
Abstract
Direct investigation of the early cellular changes induced by metastatic cells within the surrounding tissue remains a challenge. Here we present a system in which metastatic cancer cells release a cell-penetrating fluorescent protein, which is taken up by neighbouring cells and enables spatial identification of the local metastatic cellular environment. Using this system, tissue cells with low representation in the metastatic niche can be identified and characterized within the bulk tissue. To highlight its potential, we applied this strategy to study the cellular environment of metastatic breast cancer cells in the lung. We report the presence of cancer-associated parenchymal cells, which exhibit stem-cell-like features, expression of lung progenitor markers, multi-lineage differentiation potential and self-renewal activity. In ex vivo assays, lung epithelial cells acquire a cancer-associated parenchymal-cell-like phenotype when co-cultured with cancer cells and support their growth. These results highlight the potential of this method as a platform for new discoveries.
Similar content being viewed by others


Main
Cancer cell behaviour is strongly influenced by the surrounding cells in the tumour microenvironment (TME). Various cell types in the TME are known to influence cancer cell behaviour, including mesenchymal cells such as activated fibroblasts, pericytes and endothelial cells, as well as different types of inflammatory cells1.
During the early phase of metastatic growth, cancer cells generate a local TME (metastatic niche), which is distinct from the normal tissue structure and key for supporting metastatic outgrowth2. However, detailed analysis of the cellular composition of the metastatic niche, especially at early stages, is constrained by the difficulty of spatially discriminating the metastatic-niche cells within the bulk tissue. This hampers the identification of cells that might respond to early colonization by cancer cells but remain low in number as metastases grow.
In this study, we present a strategy in which metastatic cancer cells mark their neighbouring cells, thereby identifying them in the tissue and overcoming these limitations. We have applied this system to interrogate the early metastatic environment of breast cancer cells in the lung. We confirm that the system enables us to quantitatively and qualitatively distinguish known metastatic-niche cells within the tissue, and identify lung epithelial cells, in which a regenerative-like program is activated, as a component of the metastatic TME. We show that these epithelial cells acquire multi-lineage differentiation potential when co-cultured with cancer cells and support their growth. These results support the notion that, in addition to the well-characterized stromal activation, a parenchymal response might contribute to creating the metastatic microenvironment.
The mCherry niche-labelling system
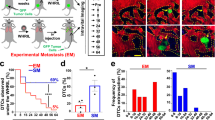
To develop a labelling system that uses metastatic cancer cells to directly identify their neighbouring cells in vivo, we generated a secreted fluorescent mCherry protein containing a modified lipid-permeable transactivator of transcription (TATk) peptide3,4 (sLP–mCherry) (Fig. 1a and Extended Data Fig. 1a). We engineered 4T1 breast cancer cells to co-express the sLP–mCherry and GFP; we refer to these cells as labelling-4T1 cells. In vitro, sLP–mCherry protein secreted by labelling-4T1 cells re-enters the cells, as indicated by changes in the intracellular localization of the red fluorescence (Extended Data Fig. 1b, c). sLP–mCherry protein is also taken up by unlabelled cells, both in co-culture with labelling-4T1 cells (Fig. 1b–d) and when cultured in medium conditioned by labelling-4T1 cells (LCM) (Extended Data Fig. 1d, e). Upon uptake into a cell, sLP–mCherry fluorescence has an intracellular half-life of 43 h (Extended Data Fig. 1f) and is localized in CD63+ multi-lamellar bodies (lysosomal-like structures) where, owing to its high photostability5, it retains high fluorescence intensity (Extended Data Fig. 1g, h). Fractionation of LCM shows that only the soluble fraction retains labelling activity, whereas the extracellular vesicles, a proportion of which contain sLP–mCherry, do not show labelling activity in vitro (Extended Data Fig. 1i–k).

a, Label design. Labelling-4T1 cells co-express the lipid-soluble cell-penetrating mCherry-fusion protein label and GFP. b, c, Representative FACS plots of naive 4T1 cells cultured alone (b) or co-cultured with labelling-4T1 cells (c). Numbers indicate the percentage of cells in the respective quadrant. d, Fluorescence image from co-cultures (scale bar, 10 μm). Data representative of two independent experiments (b–d). e–g, In vivo labelling. e, Schematic of the experiment6: labelling-4T1 cells are injected into mice; these cells metastasize in the lung and label nearby cells in the TME (niche) with mCherry. f, Representative FACS plot of a metastatic lung, n = 50 mice. g, Representative immunofluorescence images of labelling-4T1 cell metastasis (n = 8 mice). Labelling-4T1 cells are positive for both GFP and mCherry, whereas metastatic niche cells are positive for mCherry only. Blue, DAPI. Scale bars: main panels, 20 μm; enlarged insets, 10 μm. For gating strategy see Supplementary Information.
