 5 (PRMT5) inhibitors. ACS Med. Chem. Lett. 10, 1033–1038 (2019).
Hsiao, K., Zegzouti, H. & Goueli, S. A. Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 8, 321–339 (2016).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Winter, G. et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 74, 85–97 (2018).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 (2009).
Acknowledgements
We thank the staff at the National Synchrotron Light Source II (NSLS-II) beamlines 17-ID-1 and 17-ID-2 for facilitating X-ray data collection. This work was funded by grant R35-GM13170 (A.K.A) from the National Institutes of Health (NIH). NSLS-II is a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for BioMolecular Structure (CBMS) at NSLS-II is primarily supported by the NIH, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010).
Author information
Authors and Affiliations
Contributions
J.K. and A.K.A. designed the experiments. J.K. performed all of the experiments. O.R. assisted in X-ray data collection and methylation assays. R.Q.-F. assisted in MTase domain purification and methylation assays. D.S. assisted in expression of the MTase domain. A.K.A. guided the project. A.K.A. and J.K. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Evzen Boura and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Beth Moorefield and Florian Ullrich, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Structural comparison of nsp14-N7-MTases.
a. Structure of SARS-CoV-2 nsp14-N7-MTaseSAM complex. b. Cryo-EM structure of SARS-CoV-2 nsp14/nsp10 heterodimer (PDB:7N0D, chain H). c. Crystal structure of SARS-CoV nsp14/nsp10GpppA:SAH complex (PDB:5C8S, chain B). The nsp14 MTase core, ExoN domain, and the hinge region are colored cyan, slate blue and orange, respectively. The nsp10 subunit is colored purple. d. Cα trace superposition of the MTase domains of SARS-CoV-2 (cyan) and SARS-CoV (PDB: 5C8T, chain B, orange). The bound SAM is also shown.
Extended Data Fig. 2 Comparison of atomic binding details of nsp14-N7-MTase.
a. Atomic binding details of SARS-CoV-2 nsp14-N7-MTase core (left) and previously reported SARS-CoV nsp14/nsp10 (PDB: 5C8T, chain B, right) with bound SAM. Hydrogen bonds are depicted as dashed lines and water molecules are shown as red spheres. b. Fo-Fc difference density map for SAM in the SARS-Cov-2 N7-MTaseSAM (left) and SARS-CoV nsp14/10SAM structures (right) contoured at 3σ level.
Extended Data Fig. 3 Interactions between the MTase domain and ExoN domain.
The nsp14 MTase core, ExoN domain, and the hinge region are colored cyan, slate blue and orange, respectively (PDB:7N0D). ExoN stabilizes the MTase domain via polar and non-polar interactions with the hinge and N7-MTase core.
Extended Data Fig. 4 Methyltransferase assay.
The methyltransferase activity of the full length nsp14/10 complex compared with the TEL-MTase fusion and the MTase domain alone. Data represents the mean ± SD of three independent reactions.
Extended Data Fig. 5 ITC analysis of nsp14/10 complex with SAM, SAH and SFG.
ITC titration data for SAM (left), SAH (middle) and SFG (right) with the nsp14/nsp10 complex are shown. The equilibrium dissociation constants (KD) were derived from the resulting binding isotherms. The chemical structures of each ligand are also shown.
Extended Data Fig. 6 ITC analysis of TEL-MTase and MTase domain with SAM, SAH and SFG.
ITC titration data for SAM (left), SAH (middle) and SFG (right) with the TEL-MTase fusion (a) and the MTase domain alone (b). The equilibrium dissociation constants (KD) were derived from the resulting binding isotherms.
Extended Data Fig. 7 Sequence comparison.
a. Sequence alignment of the nsp14 N7-MTase domain from 7 pathogenic coronaviruses. The identical and similar residues are highlighted in dark gray and light gray, respectively. b. Sequence alignment of the nsp14 N7-MTase domain of SARS-CoV-2 and its variants. The secondary structure elements are shown above the sequence alignment. Red triangles highlight residues that interact with SAM. Blue circles highlight residues that coordinate the zinc ion.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2.
Source data
Source Data Extended Data Fig. 4
Raw data for the methyltransferase assay.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kottur, J., Rechkoblit, O., Quintana-Feliciano, R. et al. High-resolution structures of the SARS-CoV-2 N7-methyltransferase inform therapeutic development. Nat Struct Mol Biol 29, 850–853 (2022). https://doi.org/10.1038/s41594-022-00828-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-022-00828-1
