
Abstract
Photosynthetic organisms have developed various light-harvesting systems to adapt to their environments1. Phycobilisomes are large light-harvesting protein complexes found in cyanobacteria and red algae2,3,4, although how the energies of the chromophores within these complexes are modulated by their environment is unclear. Here we report the cryo-electron microscopy structure of a 14.7-megadalton phycobilisome with a hemiellipsoidal shape from the red alga Porphyridium purpureum. Within this complex we determine the structures of 706 protein subunits, including 528 phycoerythrin, 72 phycocyanin, 46 allophycocyanin and 60 linker proteins. In addition, 1,598 chromophores are resolved comprising 1,430 phycoerythrobilin, 48 phycourobilin and 120 phycocyanobilin molecules. The markedly improved resolution of our structure compared with that of the phycobilisome of Griffithsia pacifica5 enabled us to build an accurate atomic model of the P. purpureum phycobilisome system. The model reveals how the linker proteins affect the microenvironment of the chromophores, and suggests that interactions of the aromatic amino acids of the linker proteins with the chromophores may be a key factor in fine-tuning the energy states of the chromophores to ensure the efficient unidirectional transfer of energy.
Similar content being viewed by others

Main
Light absorption is the first step of photosynthesis. The membrane-extrinsic soluble phycobilisomes (PBSs) are responsible for the majority of light capture in cyanobacteria and red algae2,3,4. PBSs are composed of phycobiliproteins (PBPs) and linker proteins2, and sunlight is absorbed and the energy transferred by open-chain tetrapyrrole chromophores that covalently bind to PBPs and some linker proteins2,6. A heterodimer of two different PBP subunits (α- and β subunits) assembles into a ring-shaped (αβ)3 trimer, which serves as the basic unit for the PBS assembly2. The typical PBS consists of several peripheral rods surrounding the central core1,7. Solar photonic energy absorbed by the peripheral rods can be rapidly funnelled to the core1 and eventually to the terminal emitters—chromophores of the core–membrane linker protein (LCM)8,9,10 or allophycocyanin D (ApcD)11,12—then transferred to photosystems I and II2,10,13,14.
Four morphological types of PBS have been observed: hemidiscoidal15,16,17,18,19,20, hemiellipsoidal21,22, block-type23 and bundle-type24. We recently solved the structure of the block-shaped PBS from the red alga G. pacifica at 3.5 Å resolution, which provided the detailed architecture of the intact PBS5. However, to our knowledge there have been no high-resolution structures reported for other morphological types of PBS. Moreover, although we determined the locations of all chromophores of the G. pacifica PBS, owing to resolution limitations we could not reveal how the energies of the bilins are modulated by their surroundings. To address these questions, we resolved the structure of a typical hemiellipsoidal-shaped PBS21,22,25 from Porphyridium purpureum, one of the few unicellular red algae and a widely used model alga26,27,28. The resulting structure, determined by cryo-electron microscopy (cryo-EM) at 2.82 Å resolution, reveals how the linker proteins affect the microenvironments of chromophores.
Overall structure
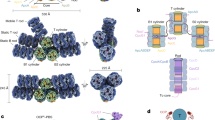
Intact PBSs were purified from P. purpureum and their subunit composition and spectroscopic properties are shown in Extended Data Fig. 1. The overall resolution of the resulting structure is 2.82 Å, with a higher resolution of 2.68 Å for the core region (Fig. 1a, Extended Data Fig. 2, Extended Data Table 1). Some long loops in LCM, which are absent in the electron microscopy map of the G. pacifica PBS, are clearly resolved in this reconstruction (Fig. 1b). The different types of bilins can be unambiguously assigned on the basis of the densities and the dihedral angles in combination with the results of published biochemical analysis29,30,31,32,33,34,35 that classified the phycocyanin in P. purpureum as R-phycocyanin, containing one phycoerythrobilin (PEB) and one phycocyanobilin (PCB) on the β subunit34 (Fig. 1c, Extended Data Fig. 3). In total, we built 706 protein subunits comprising 528 phycoerythrin subunits, 72 phycocyanin subunits, 46 allophycocyanin subunits and 60 linker proteins, and we assigned 1,598 chromophores (Extended Data Table 2a).

a, Local resolution map of the PBS from P. purpureum. The map was estimated with ResMap and generated in Chimera. b, The density (mesh) for the linker protein LCM superimposed with its atomic model (cartoon). Three loops (red)—V77 to A98, F132 to M145 and L656 to L687—are clearly resolved. c, The densities (mesh) of the representative PCB, PEB and phycourobilins (PUB) bilins (stick representation) show their different coplanarities. d, Overall structure of the PBS shown in surface representation. The rods are shown in different colours, and the core is coloured green. All extra hexamers, individual monomers and β subunits, and linker proteins are coloured light green, wheat and red, respectively. The superimposed structure of the G. pacifica PBS is coloured grey. e, Schematic showing the distribution of the individual phycoerythrin (αβ) monomers and phycoerythrin β subunits. One half (M1 and S1–S10) are coloured wheat and the other half are grey. f, Structures of all linker proteins shown in cartoon representation, from the same view as in d. They are the same colours as the hexamers in which they are located. The superimposed structures of linker proteins of the G. pacifica PBS are coloured grey.
The two-fold symmetric PBS resembles an opened fan from the face view and has an oval outline from the top view, with approximate dimensions of 610 Å (length), 390 Å (height) and 380 Å (thickness) (Fig. 1d). It contains 14 peripheral rods surrounding a pyramidal-shaped core (Extended Data Fig. 4a). The core contains one top cylinder (B) formed by two allophycocyanin trimers stacked back to back, and two bottom cylinders (A and A′), each of which is assembled by one (αβ)6 allophycocyanin hexamer and one allophycocyanin trimer (Extended Data Fig. 4b). The rods are categorized into two types according to their PBP composition: type I rods (Ra/Ra′, Rb/Rb′ and Rc/Rc′) are composed of both phycoerythrin and phycocyanin, whereas type II rods (Rd/Rd′, Re/Re′, Rf/Rf′ and Rg/Rg′) are composed entirely of phycoerythrin (Extended Data Fig. 4d). Except for rods Rf/Rf′ and Rg/Rg′, each of which contains two phycoerythrin hexamers in both PBSs, the number of phycoerythrin hexamers in each of the remaining rods is one fewer in the P. purpureum PBS than in the G. pacifica PBS (Extended Data Fig. 4c, d). The P. purpureum PBS also contains extra phycoerythrin hexamers; however, the hexamers He/He′, which are located near the surface of the outermost hexamer of rods Rb/Rb′ and Rc/Rc′ in the G. pacifica PBS, are absent owing to the short lengths of these rods in the P. purpureum PBS (Fig. 1d, Extended Data Fig. 4c). There are 2 individual phycoerythrin (αβ) monomers M1 (M1′) and 20 individual phycoerythrin β subunits S1–S10 (S1′–S10′) interspersed throughout the whole PBS (Fig. 1e). These components fill the empty spaces outside the rods, core and extra hexamers, and may stabilize the PBS. The P. purpureum PBS is aligned well with the G. pacifica PBS, but has a smaller size owing to the reduced number of phycoerythrin hexamers, indicating a similar overall organization of the rods and core (Fig. 1d, Extended Data Fig. 4c). The molecular mass of the P. purpureum PBS is 14.7 MDa, which is less than that of the G. pacifica PBS (18.0 MDa) after considering the molecular masses of the chromophores.