In vivo, intravenous injection of labelling-4T1 cells (GFP+mCherry+) into syngeneic BALB/c mice to induce lung metastases efficiently labels surrounding host tissue cells (GFP−mCherry+), penetrating approximately five cell layers (Fig. 1e–g and Extended Data Fig. 2a, b). This enables specific discrimination of host cells in close proximity to cancer cells from distal lung cells (GFP−mCherry−) using fluorescence-activated cell sorting (FACS) (Fig. 1f). Notably, when micro-metastases grow larger, the number of mCherry+-niche cells in the tissue remains proportional to the number of metastatic cells (Extended Data Fig. 2c). We detected no adaptive immunogenicity against sLP–mCherry and the local increase of CD45+ immune cells within the mCherry population was observed specifically as a response to cancer cells (Extended Data Fig. 2d−f). Thus, this mCherry-niche-marking strategy enables spatial reconstitution of the local metastatic niche within the tissue. This permits functional identification of labelled cells and direct comparison with unlabelled cells within the same lung.
Tissue spatial resolution
To demonstrate the utility of the mCherry-niche strategy to specifically interrogate the local early changes induced by cancer cells, we seeded 4T1-labelling cells in the lung via tail-vein injection. Lung tissue distant from micro-metastases remained unperturbed by primary-tumour-derived systemic changes7. To validate the mCherry-niche strategy, we first examined components known to be involved in metastatic-niche formation. CD45+ immune cells were very abundant in the mCherry+ niche and nearly exclusively derived from the myeloid lineage (CD11b+) (Extended Data Figs. 2d, 3a). Lung neutrophils have been reported to enhance metastatic growth of cancer cells8,9, and were indeed detected in the mCherry+ niche (Extended Data Fig. 3b). Because abnormalities in lung neutrophils are often associated with cancer10, we isolated mCherry+-niche neutrophils (Ly6G+) and compared their proteome to that of unlabelled neutrophils from the same lungs (Fig. 2a). The sub-pool of mCherry+-niche neutrophils exhibited an increase in translation, oxidative phosphorylation and intracellular reactive oxygen species (ROS) levels relative to unlabelled neutrophils, as determined by FACS analysis (Fig. 2b, Extended Data Fig. 3c–f and Supplementary Data). To validate the functional relevance of specific features identified in mCherry+-niche cells, we developed a 3D-scaffold co-culture system that mimics complex tissue-like cell–cell interactions. We found that lung neutrophils increased growth of actin–GFP+ mouse mammary tumour virus (MMTV)–polyoma virus middle T antigen (PyMT) breast cancer cells in a ROS-dependent manner (Fig. 2c–e and Extended Data Fig. 3g, h). Collectively, these data highlight the potential of our strategy to detect in vivo changes that are spatially restricted to the metastatic environment.

a, b, Proteomic analysis of FACS-sorted Ly6G+ cells: all differentially detected proteins (a) and proteins associated with oxidative phosphorylation (b). c–e, Three-dimensional co-culture, with or without the ROS inhibitor TEMPO, of GFP+ MMTV–PyMT cancer cells and Ly6G+ cells sorted by magnetic-activated cell sorting (MACS). c, The co-culture scheme. d, Quantification of GFP signal (n = 3 independent experiments, each with 3 to 10 technical replicates). Data are normalized to cancer cell growth and represented as mean ± s.e.m. Statistical analysis of biological replicates by two-way ANOVA. e, Representative images from three independent experiments (day 6; scale bar, 400 μm).
The non-immune mCherry+-niche signature
Whereas the contribution of immune cells to metastatic outgrowth has been widely investigated11, less is known about the role of other TME cell types during metastatic nesting. Notably, the mCherry-labelling strategy can be used to provide spatiotemporal information by applying it to different stages of metastatic progression. We generated the gene-expression profile of non-immune (CD45−) mCherry+-niche cells at the time point immediately preceding micro-metastases as well as at an advanced metastatic stage (Fig. 3a, b). The majority of alterations were detected at the early stage, but additional changes subsequently discriminated the niche of macro-metastases (Fig. 3c and Extended Data Fig. 4a, b), confirming the evolution of the metastatic TME over time. MetaCore dataset enrichment and gene-set enrichment analysis (GSEA) highlighted changes in pathways related to proliferation, inflammation and tissue remodelling (Extended Data Fig. 4b, c). We next focused on the upregulated (more than twofold) genes encoding soluble factors in the mCherry+ niche at both time points (Fig. 3d and Supplementary Data). We found many previously reported tumour-promoting factors12,13,14,15,16,17,18,19, further validating the ability of our labelling system to faithfully capture the in vivo metastatic niche. We also found WNT1-induced protein (WISP1)—which has been suggested to act as an oncogene in breast cancer20—to be widely expressed in the mCherry+ niche (Fig. 3d). Indeed, we detected upregulation of WISP1 in both cancer and metastatic-niche cells and confirmed its pro-metastatic activity by exogenous inhibition in vivo (Fig. 3e and Extended Data Fig. 5a–e).

a, Schematic of metastatic progression using labelling-4T1 cells6. b, Experimental design for RNA-seq experiments6. c, Principal component analysis (PCA) of CD45−Ter119− cell signatures from metastatic lungs at early (n = 3, 10 mice each) and late (n = 3, 5 mice each) time points. The black oval encloses the distal lung samples; red ovals enclose the mCherry+-niche samples to highlight their similarity in the PCA plot. d, Venn diagram of differentially expressed genes in the mCherry+ niche and selected factors that are common at early and late stages. Wisp1 is also known as Ccn4. e, WISP1-blocking antibody treatment in vivo (n = 10, 2 independent experiments). Box edges represent 25th and 75th percentiles, the horizontal bar is the median and the whiskers show the range of values. f, GSEA correlation from RNA-seq data comparing early (n = 3) or late (n = 3) mCherry+ samples with their respective mCherry− controls. g, Left, representative immunofluorescence images of lung tissue (n = 3 mice) showing mCherry-labelled micro-metastasis (red), SP-C (white) and DAPI (blue, middle). Right, enlarged view of areas indicated with dashed outlines. Scale bars: main panel, 100 μm; enlarged insets, 10 μm (white arrows and dashed outlines, mCherry-labelled SP-C+ cells). h, EPCAM+ cell frequency among Lin− (CD45−CD31−Ter119−) cells in distal lung (mCherry−) and mCherry+ cells estimated by FACS (n = 13 mice). i, Representative FACS plots from h. Numbers indicate the percentage of cells in the respective quadrant. Statistical analysis by unpaired two-tailed t-test with Welch’s correction (e), weighted Kolmogorov–Smirnov-like statistic with Benjamini–Hochberg correction (f) and paired two-tailed t-test (h).
We next probed the TME for other non-immune cell types, which might be difficult to resolve by standard techniques owing to their small numbers. Of note, we found pathways associated with lung epithelial cells in the metastatic-niche signature (Fig. 3f). Micro-metastases grow embedded within the alveolar compartment of the lung, and we found alveolar type II cells (AT2) expressing surfactant protein C (SP-C, encoded by Sftpc) in the metastatic niche (Fig. 3g). We also found mCherry+-niche cells expressing the epithelial cell adhesion marker EPCAM, further supporting the presence of cells of lung parenchymal origin (Fig. 3h, i).
Cancer-associated parenchymal cells
We found mCherry+-niche epithelial cells to have a higher proliferative activity compared to their normal lung counterparts (Fig. 4a). Concordantly, we detected alveolar cell clusters with increased proliferative activity at the metastatic borders of human breast cancer lung metastases, suggesting that a lung parenchymal response to metastatic growth may occur in both mouse and human (Extended Data Fig. 6a–f). Cancer cells benefit from the presence of a lung parenchymal response, as freshly isolated EPCAM+ cells from naive lungs supported the growth of actin–GFP+ MMTV–PyMT tumour cells in our 3D-scaffold co-culture system (Fig. 4b–d). Moreover, in line with the results shown in Fig. 2c–e, the presence of both lung neutrophils and epithelial cells further enhanced tumour growth (Extended Data Fig. 7a–d), highlighting the cellular complexity of the metastatic niche.

a, Ki67 staining in FACS-sorted mCherry− and mCherry+ EPCAM+ cells (n = 7 independent sorts). b–d, GFP+ MMTV–PyMT cancer cell growth in 3D co-culture with MACS-sorted EPCAM+ cells. b, The co-culture scheme. c, Representative images from 4 independent experiments (day 6; scale bar, 400 μm). d, Quantification of GFP signal (n = 4, each with 3 technical replicates, statistical analysis of biological replicates). Data are normalized to cancer cell growth. e–g, scRNA-seq analysis; t-SNE plots of CD45− cells from the mCherry+ niche (e; n = 1,473) or distal lung (f; n = 1,996). g, Right, heat map of mCherry+-niche EPCAM+ cells (ordered genes in rows and hierarchically clustered cells in columns); left, table shows established lineage markers (bold); asterisks indicate putative alveolar markers25. h, RT–qPCR analysis of EPCAM+ FACS-sorted cells (Sftpc, Aqp5, n = 9; Sftpb, Abca3, Pdpn, Ager, Vim, Cdh1, n = 8; Krt6, Cdh2, n = 7; Snai1, n = 4; Twist, n = 3). Data represented as fold change relative to mCherry− lung EPCAM+ cells (statistical analysis on the ΔCt values). i, EPCAM+SCA1+ cell frequency among Lin− (CD45−CD31−Ter119−) cells, determined by FACS (n = 13 mice). Statistical analysis by paired two-tailed t-test (a, h, i), one-sample two-tailed t-test (d). Data represented as mean ± s.e.m.
We next aimed to better define the perturbation occurring in lung epithelial cells in the proximity of cancer cells. To contextualize their presence among the other cellular components of the metastatic niche, we performed single-cell RNA sequencing (scRNA-seq) of CD45− cells. t-Distributed stochastic neighbour embedding (t-SNE) analysis of mCherry+-niche cells identified a large stromal cluster in which different stromal cells could be distinguished (Fig. 4e and Extended Data Fig. 8a–c). This is in agreement with the various known mesenchymal cell components of the TME, as well as the characterization of different fibroblast subsets21,22,23,24. Notably, specifically in the mCherry+ niche, Epcam-expressing epithelial cells are distributed in two clusters distinguished by the expression of E-cadherin (Cdh1) (Fig. 4e). We found that only mCherry+-niche Epcam+Cdh1+ cells shared the expression of alveolar genes25 with unlabelled distant lung Epcam+ cells (Fig. 4f, g). Conversely, mCherry+-niche Epcam+Cdh1− cells expressed both the progenitor markers SCA1 (encoded by Ly6a) and Tm4sf126,27,28 (Fig. 4g). As validation of this de-differentiated signature observed in the majority of epithelial cells in the metastatic niche, reverse transcription with quantitative PCR (RT–qPCR) of EPCAM-sorted mCherry+-niche cells also showed an overall reduction in expression of alveolar lineage markers (Fig. 4h). Moreover, the enrichment of EPCAM+SCA1+ cells in the mCherry+ niche of different metastatic cell types was confirmed by FACS analysis (Fig. 4i and Extended Data Fig. 9a–c). Similarly, the presence of epithelial cells expressing another lung progenitor marker, integrin β4 (also known as CD104)29, was increased in the mCherry+-niche and in ex vivo co-cultures (Extended Data Fig. 9d–i).