The skeleton formed by the linker proteins is very similar in the PBSs from both P. purpureum and G. pacifica (Fig. 1f), and both contain 17 types of linker protein. Superimposing the two PBSs indicate that they share very high structural conservation, except for the rod linker protein LR6 (Extended Data Fig. 4e). LR6 of the P. purpureum PBS contains the Pfam00427 domain, instead of the CBDγ domain that is present in LRγ6 of the G. pacifica PBS; this is in accordance with the overall tendency for the P. purpureum PBS to contain fewer bilins than the G. pacifica PBS. The roles of linker proteins in the assembly of the PBS—such as the sequential interactions between them to form the skeleton, the extensive contacts between them and the hexamers, and the α-helix-mediated interactions between LRC proteins and the core—are common between these two PBSs, highlighting their evolutionary conservation (Extended Data Fig. 5).
There are 120 PCBs, 1,430 PEBs and 48 phycourobilins in P. purpureum PBS (Extended Data Table 2a). The phycourobilin content in the P. purpureum PBS is considerably lower than that in the G. pacifica PBS; this is because all phycourobilins in the P. purpureum PBS originate solely from the LRγ proteins, whereas in the G. pacifica PBS—besides the LRγ proteins—all phycoerythrin β subunits also contain phycourobilin36. The lower phycourobilin content and the reduced number of total bilins in P. purpureum PBS are consistent with the fact that P. purpureum live at the sea surface37, where the light intensity is higher compared with that beneath the sea surface where G. pacifica are found37.
Interactions of LRγs with chromophores
The (αβ)3 trimers of the phycocyanins, phycoerythrins and allophycocyanins have very similar ring-like structures, in which the central cavity is a common feature. Three β82 chromophores are located near to the inner cavity and are subjected to interactions with linker proteins38 (Fig. 2a). The trimers Rc3I and Rd3I—from the type I rod Rc and the type II rod Rd, respectively—are used here to illustrate how the rod linkers LRγ4 and LRγ5 interact with the β82 chromophores (Fig. 2). Each of the β82 PEBs of Rc3I (denoted \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{1}^{82}\), \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{2}^{82}\) and \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{3}^{82}\)) is bound by two hydrogen bonds formed between the nitrogen atoms of the pyrrole rings B and C and the carboxyl group of the D85 residue of the β subunit (Fig. 2b–d), in agreement with the crystal structure of R-phycoerythrin39. In particular, three aromatic residues of LRγ4 (F80, F139 and F124) are located close to rings D of \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{1}^{82}\), \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{2}^{82}\) and \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{3}^{82}\), respectively, which will stabilize ring D and may expand the conjugation of the system owing to π–π interactions (Fig. 2b–d). Moreover, an extra PEB from LRγ4 \(({{\rm{\gamma }}}_{{{\rm{L}}}_{{\rm{R}}}{\rm{\gamma }}4}^{152})\) is adjacent—and very close—to the chromophore \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{2}^{82}\): the distance between their nearest two atoms is only 2.9 Å (Fig. 2b). Therefore, the chromophore pair may further downgrade the energy level of \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{2}^{82}\) owing to excited-state coupling39,40, with the result that \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{2}^{82}\) is probably at a lower energy level compared with that of \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{1}^{82}\) and \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{3}^{82}\). Notably, the trimer Rd3I in the type II rod Rd displays similar structural features: one aromatic residue is close to each of the β82 PEBs, and an additional bilin from the linker LRγ5 \(({{\rm{\gamma }}}_{{{\rm{L}}}_{{\rm{R}}}{\rm{\gamma }}5}^{135})\) also resides close to the \({}^{{\rm{Rd3I}}}{{\rm{\beta }}}_{2}^{82}\) (Extended Data Fig. 6a–d). Moreover, structural superimposition reveals that the LRγ linker proteins in the outmost hexamers of various rods of PBSs from both P. purpureum and G. pacifica also have similar structures (Extended Data Fig. 7a, b). These key aromatic residues, and the cysteine residues that are used to link the bilins, are well conserved in red algae (Extended Data Fig. 7c). The structural features of the interaction between LRγ and β82 PEBs therefore suggest that \({}^{{\rm{3I}}}{{\rm{\beta }}}_{2}^{82}\) PEB is in the lowest energy state among the three β82 PEBs, and that energy migration through \({}^{{\rm{3I}}}{{\rm{\beta }}}_{2}^{82}\) PEB could be the major route by which to pass energy through the rod.

a, Bottom, overall structure of the rod Rc with the hexamers shown in surface representation and the linker proteins shown in cartoon representation. Top, structure of the layer Rc3I. Proteins and bilins are shown in cartoon and sphere representations, respectively. Three β subunits are coloured differently and the β82 PEBs are boxed and analysed in detail in b–d. b, The interactions between the residue F80 and the bilin \({{\rm{\gamma }}}_{{{\rm{L}}}_{{\rm{R}}}{\rm{\gamma }}4}^{152}\) from LRγ4 with the bilin \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{2}^{82}\). c, The interaction between F139 from LRγ4 and the bilin \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{1}^{82}\). d, The interaction between F124 from LRγ4 and the bilin \({}^{{\rm{Rc3I}}}{{\rm{\beta }}}_{3}^{82}\). e, A focused view of the structure of the layer Rc1I showing the central triangle area. PBPs, the linker protein, bilins and residues are shown in surface, cartoon, ball-and-stick and stick representations, respectively. Two β82 PCBs are boxed and analysed in detail in f and g. f, The strong interaction between H58 from LRC1 and the bilin \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\). g, The interaction between Y104 from LRC1 and the bilin \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{1}^{82}\). h, \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\) has the shortest distance (33.0 Å, red line) between the rod and the core compared to \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{1}^{82}\) (57.1 Å, grey line) and \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{3}^{82}\) (55.2 Å, grey line).
Interactions of LRCs with chromophores
Energy is then transferred along the rods to the triangular area of the core-proximal hexamer (Fig. 2e). Two types of rod use different rod–core linkers to associate with the core. For Rc—which uses the linker LRC1c—a heterocyclic residue (H58) from LRC1c is located close to \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\), with a minimum distance of 2.8 Å (Fig. 2f). The pyrrole group of H58 can form a strong π–π interaction with rings B and C of \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\) (ref. 41). However, just one aromatic residue (Y104) from LRC1c forms a relatively weak π–π interaction with ring D of \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{1}^{82}\)—because of the longer distance (4.3 Å) compared with that between H58 and \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\)—and no aromatic residues interact with \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{3}^{82}\) (Fig. 2e, g). Therefore, modified by the specific surroundings, \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\) may be in the lowest-energy state among the three β82 chromophores. Moreover, \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{2}^{82}\) has the shortest distance to the core compared with \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{1}^{82}\) and \({}^{{\rm{Rc1I}}}{{\rm{\beta }}}_{3}^{82}\) (Fig. 2h), which further suggests that it may act as an energy-transit station, converging the energy absorbed by the rod and transferring it to the core. Similar situations are found for another two rods of type I and the three type I rods in G. pacifica (Extended Data Fig. 7d). This histidine residue is conserved completely across different red algal and cyanobacterial species, which is indicative of its functional importance (Extended Data Fig. 7e).
For Rd—which uses LRC2—two aromatic residues from LRC2 form parallel-displaced and T-shaped π–π interactions with \({}^{{\rm{Rd1I}}}{{\rm{\beta }}}_{2}^{82}\), respectively (Extended Data Fig. 6f). By contrast, each of another two β82 PEBs interacts with only one aromatic residue (Extended Data Fig. 6g, h). Superimposition of the LRC proteins (LRC2 and LRC3) of type II rods from both P. purpureum and G. pacifica shows that two such aromatic residues exist in all cases (Extended Data Fig. 7f). Sequence alignment also shows that these aromatic residues are conserved, which suggests their importance in fine-tuning the energies of the bilins (Extended Data Fig. 7g).
Key chromophores in the core
Previous studies have shown that three PCB chromophores in ApcD, ApcF and the α subunit of LCM (\({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\), \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) and \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\)) perform critical functions in energy transfer in the core. However, why each of these chromophores have unique functions remains to be clarified. Here we use our high-resolution structure to analyse the immediate surroundings of these key core chromophores in their native states.