In summary, we describe a parenchymal response to lung metastasis involving de-differentiated pools of epithelial cells in the niche, which we define as cancer-associated parenchymal cells (CAPs).
CAPs are activated AT2 cells
To functionally characterize CAPs, we tested their lineage differentiation potential ex vivo using a 3D Matrigel-based organoid co-culture system27 (Fig. 5a). Unlabelled resident lung EPCAM+ cells are predominantly alveolar27, and formed mainly alveolar organoids when co-cultured with CD31+ cells (Fig. 5b–d). mCherry+-niche EPCAM+ cells favoured the bronchiolar lineage and showed a remarkable capacity to generate multi-lineage bronchioalveolar organoids (Fig. 5d). Despite the bias in organoid formation towards the bronchial lineage, we did not detect mCherry-labelled cells expressing bronchial markers in vivo (Extended Data Fig. 10a). CAPs also retained high self-renewal capacity over multiple passages (Fig. 5e).

a–e, Lung organoids: co-culture scheme (a); representative bright-field images (b; scale bar, 100 μm); representative immunofluorescence of organoid sections stained with the indicated markers (c; scale bar, 50 μm); quantification (d) and organoid formation efficiency after passaging (e). Ac-tubulin, acetyl tubulin. f–h, Lung organoid cultures with or without labelling-4T1 cells: co-culture scheme (f); representative bright-field images (g; scale bar, 100 μm) and quantification (h). i, j, Lung organoids with Sftpc-CreERT2 lineage cells with or without non-labelling 4T1-GFP cells: quantification (i) and representative bright-field images (j; scale bar, 150 μm). Images are representative of six (b, c, g) or three (j) organoid cultures. Data generated from independent sorts (d, h, i) and represented as cumulative percentage using the mean ± s.d. of three co-cultures per sorting. k, Representative staining of lineage cells in metastatic lungs from Sftpc-CreERT2 mice injected with E0771 (n = 3; scale bar, 50 μm) or MMTV–PyMT (n = 3; scale bar, 100 μm) cancer cells. Statistical analysis by unpaired two-tailed t-test (d, e, h) and one-sample two-tailed t-test (i) on original non-cumulative values.
Next, we tested whether tumour cells could directly induce the CAP phenotype. When EPCAM+ cells from unlabelled distal micro-metastatic lungs or naive lungs were co-cultured with metastatic cells, they generated a higher proportion of bronchiolar and bronchioalveolar organoids (Fig. 5f–h and Extended Data Fig. 10b, c). Similar alterations were induced by cancer cells when the assay was performed using mouse lung fibroblasts (MLg cells) instead of CD31+ cells (Extended Data Fig. 10b, c).
Although lung EPCAM+ cells are predominantly alveolar, they also contain epithelial progenitors that could be enriched by cancer cells to generate increased plasticity27,30. Therefore, we performed organoid cultures using lineage-labelled AT2 (Sftpc-lineage) cells. Sftpc-lineage cells, which show no plasticity in co-culture with CD31+ cells, generated multi-lineage bronchioalveolar organoids when exposed to cancer cells, supporting the idea of a reprogramming activity driven by cancer-cell-derived factors ex vivo (Fig. 5i, j). Despite the potential of cancer cells to modulate the organoid formation ability of lineage-labelled club cells (Scgb1a1 lineage), only rare single Scgb1a1-lineage cells were found in proximity to lung metastases (Extended Data Fig. 10d–f). Conversely, metastases growing in Sftpc-lineage lungs demonstrated the alveolar (AT2) origin of the CAPs (Fig. 5k).
Recently, a rare population of AT2 cells expressing Axin2 with stem cell and repair activity (AT2 stem cells), was described in the lung alveoli31. Whereas a small proportion of Axin2-expressing cells was found in the unlabelled epithelial cluster, Axin2 was undetectable in the mCherry+-niche EPCAM clusters (data not shown). Therefore, even if cancer cell seeding could trigger lung injury, this phenomenon does not appear to specifically maintain an Axin2+ AT2 population in the metastatic niche.
Collectively, these data demonstrate the alveolar origin of CAPs and the ability of cancer cells to induce multi-lineage differentiation potential of epithelial cells ex vivo.
Discussion
This study introduces the mCherry-niche labelling system and demonstrates its ability to resolve the host tissue cellular environment in regions surrounding cancer cells. We report the presence of a lung epithelial compartment within the metastatic niche, which originates from AT2 cells. We define this TME component as CAPs and describe their activated regenerative state by showing their de-differentiated signature, tissue stem-cell-like features, multi-lineage differentiation potential and increased self-renewal activity.
Parenchymal cells have been described as triggering a tissue-wide pro-tumorigenic inflammatory response to systemic primary tumour signals32,33. In addition to these systemic effects, we here show that a regenerative-like activation in the lung parenchyma occurs as a direct local response during breast cancer metastasis. This parenchymal response, combined with the stromal activation, is potentially a key orchestrator of tumour-niche formation.
Together these results consolidate the mCherry-niche system as a platform for discoveries with the potential to identify, isolate and functionally test cells from the metastatic niche with high spatial resolution.
Methods
Sample sizes were estimated based on previous experiments conducted in our laboratory, providing sufficient numbers of mice in each group to yield a two-sided statistical test, with the potential to reject the null hypothesis with a power (1 − β) of 80%, subject to α = 0.05. No further statistical methods were used to predetermine sample size. Most experiments were not randomized: only the experiment involving treatment was randomized. Whenever possible, investigators were blinded to allocation during outcome assessment.
Statistical analysis
Statistical analyses were performed using Prism v.7.0c (GraphPad Software). P values were obtained from two-tailed Student’s t-tests with paired or unpaired adjustment. When needed, unpaired t-tests were adjusted using Welch’s correction for unequal variance. In one instance (Fig. 4i), data in one of the groups did not pass the D’Agostino and Pearson normality test, therefore a Wilcoxon matched-pairs signed-rank test was performed. Single-sample tests were also used for comparisons of co-cultured cancer cell growth on scaffolds to the normalized value of cancer cells alone. For comparisons between two scaffold conditions of growth over time or to perform multiple analysis between experimental groups, two-way ANOVA was used.
Mouse strains
All mice used are available from the Jackson Laboratory. MMTV–PyMT mice34 are on a FVB and C57BL/6 background, actin–GFP mice35 and Rag1 KO mice are on the FVB background (gift from J. Huelsken laboratory (EPFL, Lausanne, Switzerland)). Sftpc-CreERT236, Rosa26R-YFP37 (Sftpc-CreERT2;R26R-YFP) mice are on a C57BL/6 background. BALB/cJ mice and the above-mentioned lines were bred and maintained under specific-pathogen-free conditions by The Francis Crick Biological Research Facility and female mice were used between 6 and 10 weeks of age. Breeding and all animal procedures were performed at the Francis Crick Institute in accordance with UK Home Office regulations under project license P83B37B3C.
For ex vivo organoid lineage-tracing experiments, Scgb1a1-CreERT2 and Rosa26R-fGFP38, Sftpc-CreERT2 (Sftpc-CreERT2;R26R-fGFP and Scgb1a1-CreERT2;R26R-fGFP) mice on a C57BL/6 background were bred and maintained under specific-pathogen-free conditions at the Gurdon Institute of the University of Cambridge in accordance with UK Home Office project licence PC7F8AE82. All animal work was conducted under UK Home Office regulations, project licenses P83B37B3C and PC7F8AE82.
Tamoxifen administration
Tamoxifen (Merck Sigma-Aldrich) was dissolved in Mazola corn oil (Merck Sigma-Aldrich) in a 20 mg ml−1 stock solution. Two doses of tamoxifen (0.2 mg per g body weight) were given via oral gavage every other day and lung tissues were collected two days after tamoxifen administration to isolate cells for lung organoids. For in vivo lineage tracing three doses of tamoxifen (0.2 mg per g body weight) were given via oral gavage over consecutive days and mice were injected two weeks later.
Cells
MLg cells were purchased from ATCC. Cancer-associated fibroblasts (CAFs) isolated from MMTV–PyMT tumours and human normal fibroblasts (hNLFs) were a gift from E. Sahai. MMTV–PyMT cells were isolated from MMTV–PyMT tumours as previously described19. All other cell lines were provided by the Cell Services Unit of The Francis Crick Institute. All cell lines were authenticated and tested for mycoplasma by the Cell Services Unit of The Francis Crick Institute. MMTV–PyMT cells were cultured on collagen-solution-coated dishes in DMEM/F12 (Thermo Fisher Scientific) with 2% fetal bovine serum (FBS; Labtech), 100 U ml−1 penicillin–streptomycin (Thermo Fisher Scientific), 20 ng ml−1 EGF (Thermo Fisher Scientific) and 10 μg ml−1 insulin (Merck Sigma-Aldrich). The collagen solution was made with 30 μg ml−1 PureCol collagen (Advanced Biomatrix), 0.1% bovine serum albumin (BSA), 20 mM HEPES in HBSS (Thermo Fisher Scientific). HC11 cells were cultured in RPMI (Thermo Fisher Scientific) supplemented with 10% FBS, 100 U ml−1 penicillin–streptomycin, 10 ng ml−1 EGF (Thermo Fisher Scientific) and 5 μg ml−1 insulin. All other cell lines were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS and 100 U ml−1 penicillin–streptomycin. All cells were cultured at 37 °C and 5% CO2.