Functionally, ApcD is the main protein responsible for energy transfer to photosystem I13. In our structure, two aromatic residues—W87 from ApcD and Y73 from the β subunit of the core trimer A3—form T-shaped and parallel-displaced π–π interactions with \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\), respectively; this enhances the tight fitting of ring D (Fig. 3a), which is consistent with the crystal structure of ApcD from Synechocystis PCC 6803 (PDB: 4PO5)12. Notably, we observed that W87 was surrounded by R83 and R90 from ApcD and Y73 from the core A3, which provided two cation–π interactions and one T-shaped π–π interaction to W87, respectively (Fig. 3a). It can therefore be inferred that the presence of these three residues is necessary to stabilize the orientation of W87, which is critical for the conformation of \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\). In addition, more π-related interactions between residues and \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\) are extracted from the high-resolution structure of the entire PBS. The cationic side chain of R83 extends to the top of ring C of \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\), forming the cation–π interaction42. F59 and Y65 may contribute two additional π–π interactions to ring A of \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\) (Fig. 3a). We then superimposed ApcDs from P. purpureum, G. pacifica and Synechocystis PCC 6803 with the α subunit of the core A3, and found that W87, F59 and Y65 are common to all ApcD molecules, although Y65 was replaced by V65 in the A3 α subunit (Extended Data Fig. 8a).

a, The surrounding residues of the bilin \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\). The residues and bilin are shown in stick and ball-and-stick representations, respectively. b, The interactions between \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) and ApcF. The residues and bilin are shown in stick and ball-and-stick representations, respectively. c, \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) is buried by a contiguous hydrophobic cap formed by the linker protein LCM. LCM is shown in cartoon representation in red and the cap is displayed in surface representation. \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) is shown in sphere representation. d, The interactions between the cap and \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\). The hydrophobic residues in the cap are shown in stick representation in red.
ApcF plays a crucial role in energy migration to the terminal chromophore of LCM14,43. Analysis of the PCB pocket of ApcF showed that the positively charged R89 formed one cation–π interaction with ring C of \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\), and Y93 and Y97 formed one T-shaped π–π and one parallel-displaced π–π interaction with ring D, respectively. Moreover, R89, Y93 and Y97 interact with each other by either cation–π or π–π interactions (Fig. 3b). The superimposition of ApcF molecules from P. purpureum and G. pacifica with the β subunit from the core A2 shows that these three residues exist in the same position in all proteins, suggesting their importance for the stability of the PCB (Extended Data Fig. 8b). Except for these common features, an aromatic residue from ApcF (F60) is located above ring A of \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) (Fig. 3b); an aromatic residue was also found in the ApcF of G. pacifica (Y60), whereas this residue is replaced by L60 in other β subunits (Extended Data Fig. 8b). This aromatic residue may therefore form additional π–π interactions with \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) and hence lower its energy. Another notable feature in our structure of the complete PBS is that LCM is directly involved in the interaction with \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\). Several hydrophobic residues of LCM are located at the ApcF/LCM interface and within 4 Å of \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\)—this creates a contiguous hydrophobic ‘cap’ that buries \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) (Fig. 3c, d, Extended Data Fig. 8c), which can enhance the stability of the conformation of \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\). The residues present around the chromophores from other β subunits (\({}^{{\rm{A2}}}{{\rm{\beta }}}_{1}^{81}\) and \({}^{{\rm{A2}}}{{\rm{\beta }}}_{2}^{81}\)) are less hydrophobic (Extended Data Fig. 8d). Similarly, this hydrophobic cap is also found around the PCB of ApcF from the G. pacifica PBS (Extended Data Fig. 8e).
The terminal chromophore PCB in LCM \(({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186})\) exhibits fluorescence with similar emission wavelengths to those of the intact PBS, and is at a lower energy than the upstream PCBs10. Although the overall structure of the α-subunit domain of LCM \(({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}})\) overlapped well with the recombinant \({{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}\) (PDB: 4XXI)10 (Fig. 4a), some differences and new structural information are revealed in this study of the native PBS. In structure 4XXI, two different conformations of W164 are found above \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\): one is parallel to ring D and the other is nearly perpendicular10. However, in our structure, W154 at the same position displays only one conformation, parallel to ring D (Fig. 4b)—this indicates that the native LCM has a unique preference for how the side chain of such a residue is positioned. Moreover, compared with 4XXI, two extra loops (V77–A98 and F132–M145) were resolved in our \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) (Figs. 1c, 5a). Two residues in this loop, Y140 and R144, are in direct contact (less than 4 Å) with \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\) (Fig. 4b). In particular, the side chain of Y140 is oriented towards the inside of the \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\) pocket (Fig. 4b). In such a conformation there will exist a steric clash between Y140 and the ZZZasa configuration of \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\), thus providing a driving force for the formation of the ZZZssa configuration (Fig. 4c, Extended Data Fig. 8f). Comparison of \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) with five other similar subunits reveals that the orientation of this tyrosine in \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) is opposite to that in other subunits (Extended Data Fig. 8g). Therefore, Y140 of LCM is another factor that causes \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\) to uniquely adopt the ZZZssa geometry, which exhibits enhanced coplanarity of rings A and B compared with other PCBs in the α subunits of the core (Fig. 4d, Extended Data Fig. 3c).

a, Structural alignment of the α subunit of LCM \(({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}})\) in the PBS and the recombinant α subunit of LCM (PDB: 4XXI). \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) and 4XXI are coloured red and pale yellow, respectively. The bilin in \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) is shown in ball-and-stick representation. Two extra loops (V77–A98 and F132–M145) are displayed in sausage representation. b, Structural differences between \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) and 4XXI. Two different conformations of tryptophan (W154 in LCM and W164 in 4XXI) are shown in stick representation in red and yellow. Y140 and R144 from one loop are shown in surface representation in red. c, Steric clashing is observed between Y140 from LCM and the ZZZasa configuration of \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\) (grey), but is absent between Y140 and the ZZZssa configuration of \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\) (red). d, Cryo-EM densities (mesh) of the bilins (stick) in the α subunit of the core (Core_PCB, yellow) and \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) \(({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186},\,{\rm{red}})\) show the enhanced coplanarity of rings A and B of \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\).

a, Different views of the core with the bilins in different core layers shown in different colours. Bilins in \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\) \(({{\rm{\alpha }}}^{{{\rm{L}}}_{{{\rm{CM}}}^{{\prime} }}})\), ApcD (ApcD′) and ApcF (ApcF′) are coloured red, orange and brown, respectively. Bilins in rods are the same colours as the rods in which they are located according to the colouring scheme in Fig. 1d. Bilins are shown in stick representation, and key bilins are shown as thicker sticks. The numbers indicate the distances (Å) between the bilin pairs. b, Interaction between F361 from LCM and the bilin \({}^{{\rm{A2}}}{{\rm{\beta }}}_{1}^{81}\). c, Interaction between F850 from LCM and the bilin \({}^{{\rm{B1}}}{{\rm{\beta }}}_{3}^{81}\). d, Interactions between Y416 and F420 from LCM with the bilin \({}^{{\rm{A2}}}{{\rm{\beta }}}_{2}^{81}\). e, Interactions between Y443, Y583 and F610 from LCM′ with the bilin \({}^{{{\rm{A}}}^{{\prime} }1}{{\rm{\beta }}}_{3}^{81}\). f, Interaction between Y63 from LC and the bilin \({}^{{\rm{A3}}}{{\rm{\beta }}}_{2}^{81}\). g, The interaction between F454 from LCM and the bilin \({}^{{\rm{A1}}}{{\rm{\beta }}}_{1}^{81}\).