Human samples
Human pulmonary breast cancer metastases from independent patients were obtained from the Grampian Biorepository, Aberdeen Royal Infirmary (REC approval: 16/NS/0055). Four samples were stained by immunohistochemistry and immunofluorescence and proliferation of epithelial cells was quantified. Further information about the human samples used is provided in the Supplementary Information.
Labelling system
A soluble peptide (SP)3 and a modified TAT peptide4 were cloned upstream of the mCherry cDNA, under the control of a mouse PGK promoter (sLP–mCherry, see Supplementary Data for sequence). The sLP–mCherry sequence was cloned into a pRRL lentiviral backbone. 4T1, Renca, CT26 and HC11 cells were stably infected with sLP–mCherry and pLentiGFP lentiviral particles and subsequently sorted to isolate mCherry+GFP+ cells.
Induction of experimental metastases
Procedures were performed at the Francis Crick Institute in accordance with UK Home Office regulations under project license P83B37B3C. Cancer cells were injected intravenously to generate metastases in the lung: 4T1 (1,000,000), Renca (500,000) or CT26 (200,000) cells were resuspended in 100 μl PBS and injected into the tail vein of BALB/cJ mice. Mice were euthanized on the basis of a time period rather than on the basis of their clinical signs. Therefore, the experimental end point (time controlled, seven days unless otherwise specified) most likely occurred before a humane end point (as determined by deterioration of heath conditions). All animals were monitored daily for unexpected clinical signs following the P83B37B3C licence guidelines and the principles set out in the NCRI Guidelines for the Welfare and Use of Animals in Cancer Research (UK). Deterioration of health conditions—such as reduction in food and water consumption, changes in the general appearance of the animal, or weight loss of 10% over a 24-h period—would result in animals being euthanized before the experimental end point.
In vivo lineage-tracing experiments
Sftpc-CreERT2 and Scgb1a1-CreERT2 mice on a C57BL/6 background were injected into the tail vein with 175,000 MMTV–PyMT C57BL/6 cells and lungs were collected 4 weeks later, or with 700,000 E0771 cells and lungs were collected 12 days later.
Tissue digestion for cell isolation or analysis
Lung tissues were dissociated as previously described19. In brief, lungs were removed at day 7 after tumour cell injection (unless otherwise specified), minced manually and then digested for 30 min in a shaker at 37 °C with a mixture of DNase I (Merck Sigma-Aldrich) and Liberase TM and TH (Roche Diagnostics) in HBSS solution. Samples were then washed, passed through a 100-μm filter and incubated in Red Blood Cell Lysis buffer (Miltenyi Biotec) for 3–5 min at room temperature. After a wash with MACS buffer (0.5% BSA and 250 mM EDTA in PBS), samples were passed through a 40-μm filter and a 20-μm strainer-capped flow cytometry tube to generate a single-cell suspension to use for flow cytometric analysis or further purification.
FACS analysis and cell sorting
Prepared single-cell suspensions of mouse lung tissues and in vitro cell lines were incubated with mouse FcR Blocking Reagent (Miltenyi Biotec) for 10 min at 4 °C followed by an incubation with a mix of pre-labelled antibodies (antibody information is provided in the Supplementary Information) for 30 min at 4 °C. After two washes with MACS buffer, dead cells were stained with DAPI. Flow cytometry analyses were carried out on a BD LSR-Fortessa (BD Biosciences) and FlowJo v.10.4.2 (FlowJo, LCC 2006-2018) was used for further analysis. All cell-sorting experiments were carried out using a BD Influx cell sorter (BD Biosciences).
Tissue digestion and FACS analysis in ex-vivo lineage-tracing experiments
Lung tissues were dissociated with a collagenase–dispase solution as previously described27. In brief, after lungs were cleared by perfusion with cold PBS through the right ventricle, 2 ml dispase (50 U ml−1, BD Biosciences) was instilled into the lungs through the trachea until the lungs inflated, followed by instillation of 1% low melting agarose (Bio-Rad Laboratories) through the trachea to prevent leakage of dispase. Each lobe was dissected and minced into small pieces in a conical tube containing 3 ml PBS, 60 μl collagenase–dispase (Roche) and 7.5 μl of 1% DNase I (Merck Sigma-Aldrich) followed by rotating incubation for 45 min at 37 °C. The cells were then filtered sequentially through 100- and 40-μm strainers and centrifuged at 1,000 r.p.m. for 5 min at 4 °C. The cell pellet was resuspended in 1 ml of ACK lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) and lysed for 90 s at room temperature. Six millilitres of basic F12 medium (Thermo Fisher Scientific) was added and 500 μl FBS (Fisher Scientific) was slowly added in the bottom of the tube. Cells were centrifuged at 1,000 r.p.m. for 5 min at 4 °C. The cell pellet was resuspended in PF10 buffer (PBS with 10% FBS) for further staining. The antibodies used were as follows: CD45 (30-F11)–APC (BD Biosciences), CD31 (MEC13.3)–APC (BD Biosciences) and EPCAM (G8.8)–PE-Cy7 (BioLegend). For antibody list see Supplementary Information. The MOFLO system (Beckman Coulter) was used for the sorting at Wellcome–MRC Stem Cell Institute Flow Cytometry Facility.