In addition to \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{{\rm{ApcD}}}^{81}\), \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) and \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\), the energy states of some other chromophores in the core are subjected to modification by the linker proteins. The shortest distance between rod Ra and the core was found between \({}^{{\rm{1I}}}{{\rm{\beta }}}_{2}^{82}\)/Ra and \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{2}^{81}\) (31 Å) (Fig. 5a), which may facilitate energy transfer from Ra to the core. The bilin nearest to \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{2}^{81}\) is \({}^{{\rm{A2}}}{{\rm{\beta }}}_{1}^{81}\), rings C and D of which form a parallel-displaced π–π interaction with F361 from LCM (Fig. 5b), and thus may mediate energy transfer to \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\). The energy absorbed by rods Rb and Rc′ may travel through the core layer B1 to \({}^{{\rm{B1}}}{{\rm{\alpha }}}_{1}^{81}\), the nearest bilin to the basal cylinders (Fig. 5a). Bilin \({}^{{\rm{B1}}}{{\rm{\beta }}}_{3}^{81}\) may play an essential role in this process because it is subjected to a parallel-displaced π–π interaction with F850 from LCM (Fig. 5c). In the basal cylinders, the two bilins \({}^{{\rm{A2}}}{{\rm{\beta }}}_{2}^{81}\)/\({}^{{{\rm{A}}}^{{\prime} }2}{{\rm{\beta }}}_{2}^{81}\) and \({}^{{\rm{A1}}}{{\rm{\beta }}}_{3}^{81}\)/\({}^{{{\rm{A}}}^{{\prime} }1}{{\rm{\beta }}}_{3}^{81}\)—which are adjacent to the bilins on ApcF/ApcF′ and separated from them by 34.8 Å and 25.7 Å, respectively—have special microenvironments (Fig. 5a). \({}^{{\rm{A2}}}{{\rm{\beta }}}_{2}^{81}\) is affected by the π–π interactions with Y416 and F420 from LCM (Fig. 5d) and \({}^{{{\rm{A}}}^{{\prime} }1}{{\rm{\beta }}}_{3}^{81}\) is affected by several π–π interactions between its rings C and D with Y443, Y583 and F610 from LCM′ (Fig. 5e)—this suggests that these two bilins may facilitate energy flow to \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\)/\({}^{{{\rm{A}}}^{{\prime} }2}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\). The bilin pair \({}^{{\rm{Rd1I}}}{{\rm{\alpha }}}_{1}^{84}\) and \({}^{{\rm{A3}}}{{\rm{\alpha }}}_{3}^{81}\) shows the shortest distance (26.1 Å) between rod Rd and the core (Fig. 5a). The bilin \({}^{{\rm{A3}}}{{\rm{\beta }}}_{2}^{81}\) may mediate further energy transfer because it is subjected to π–π interactions with Y63 from LC (Fig. 5f). For Re, both hexamers Re1 and Re2 attach to the core layer A1; as such, the energy could flow from either Re1 or Re2 to the core. The shortest distance between Re2 and the core is from \({}^{{\rm{2I}}}{{\rm{\alpha }}}_{1}^{84}\)/Re to \({}^{{\rm{A1}}}{{\rm{\alpha }}}_{3}^{81}\) (33 Å) (Fig. 5a). The energy could then travel via the \({}^{{\rm{A1}}}{{\rm{\beta }}}_{1}^{81}\), because this bilin has the shortest distance to \({}^{{\rm{A1}}}{{\rm{\alpha }}}_{3}^{81}\) and is affected by F454 from LCM through two parallel-displaced π–π interactions with rings C and D (Fig. 5g). Together, our results show that core linker proteins are extensively involved in the modulation of the energy states of core bilins to ensure the efficient unidirectional transfer of energy. These findings provide the framework for a detailed examination of energy transfer in future studies.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Preparation of phycobilisomes
P. purpureum (From UTEX Culture Collection of Algae, UTEX 2757) was cultured in Bold 1NV: Erdshreiber (1:1) half-seawater medium, bubbled with sterilizing filtered air at 22 °C, under a 16 h:8 h light–dark cycle, with a white-light flux of about 37 μmol photons per m2 per second. Algal cells were collected by centrifugation for 10 min at 6,000g, and resuspended in Buffer A (0.65 M Na/KPO4 buffer with 0.5 M sucrose and 10 mM EDTA, pH 7.0) at 0.3 g of wet weight per ml. Then cells were homogenized twice at 4 °C using a French Press (EmulsiFlex-C3, Avestin) at 4,000 p.s.i., and phenylmethylsulfonyl fluoride was added to a final concentration of 1 mM. After 30 min of incubation with lauryldimethylamine N-oxide (Sigma) (48 mg g−1 wet algal cells), debris and supernatant chlorophyll were removed by centrifugation at 20,000g for 30 min at 18 °C. The middle aqueous violet solution was loaded in a discontinuous sucrose gradient (2 ml of 0.5 M, 2 ml of 0.75 M, 2 ml of 1.0 M, 2 ml of 1.5 M, 1 ml of 2.0 M, all in Buffer B: 0.75 M K/NaPO4 buffer with 10 mM EDTA, pH 7.0) and spun at 120,000g for 4 h at 18 °C using a SW41 rotor on Optima XPN-100 centrifuge (Beckman Coulter). Three visible bands were obtained after centrifugation and violet band 1 is the main layer of intact PBSs (Extended Data Fig. 1a).
Absorption and fluorescence spectrum measurement
Absorption of the intact PBS was measured between 300–800 nm using an Ultrospec 2100 Pro ultraviolet–visible spectrophotometer (Biochrom).
Fluorescence emission spectra were recorded using a Hitachi FL-4500 fluorescence spectrophotometer at room temperature. After exciting at 450 nm, fluorescence emission was monitored from 500 to 700 nm.
Mass spectrometry analysis
Mass spectrometry analysis was performed as previously described5. In brief, the purified PBS was separated by 4–12% Bis-Tris SDS–PAGE in MES buffer and the gel was stained with ZnSO4 to detect bilin-containing proteins with ultraviolet light by Zn-enhanced fluorescence. Then, four fluorescence bands with molecular mass greater than 25 kDa were excised for in-gel digestion and proteins were identified by mass spectrometry (Extended Data Fig. 1b). The intact PBS complex in solution was also subjected to mass spectrometric analysis. Finally, all 25 protein components of PBS were identified in the samples (Extended Data Fig. 1e).
Cryo-EM sample preparation and data collection
We used holey-carbon copper grids (Quantifoll R2/2, 400 mesh) covered with homemade ultrathin carbon for cryo-EM sample preparation. Cryo-EM grids were prepared with Vitrobot Mark IV (FEI Company) at 16 °C and 100% humidity. The grids were glow-discharged after adding 50 μl amylamine to a glass culture dish in the plasma cleaner and vapouring into the air. We added a 1.5 μl aliquot of protein with a concentration of 1.5 mg ml−1 to the grids and waited for 60 s, and then added 3.5 μl of 50 mM Tris buffer (pH 8.0) to the grids and quickly mixed with the sample twice to reduce the salt concentration. The grids were then blotted for 3.5 s and plunged into liquid ethane cooled by liquid nitrogen.
The cryo-EM data were collected using a Titan Kiros Microscope (FEI) operated at a voltage of 300 kV and equipped with a Cs corrector, a GIF Quantum energy filter (Gatan) and a K2 Summit direct electron detector (Gatan). A preset defocus range of −1.2 μm to −2.2 μm was used. All cryo-EM images were recorded at a nominal magnification of 105,000× in super-resolution mode. Each stack was exposed for 5.6 s with an exposure time of 0.175 s per frame and recorded as a movie of 32 frames, resulting in the total dose rate of approximately 48 electrons per Å2 for each stack. GIF was set to a slit width of 20 eV. The data were collected automatically using the software AutoEMation44. The stacks were motion-corrected with MotionCor245 and binned twofold, resulting in a pixel size of 1.091 Å per pixel.
Cryo-EM data analysis
A total of 16,218 micrographs were collected. Micrograph screening, manual particle picking and normalization were performed using EMAN246 and RELION3.0 beta47,48,49. The contrast transfer function parameters of each micrograph were estimated using CTFFIND450,51 and automatic particle picking; all the 2D, 3D classification, 3D refinement and local defocus calculation were performed with RELION3.0 beta47,48,49.
The workflow of the data analysis is shown in Extended Data Fig. 2f. Two batches of data were collected and processed individually at the beginning. Particles were first manually picked from a small set of micrographs to produce templates for autopicking. Then particles were autopicked on all micrographs and manually screened to eliminate aggregation and ice contamination. Finally 322,889 and 363,480 particles were selected for the next 2D classification. After several rounds of 2D classification, 299,888 and 333,012 particles were left for the 3D classification. After 3D classification, two classes from each dataset with good quality were selected for the final reconstruction. At this point, we calculated the local defocus values for each particle and re-extracted particles from the dose-weighted micrographs52. Then two batches of data were merged to perform the 3D refinement. The final resolution of the 3D auto-refinement after post-processing was 2.82 Å with a final particle number of 191,825 after imposing the C2 symmetry. Application of a mask for the core region during refinement further improved the resolution of this region to 2.68 Å. We also applied local masks for each rod and extra phycoerythrin hexamer, which resulted in improved quality of local maps with resolutions ranging between 2.77 Å and 3.56 Å. The maps for the target regions were extracted from the overall map by Chimera53, and the masks were created by RELION3.0 beta47,48,49. All resolutions were estimated with the gold-standard Fourier shell correlation 0.143 criterion with high-resolution noise substitution. All the local resolution maps were calculated using ResMap54.
Model building and refinement
We searched the published genome and transcriptome database of P. purpureum27 against the 25 protein sequences of the G. pacifica PBS using the Basic Local Alignment Search Tool (BLAST). A total of 24 homologues, including eight PBP proteins and 16 linker proteins were obtained by this procedure, and these proteins are used as the candidates for model building. Local maps generated by the different masks described above were used to facilitate the model building process.
Because the sequences of P. purpureum and G. pacifica have high homology with each other, we first docked the structures of the G. pacifica PBS proteins (PDB: 5Y6P)5 into the electron microscopy maps using Chimera53. All the PBP proteins and most of the linker proteins were fitted well. Then the sequence assignments were guided by well-resolved bulky residues such as phenylalanine, tyrosine, tryptophan and arginine, and the sequences of the G. pacifica PBS were replaced with corresponding residues in the P. purpureum PBS in Coot55 and every residue was examined and manually adjusted to better fit in the map. Some of the LRγ4 proteins could not be fitted well at the N-terminal region. We first built the C-terminal CBDγ domain as described above, and then performed the de novo building in Coot55 with bulky residues as land markers as most of these residues were clearly visible in our cryo-EM maps. The linker protein located at the centre cavity of the hexamer Hd is LRγ6 in the G. pacifica PBS that contains the CBDγ domain; however, the density at this region in the P. purpureum PBS shows recognizable structural features of the Pfam00427 domain. Therefore, we named this linker protein LR6 and first docked the structure of the Pfam00427 domain from LR2 into the density. By carefully examining the densities outside the Pfam00427 domain of LR6, a YYW motif was unambiguously identified according to the clear side-chain densities. Then we obtained the full-length sequence of LR6 by searching the published genome and transcriptome database of P. purpureum for the protein containing both the Pfam00427 domain and the YYW motif. The sequence of LR2 was replaced with corresponding residues in LR6 in Coot55 and de novo atomic model building was conducted for the rest of the sequence in Coot55. Finally, 25 protein sequences were identified and confirmed by good agreement of the side-chain information between the sequences and the density maps (Supplementary Table 1).
The initial model was completed via iterative rounds of manual building with Coot55 and refinement with phenix.real_space_refine56,57. During this process, each part of the whole PBS model corresponding to each local map was refined against the local map with secondary structure and geometry restraints to prevent overfitting. Then, all parts were merged into a whole PBS model and this overall model was refined again against the overall 2.8 Å map using phenix.real_space_refine56,57. The atomic model was cross-validated according to previously described procedures58. In brief, atoms in the final model were randomly shifted by up to 0.5 Å, and the new model was then refined against one of two half-maps generated during the final 3D reconstruction. FSC values were calculated between the map generated from the resulting model and the two half-maps, as well as the averaged map of two half-maps. We did not observe notable separation between FSCwork and FSCfree, indicating that our model was not over-refined (Extended Data Fig. 2e). The data collection, model refinement and validation statistics are presented in Extended Data Tables 1, 2b. The statistics of the geometries of the models were generated using MolProbity59. All the figures were prepared in PyMOL (http://pymol.org) or Chimera53. The sequence alignments were performed by ClustalX260 and created by ESPript61.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The atomic coordinates have been deposited in the Protein Data Bank with the accession code 6KGX. The electron microscopy maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-9976 for the overall map and EMD-9977 through to EMD-9988 for the 12 local maps. The raw electron microscopy images used to build the 3D structure are available from the corresponding authors upon request.
References
Bryant, D. A. & Canniffe, D. P. How nature designs light-harvesting antenna systems: design principles and functional realization in chlorophototrophic prokaryotes. J. Phys. B 51, 033001 (2018).
Adir, N., Dines, M., Klartag, M., McGregor, A. & Melamed-Frank, M. in Complex Intracellular Structures in Prokaryotes (ed. Shively, J. M.) 47–77 (Springer, 2006).
Glazer, A. N. Light guides. Directional energy transfer in a photosynthetic antenna. J. Biol. Chem. 264, 1–4 (1989).
Adir, N., Bar-Zvi, S. & Harris, D. The amazing phycobilisome. Biochim. Biophys. Acta Bioenerg. https://doi.org/10.1016/j.bbabio.2019.07.002 (2019).
Zhang, J. et al. Structure of phycobilisome from the red alga Griffithsia pacifica. Nature 551, 57–63 (2017).
Sidler, W. A. in The Molecular Biology of Cyanobacteria (ed. Bryant, D. A.) 139–216 (Springer, 1994).
Singh, N. K., Sonani, R. R., Rastogi, R. P. & Madamwar, D. The phycobilisomes: an early requisite for efficient photosynthesis in cyanobacteria. EXCLI J. 14, 268–289 (2015).
Gao, X. et al. Molecular insights into the terminal energy acceptor in cyanobacterial phycobilisome. Mol. Microbiol. 85, 907–915 (2012).
Lundell, D. J., Yamanaka, G. & Glazer, A. N. A terminal energy acceptor of the phycobilisome: the 75,000-dalton polypeptide of Synechococcus 6301 phycobilisomes—a new biliprotein. J. Cell Biol. 91, 315–319 (1981).
Tang, K. et al. The terminal phycobilisome emitter, LCM: a light-harvesting pigment with a phytochrome chromophore. Proc. Natl Acad. Sci. USA 112, 15880–15885 (2015).
Glazer, A. N. & Bryant, D. A. Allophycocyanin B (λ max 671, 618 nm): a new cyanobacterial phycobiliprotein. Arch. Microbiol. 104, 15–22 (1975).
Peng, P. P. et al. The structure of allophycocyanin B from Synechocystis PCC 6803 reveals the structural basis for the extreme redshift of the terminal emitter in phycobilisomes. Acta Crystallogr. D 70, 2558–2569 (2014).