Lung organoid assay
Lung organoid co-culture assays were previously reported27,39. In brief, freshly sorted epithelial cells (EPCAM+CD45−CD31−Ter119−GFP−) from either the metastatic niche or the distal lung were resuspended in 3D basic medium (DMEM/F12, supplemented with 10% FBS, penicillin–streptomycin, 1 mM HEPES and insulin–transferrin–selenium (ITS) (Merck Sigma-Aldrich), and mixed with MACS-sorted CD31+ lung stromal cells or MLg cells followed by resuspension in growth factor-reduced (GFR) Matrigel (BD Biosciences) at a ratio of 1:1. One hundred microlitres of mixture was then placed in a 24-well transwell insert with a 0.4-μm pore (Corning). Distal lung or niche epithelial cells (103 to 2.5 × 103 cells) and 25,000 CD31+ or MLg cells were seeded in each insert. Five hundred microlitres of 3D basic medium was placed in the lower chamber and medium was changed every other day. In addition, freshly sorted Scgb1a1-lineage labelled cells or Sftpc-lineage labelled cells were resuspended in 3D basic medium followed by mixing with GFR Matrigel retaining CD31+ stromal cells as described above. For co-culture of lung epithelial cells with tumour cells, a mixture of 103 to 2.5 × 103 distal lung epithelial cells and 25,000 CD31+ cells in Matrigel was placed in the Transwell insert, and 2,000 tumour cells were FACS-sorted from metastatic lungs and seeded in the lower chamber. Plates were scored for colony number after 14 days. Organoid-forming efficiency was calculated as the number of organoids formed per number of cells plated per well as a percentage. Quantification of distinct types of differentiated organoids was performed by scoring the organoids expressing SOX2 or SP-C and HOPX by immunofluorescence from at least five step sections (20 μm apart) per individual well. Bright-field images were acquired after 14 days using an EVOS microscope (Thermo Fisher Scientific).
3D cell culture
Primary MMTV–PyMT actin–GFP cells were seeded at a density of 5,000 cells per well in a collagen-solution-coated Alvetex Scaffold 96-well plate (ReproCELL). The following day, Ly6G+ lung cells and/or Epcam+ lung epithelial cells were sorted by MACS and seeded on top of the cancer cells at a density of 50,000 cells per well. In selected experiments, wells were supplemented with 4-hydroxy-TEMPO (200 μM, Merck Sigma-Aldrich) or mouse WISP1 antibody (250 ng ml−1, MAB1680, R&D Systems). The growth of GFP+ cells was monitored daily for 6 days using the SteREO LumarV12 stereomicroscope (Zeiss), and images were quantified using ImageJ (NIH). For quantification, the Li’s minimum cross entropy thresholding algorithm was performed on the stacked images.
For the CD104 staining experiment, EPCAM+ lung cells were sorted from mouse lung tissues by MACS and seeded at a density of 1,500,000 cells per well on collagen-solution-coated Alvetex Scaffold 12-well inserts. After 48 h, MMTV–PyMT actin–GFP cells were seeded on top of the EPCAM+ cells at a density of 2,000 cells per scaffold insert.
Immunofluorescence and immunohistochemistry
Mouse lungs were fixed in 4% PFA in PBS for 24 h and embedded in paraffin blocks. Four-micrometre-thick tissue sections were cut, deparaffinized and rehydrated using standard methods. After heat-mediated antigen retrieval in citrate buffer (unless stated otherwise), sections were blocked with a solution of 1% BSA, 10% donkey serum in PBS. For antibody list, see Supplementary Information.
mCherry and GFP staining
An overnight incubation at 4 °C with goat GFP and rabbit mCherry antibodies was followed by 1 h incubation at room temperature with anti-goat Alexa Fluor 488- and anti-rabbit Alexa Fluor 555-conjugated antibodies (1:400; Thermo Fisher Scientific). Next, the slides were incubated with Sudan Black B for 20 min and mounted with Vectashield mounting medium with DAPI (Vector Laboratories).
Lineage staining
An overnight incubation at 4 °C with goat GFP antibody was followed by 45-min incubation at room temperature with secondary biotinylated antibodies. Next, the Vectastain Elite ABC kit (Vector Laboratories) was used according to the manufacturer’s instructions. Cell nuclei were visualized with haematoxylin and analysis was performed on a Nikon Eclipse 90i light microscope and with NIS-elements software (Nikon).
WISP1 staining
An overnight incubation at 4 °C with goat GFP and rabbit WISP1 antibodies was followed by 30-min incubation at room temperature with anti-goat Alexa Fluor 488 and anti-rabbit Alexa Fluor 555 (1:500; Thermo Fisher Scientific). Next, the slides were incubated with Sudan Black B for 20 min and mounted with Vectashield mounting medium with DAPI (Vector Laboratories).
Ki67 staining
EPCAM+CD45−CD31−Ter119−GFP− cells were sorted from lung suspensions, plated on polylysine-coated glass coverslips for 15 min at room temperature and fixed in 4% PFA in PBS for 10 min. After fixation, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min and incubated with a blocking solution (1% BSA, 10% goat serum, 0.3 M glycine and 0.1% Tween-20 in PBS) for 1 h at room temperature. Next, cells were incubated overnight with a rabbit Ki67 antibody diluted in blocking solution followed by a 1 h incubation with a goat anti-rabbit Alexa Fluor 488 antibody (1:500; Thermo Fisher Scientific). Finally, cells were mounted with Vectashield mounting medium with DAPI for imaging.