Dong, C. et al. ApcD is necessary for efficient energy transfer from phycobilisomes to photosystem I and helps to prevent photoinhibition in the cyanobacterium Synechococcus sp. PCC 7002. Biochim. Biophys. Acta 1787, 1122–1128 (2009).
Ashby, M. K. & Mullineaux, C. W. The role of ApcD and ApcF in energy transfer from phycobilisomes to PS I and PS II in a cyanobacterium. Photosynth. Res. 61, 169–179 (1999).
Bryant, D. A., Guglielmi, G., de Marsac, N. T., Castets, A.-M. & Cohen-Bazire, G. The structure of cyanobacterial phycobilisomes: a model. Arch. Microbiol. 123, 113–127 (1979).
Williams, R. C., Gingrich, J. C. & Glazer, A. N. Cyanobacterial phycobilisomes. Particles from Synechocystis 6701 and two pigment mutants. J. Cell Biol. 85, 558–566 (1980).
Yamanaka, G., Glazer, A. N. & Williams, R. C. Molecular architecture of a light-harvesting antenna. Comparison of wild type and mutant Synechococcus 6301 phycobilisomes. J. Biol. Chem. 255, 11104–11110 (1980).
Ducret, A., Sidler, W., Wehrli, E., Frank, G. & Zuber, H. Isolation, characterization and electron microscopy analysis of a hemidiscoidal phycobilisome type from the cyanobacterium Anabaena sp. PCC 7120. Eur. J. Biochem. 236, 1010–1024 (1996).
Arteni, A. A., Ajlani, G. & Boekema, E. J. Structural organisation of phycobilisomes from Synechocystis sp. strain PCC6803 and their interaction with the membrane. Biochim. Biophys. Acta 1787, 272–279 (2009).
Chang, L. et al. Structural organization of an intact phycobilisome and its association with photosystem II. Cell Res. 25, 726–737 (2015).
Gantt, E. & Lipschultz, C. A. Phycobilisomes of Porphyridium cruentum. I. Isolation. J. Cell Biol. 54, 313–324 (1972).
Arteni, A. A. et al. Structure and organization of phycobilisomes on membranes of the red alga Porphyridium cruentum. Photosynth. Res. 95, 169–174 (2008).
Gantt, E. & Lipschultz, C. A. Structure and phycobiliprotein composition of phycobilisomes from Griffithsia pacifica (Rhodophyceae). J. Phycol. 16, 394–398 (1980).
Guglielmi, G., Cohen-Bazire, G. & Bryant, D. A. The structure of Gloeobacter violaceus and its phycobilisomes. Arch. Microbiol. 129, 181–189 (1981).
Lange, W., Wilhelm, C., Wehrmeyer, W. & Mörschel, E. The supramolecular structure of photosystem II–phycobilisome-complexes of Porphyridium cruentum. Bot. Acta 103, 250–257 (1990).
Gantt, E. & Conti, S. F. The ultrastructure of Porphyridium cruentum. J. Cell Biol. 26, 365–381 (1965).
Bhattacharya, D. et al. Genome of the red alga Porphyridium purpureum. Nat. Commun. 4, 1941 (2013).
Tajima, N. et al. Analysis of the complete plastid genome of the unicellular red alga Porphyridium purpureum. J. Plant Res. 127, 389–397 (2014).
Gantt, E. & Lipschultz, C. A. Phycobilisomes of Porphyridium cruentum: pigment analysis. Biochemistry 13, 2960–2966 (1974).
Glazer, A. N. & Hixson, C. S. Subunit structure and chromophore composition of rhodophytan phycoerythrins. Porphyridium cruentum B-phycoerythrin and b-phycoerythrin. J. Biol. Chem. 252, 32–42 (1977).
Redlinger, T. & Gantt, E. Phycobilisome structure of Porphyridium cruentum: polypeptide composition. Plant Physiol. 68, 1375–1379 (1981).
Ficner, R. & Huber, R. Refined crystal structure of phycoerythrin from Porphyridium cruentum at 0.23-nm resolution and localization of the gamma subunit. Eur. J. Biochem. 218, 103–106 (1993).
Ducret, A., Sidler, W., Frank, G. & Zuber, H. The complete amino acid sequence of R-phycocyanin-I α and β subunits from the red alga Porphyridium cruentum. Structural and phylogenetic relationships of the phycocyanins within the phycobiliprotein families. Eur. J. Biochem. 221, 563–580 (1994).
Glazer, A. N. & Hixson, C. S. Characterization of R-phycocyanin. Chromophore content of R-phycocyanin and C-phycoerythrin. J. Biol. Chem. 250, 5487–5495 (1975).
Camara-Artigas, A. et al. pH-dependent structural conformations of B-phycoerythrin from Porphyridium cruentum. FEBS J. 279, 3680–3691 (2012).
Ritter, S., Hiller, R. G., Wrench, P. M., Welte, W. & Diederichs, K. Crystal structure of a phycourobilin-containing phycoerythrin at 1.90-Å resolution. J. Struct. Biol. 126, 86–97 (1999).
Lüning, K. in Seaweeds: Their Environment, Biogeography, and Ecophysiology. (ed. Yarish, C. and Kirkman, H.) (John Wiley & Sons, 1990).
Liu, L.-N., Chen, X.-L., Zhang, Y.-Z. & Zhou, B.-C. Characterization, structure and function of linker polypeptides in phycobilisomes of cyanobacteria and red algae: an overview. Biochim. Biophys. Acta 1708, 133–142 (2005).
Jiang, T., Zhang, J. & Liang, D. Structure and function of chromophores in R-phycoerythrin at 1.9 Å resolution. Proteins 34, 224–231 (1999).
Cantor, C. R. & Schimmel, P. R. Biophysical Chemistry: Part II Techniques for the Study of Biological Structure and Function (W. H. Freeman, 1980).
Gervasio, F. L., Chelli, R., Marchi, M., Procacci, P. & Schettino, V. Determination of the potential of mean force of aromatic amino acid complexes in various solvents using molecular dynamics simulations: the case of the tryptophan−histidine pair. J. Phys. Chem. B 105, 7835–7846 (2001).
Gallivan, J. P. & Dougherty, D. A. Cation–π interactions in structural biology. Proc. Natl Acad. Sci. USA 96, 9459–9464 (1999).
Gindt, Y. M., Zhou, J., Bryant, D. A. & Sauer, K. Spectroscopic studies of phycobilisome subcore preparations lacking key core chromophores: assignment of excited state energies to the Lcm, β18 and αAP-B chromophores. Biochim. Biophys. Acta 1186, 153–162 (1994).
Lei, J. & Frank, J. Automated acquisition of cryo-electron micrographs for single particle reconstruction on an FEI Tecnai electron microscope. J. Struct. Biol. 150, 69–80 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Scheres, S. H. W. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012).
Scheres, S. H. W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, 163 (2018).
Mindell, J. A. & Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Grant, T. & Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife 4, e06980 (2015).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Amunts, A. et al. Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489 (2014).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
We thank the staff at the Tsinghua University Branch of the National Protein Science Facility (Beijing) for technical support on the Cryo-EM and High-Performance Computation platforms; J. Wang for model validation; D. Liu for model building and structure refinement; H. Lin and X. Pan for discussion; and X. Li and H.-W. Wang for recommendations for the computation. This work was supported by the National Basic Research Program (grants 2016YFA0501101 and 2017YFA0504600 to S.-F.S.) and the National Natural Science Foundation of China (grants 31670745 and 31861143048 to S.-F.S.).
Author information
Authors and Affiliations
Contributions
S.-F.S. supervised the project; J.M. prepared the samples, collected and analysed the electron microscopy data, performed the initial model building and the biochemical and biophysical analyses; X.Y. performed the model building and the structure refinement; J.M., X.Y., S.S. and S.-F.S. analysed the structure; X.W. helped with the electron microscopy data collection and the biochemical and biophysical analyses; S.Q. contributed to the sample selection; J.M. and X.Y. wrote the initial draft; and S.S. and S.-F.S. edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Preparation and characterization of the PBS from P. purpureum.
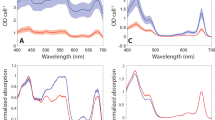
a, Isolation of PBSs using sucrose density gradient centrifugation. Three visible bands were observed. Band 1 is the sample of PBSs used for single-particle analysis in this study. The purification of PBS was repeated independently at least three times with similar results. b, Analysis of the protein composition of band 1 by SDS–PAGE stained with ZnSO4 to enable the detection of bilin-containing proteins with ultraviolet light by Zn-enhanced fluorescence. The bands of LRγ4, 5, 7, 8 and PBPs identified by mass spectrometric analysis are indicated. For gel source data, see Supplementary Fig. 1. The purification and characterization of the protein composition was repeated independently at least three times with similar results. c, Absorption spectrum of band 1 and the PBS from G. pacifica. The peaks at 498 nm, 620 nm and 650 nm are from phycourobilins, PCBs of phycocyanins and PCBs of allophycocyanins, respectively. The peaks at 540 nm and 565 nm are from PEBs. The reduced absorption of the P. purpureum PBS compared with the G. pacifica PBS at 498 nm indicates that the phycourobilin content of P. purpureum is much lower than that of G. pacifica. d, Fluorescence emission spectra of the three bands. Emission maxima at 580 nm and 676 nm represent the disassembled phycoerythrin hexamer and the terminal emitter in the intact PBS, respectively. Band 1 has an emission peak at 676 nm, band 2 at 580 nm and band 3 has two emission peaks at 676 nm and 580 nm, indicating that band 1 contains intact PBSs, band 2 contains free PBPs and band 3 contains partially disassembled PBSs. e, Results of the mass spectrometric analysis of purified PBSs. Two batches of sample were analysed. The similar results confirmed the consistency of our purification method.
Extended Data Fig. 2 Cryo-EM analysis of the PBS from P. purpureum.
a, A representative motion-corrected electron micrograph of PBSs. Scale bar, 50 nm. Tens of thousands of micrographs were collected with similar results. b, Fourier power spectrum of the micrograph showing the Thon ring extending to 2.25 Å. Tens of thousands of micrographs were collected with similar results. c, Typical good, reference-free 2D class averages from single-particle PBS images. Scale bar, 20 nm. More than three rounds of 2D class average were performed with similar results. d, Gold-standard Fourier shell correlation (FSC) curves for the 3D electron microscopy reconstructions of the PBS. Blue curve, FSC curve for the overall structure; green curve, FSC curve for the core region that was masked during refinement. e, FSC curves for the cross-validation of the atomic model. The small difference between work and free FSC curves suggested that the model was not overfitted. f, The workflow for the 2D and 3D classifications for cryo-EM data processing. The masking strategy for dealing with sub-regions of PBS is enclosed within dashed lines. For details, see ‘Cryo-EM data analysis’ in Methods.
Extended Data Fig. 3 Characterization of different types of chromophore.
a, Cryo-EM densities (mesh) of bilins (stick) bound to LRγ4 in the rod Rb, LRγ5 in rod Rd, LRγ7 in the hexamer Ha and LRγ8 in the rod Rd. b, The densities (mesh) of some PCB and PEB bilins (stick) in R-phycocyanins and phycoerythrins from rods Ra and Rb to show their different coplanarities. All of the density maps of PCB bilins showed a very flat conformation of rings B, C and D, consistent with the carbon–carbon double bond between rings C and D in PCB that constraints the movement of ring D, so that ring D is coplanar with the B–C plane. Conversely, most of the density maps of PEB displayed a curved conformation of rings B, C and D owing to the single carbon–carbon bond between rings C and D in PEB that allows the rotation of ring D, so that ring D deviates from the B–C plane. However, some PEBs in R-phycocyanin also showed a planar conformation—such as \({}^{{\rm{Ra1I}}}{{\rm{\beta }}}_{3}^{153}\) and \({}^{{\rm{Rb1I}}}{{\rm{\beta }}}_{1}^{153}\)—although to a lesser extent than that for a typical PCB molecule. c, Dihedral angles of three kinds of chromophore. The dihedral angles Φ1, Ψ1, Φ2 . . . are defined by the atoms NA–C(4)–C(5)–C(6), C(4)–C(5)–C(6)–NB, NB–C(9)–C(10)–C(11) . . . etc.
Extended Data Fig. 4 Overall structure of the PBS from P. purpureum and comparison with that from G. pacifica.
a, Schematic diagram showing the organization of the rods and the core from two perpendicular views. The colouring scheme is the same as in Fig. 1e. b, Structure of the core from two perpendicular views shows the assembly and arrangement of the core layers. c, Overall structure of the PBS overlapped with the G. pacifica PBS displayed in surface representation from three perpendicular views. The additional hexamers in the G. pacifica PBS are coloured white and labelled. d, Schematic model of the PBS architecture. The connections between PBS components are shown. Dark and light colours show C2 symmetric parts of rods. Dark and light salmon, phycoerythrin hexamers in rod; dark and light brown, extra phycoerythrin hexamers; dark and light forest green, phycocyanin hexamers; blue, allophycocyanin trimer; large rectangular box, Pfam00427 domains; small rectangular box, Pfam01383 domains; square box, CBDγ. e, Comparison of linker proteins from P. purpureum with those from G. pacifica. Structures of the 19 well-resolved linker proteins (magenta) are superimposed with those from the G. pacifica PBS (cyan). The linker proteins share very high structural conservation—such as the Pfam00427 domain in the rod–core linker (LRC)1–3/LRC1′–3′, the rod linker (LR)1–3/LR1′–3′ and LCM/LCM′, the Pfam01383 domain in the core linker (LC)/LC′ and LR1/ LR1′, the FAS1 domain in LRC6/LRC6′ and LR9/LR9′, the CBDγ domain in LRγ4–5/LRγ4′–5′ and LRγ7–8/ LRγ7′–8′, the coiled-coil motif at the C termini of LRC2–3/LRC2′–3′, and the long α-helix in the middle of the LRC4–5/LRC4′–5′. Note that LR6 from the P. purpureum PBS is different from LRγ6 from the G. pacifica PBS, therefore they are not aligned. Domains of \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\), Pfam00427 (00427), Pfam01383 (01383), CBDγ, and FAS1 are labelled.
Extended Data Fig. 5 Interactions between LRC proteins and the core.
a, Organization of LRC proteins LRC1–3/LRC1′–3′ and the core. The grooves on the α subunits that contact the linker helices are shown in red. b, Structural similarity and differences among LRC1a, LRC1b and LRC1c. These rod–core linkers are superimposed relative to the Pfam00427 domain. The helices that interact with the core are boxed. c, Structural similarity of LRC2 and LRC3, as demonstrated by superimposition of the Pfam00427 domain at the N termini and the coiled-coil motif at the C termini. The helices interacting with the core are boxed. d–f, Interactions between the αAPC subunit and the helices of LRC1b (d), LRC2 (e) and LRC3 (f). The residues involved in the interaction of LRC proteins are coloured green and shown in stick representation. The αAPC are shown in surface representation, and the residues involved in the interaction are red.
Extended Data Fig. 6 Interactions of the linker proteins LRγs and LRCs with chromophores in the rod Rd.
a, Bottom, overall structure of the rod Rd with the hexamers shown in surface representation and the linker proteins shown in cartoon representation. Top, structure of the layer Rd3I. Proteins and bilins are shown in cartoon and sphere representations, respectively. Three β subunits are coloured differently and the β82 PEBs are boxed and analysed in detail in b–d. b, The interactions between the residue Y63 and the bilin \({{\rm{\gamma }}}_{{{\rm{L}}}_{{\rm{R}}}{\rm{\gamma }}5}^{135}\) from LRγ5 with the bilin \({}^{{\rm{Rd3I}}}{{\rm{\beta }}}_{2}^{82}\). c, The interaction between F122 from LRγ5 and the bilin \({}^{{\rm{Rd3I}}}{{\rm{\beta }}}_{1}^{82}\). d, The interaction between F107 from LRγ5 and the bilin \({}^{{\rm{Rd3I}}}{{\rm{\beta }}}_{3}^{82}\). e, A focused view of the structure of the layer Rd1I showing the central triangle area. PBPs, the linker protein, bilins and residues are shown in surface, cartoon, ball-and-stick and stick representations, respectively. Three β82 PCBs are boxed and analysed in detail in f–h. f, The interactions between Y201 and F207 from LRC2 and the bilin \({}^{{\rm{Rd1I}}}{{\rm{\beta }}}_{2}^{82}\). g, The interaction between Y90 from LRC2 and the bilin \({}^{{\rm{Rd1I}}}{{\rm{\beta }}}_{1}^{82}\). h, The interaction between Y137 from LRC2 and the bilin \({}^{{\rm{Rd1I}}}{{\rm{\beta }}}_{3}^{82}\).
Extended Data Fig. 7 Comparisons of linker proteins from both P. purpureum and G. pacifica.
a, b, Structural alignment of LRγ linker proteins in the outmost hexamers of various rods from the P. purpureum PBS (a) and the G. pacifica PBS (b). β82 PEBs and residues of LRγ linker proteins are shown in ball-and-stick and stick representations, respectively. Note that an aromatic residue from the LRγ linker is present near to each β82 PEB to form π–π interactions, and one bilin from the LRγ linker \(({{\rm{\gamma }}}_{{{\rm{L}}}_{{\rm{R}}}{\rm{\gamma }}})\) always provides additional π electrons to the conjugation system of the \({{\rm{\beta }}}_{2}^{82}\) PEB. These aromatic residues and the bilins from LRγ linker proteins are conserved in both P. purpureum and G. pacifica. c, Sequence alignment of LRγ4–5 from P. purpureum and other red algae. Three aromatic residues interacting with the β82 PEBs and the cysteine residues used to link the bilins close to the β82 PEBs are marked by stars. LRgamma4_GP and LRgamma5_GP, LRγ4–5 from G. pacifica; PXF41621.1, γ-subunit from Gracilariopsis chorda; XP_005715244.1, γ-subunit from Chondrus crispus; OSX79262, γ-subunit from Porphyra umbilicalis; AAN39000.1, γ-subunit from Griffithsia japonica; AXQ05179.1, γ-subunit from Agarophyton chilense. d, Structural alignment of LRC1 linker proteins from P. purpureum and G. pacifica in the phycocyanin hexamer showing the bilin \({{\rm{\beta }}}_{2}^{82}\) and the surroundings. The key histidine residue close to the \({{\rm{\beta }}}_{2}^{82}\) PCB is conserved. e, Sequence alignment of LRC1 from P. purpureum and other red algal and cyanobacterial species. The key histidine residue close to the β82 PCBs is marked with a star. LRC1_GP, LRC1 from G. pacifica; YP_009294673.1, LRC1 from red algal G. chorda; YP_007627464.1, LRC1 from red algal C. crispus; YP_009413376.1, LRC1 from red algal P. umbilicalis; YP_009244497.1, LRC1 from red algal A. chilense; WP_006617749.1, LRC1 from cyanobacteria Arthrospira platensis; WP_009783358.1, LRC1 from cyanobacteria Lyngbya sp. PCC 8106; WP_017720249.1, LRC1 from cyanobacteria Oscillatoria sp. PCC 10802; WP_071516454.1, LRC1 from cyanobacteria Geitlerinema sp. PCC 9228. f, Structural alignment of the LRC2 and LRC3 linker proteins from P. purpureum and G. pacifica in the phycoerythrin hexamer proximal to the core showing the bilin \({{\rm{\beta }}}_{2}^{82}\) and the surroundings. Two aromatic residues near to the \({{\rm{\beta }}}_{2}^{82}\) PEB are conserved in both P. purpureum and G. pacifica. g, Sequence alignment of LRC2–3 from P. purpureum and other red algae. Two aromatic residues close to the β82 PEBs are marked with stars. LRC2_GP and LRC3_GP are from G. pacifica. PXF39827.1, XP_005715536.1 and OSX69059.1 are from G. chorda, C. crispus and P. umbilicalis, respectively.
Extended Data Fig. 8 Characterization of ApcD, ApcF and the α subunit domain of LCM.
a, Magnified view of the superimposition of ApcD proteins from P. purpureum, G. pacifica (GP_ApcD), Synechocystis PCC 6803 (4PO5_ApcD) and the α subunit of the core layer A3 (α_CoreA3). Bilins and residues are shown in ball-and-stick and stick representations, respectively. Three aromatic residues near to the PCB are conserved in all ApcD proteins, but not in the α subunit of the core layer A3. b, Magnified view of the superimposition of ApcF proteins from P. purpureum and G. pacifica (GP_ApcF), and the β subunit of the core A2 (β_CoreA2). \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) is shown in ball-and-stick representation in sand. c, A schematic of interactions between \({}^{{\rm{A2}}}{{\rm{\beta }}}_{{\rm{ApcF}}}^{87}\) and the hydrophobic cap. d, Magnified view of the PCB pocket of ApcF (left), \({}^{{\rm{A2}}}{{\rm{\beta }}}_{1}^{81}\) (middle) and \({}^{{\rm{A2}}}{{\rm{\beta }}}_{2}^{81}\) (right). The protein is shown in surface representation and coloured on the basis of amino acid hydrophobicity (see colour bar). The side chains of hydrophobic residues within 5 Å of the PCB are shown in stick representation. e, Magnified view of the structural alignment of the hydrophobic caps formed by LCM proteins from P. purpureum and G. pacifica. f, Schematic of the steric hindrance experienced by Y140/LCM and the ZZZasa configuration of \({}^{{\rm{A2}}}{{\rm{\alpha }}}_{{{\rm{L}}}_{{\rm{CM}}}}^{186}\). g, Structural alignment of \({{\rm{\alpha }}}^{{{\rm{L}}}_{{\rm{CM}}}}\), ApcD, ApcF, the α subunit (ApcA_A2) and the β subunit (ApcB_A2) in the core. The PCB pockets are indicated in the magnified view on the right.
Supplementary information
41586_2020_2020_MOESM1_ESM.pdf
Supplementary Figure 1 | Uncropped SDS-PAGE gels for Extended Data Fig. 1b a, The SDS-PAGE gel stained by the coomassie brilliant blue to show the protein marker. b, The SDS-PAGE gel stained by ZnSO4 to show the bilin-containing proteins.
Rights and permissions
About this article

Cite this article
Ma, J., You, X., Sun, S. et al. Structural basis of energy transfer in Porphyridium purpureum phycobilisome. Nature 579, 146–151 (2020). https://doi.org/10.1038/s41586-020-2020-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2020-7
This article is cited by
-
ApcE plays an important role in light-induced excitation energy dissipation in the Synechocystis PCC6803 phycobilisomes
Photosynthesis Research (2024)
-
Regulation of different light conditions for efficient biomass production and protein accumulation of Spirulina platensis
Journal of Oceanology and Limnology (2024)
-
Cryo-EM and femtosecond spectroscopic studies provide mechanistic insight into the energy transfer in CpcL-phycobilisomes
Nature Communications (2023)
-
A structure of the relict phycobilisome from a thylakoid-free cyanobacterium
Nature Communications (2023)
-
In situ structure of the red algal phycobilisome–PSII–PSI–LHC megacomplex
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