E-cadherin staining
CD49f+CD104+CD45−CD31−Ter119−GFP− cells were sorted from lung suspensions, cytospun on glass slides and fixed in 4% PFA in PBS for 10 min. Next, cells were permeabilized with 0.5% Triton X-100 for 30 min and incubated in blocking solution (4% BSA, 0.05% Tween-20 in PBS) for 45 min at room temperature. Then, cells were incubated with a rat E-cadherin antibody in blocking solution overnight at 4 °C followed by an incubation with a goat anti-rat Alexa Fluor 647 antibody (1:500; Thermo Fisher Scientific). Finally, cells were mounted with Vectashield mounting medium with DAPI for imaging.
CD104 staining
EPCAM+ cells were sorted by MACS and plated on Alvetex scaffold inserts as described above. Seven days after plating the whole scaffold was collected, washed with PBS and incubated in blocking solution (10% goat serum in PBS) for 1 h at room temperature. Next, the samples were incubated with a conjugated CD104–eFluor660 antibody (1:100 in PBS with 1:10 FcR blocking (Miltenyi Biotec)) for 1 h at room temperature. Then, the samples were fixed with 4% PFA in PBS for 10 min and mounted with Vectashield mounting medium with DAPI. Images were captured with the Axio Scan.Z1 slide scanner (Zeiss, Germany).
Lung organoid staining
Cultured organoids were fixed with 4% PFA in PBS for 2–4 h at room temperature followed by immobilization with Histogel (Thermo Fisher Scientific) for paraffin embedding. At least five step sections (20 μm apart) per individual well were stained. Fluorescence images were acquired using a confocal microscope Leica TCS SP5 (Leica Microsystems). All the images were further processed with Fiji software.
TTF1 and Ki67 co-staining
Target retrieval solution pH 9 (Agilent DAKO) was used for antigen retrieval. For histology, 1-h incubation at room temperature with mouse TTF1 antibody was followed by 45-min incubation at room temperature with secondary biotinylated antibodies. Next, the Vectastain Elite ABC kit (Vector Laboratories) was used according to the manufacturer’s instructions. Cell nuclei were visualized with haematoxylin and analysis was performed on a Nikon Eclipse 90i light microscope and with NIS-elements software (Nikon). For immunofluorescence, 1 h incubation at room temperature with mouse TTF1 and rabbit Ki67 antibodies was followed by 45 min incubation at room temperature with anti-mouse Alexa Fluor 555 and anti-rabbit Alexa Fluor 488 (1:250; Thermo Fisher Scientific). Next, the slides were incubated with Sudan Black B for 20 min and mounted with Vectashield mounting medium with DAPI (Vector Laboratories).
All images were captured with a Zeiss Upright710 confocal microscope or a Zeiss Upright780 confocal microscope unless otherwise stated.
RT–qPCR
RNA preparation was performed using the MagMax-96 Total RNA Isolation Kit (Thermo Fisher Scientific). cDNA synthesis was performed using a SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) according to the manufacturer’s protocol. Quantitative real-time PCR samples were prepared with 50–100 ng total cDNA for each PCR reaction. The PCR, data collection and data analysis were performed on a 7500 FAST Real-Time PCR System (Thermo Fisher Scientific). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal expression reference. A list of primers used can be found in the Supplementary Information.
Anti-WISP1 treatment in vivo
BALB/cJ female mice (6–8 weeks old) were administered with WISP1 antibody or a control-IgG antibody (5 μg AF1680 and 5 μg MAB1680, R&D Systems) via intra-tracheal injection (50 μl per mouse). The following day, mice were intravenously injected with 250,000 4T1 cells. Anti-WISP1 or control-IgG treatment was repeated daily via a second intra-tracheal injection on day 4 and intra-peritoneal injections on days 2, 3, 5 and 6. Mice were collected 7 days after the first treatment and lungs were embedded, cut and stained with haemotoxylin and eosin (H&E). The lung metastatic burden was assessed by counting the number of metastases on four levels (100-μm intervals) from two lung lobes (n = 10 per group).
EdU in vitro proliferation assay
MMTV–PyMT actin–GFP cells were seeded at a density of 10,000 cells per well into collagen-solution-coated six-well plates. The following day, Ly6G+ lung cells and/or EPCAM+ lung cells were sorted by MACS and added to the wells at a density of 100,000 cells per well. After 60 h, wells were supplemented with 20 μM EdU (5-ethynyl-2′-deoxyuridine). Cells were collected 6 h later and EdU incorporation was assessed using the Click-iT Plus EdU Flow Cytometry Assay Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Sample data were acquired on a BD LSR-Fortessa flow cytometer and analysed using FlowJo 10 software.
Conditioned medium preparation and vesicle isolation
Labelling-4T1 cells were plated on 10-cm Petri dishes. When cells were 80% confluent, 10 ml DMEM with 10% FCS was added to be conditioned for 48 h. The conditioned medium preparation and vesicle isolation were performed as previously described40. In brief, the medium was collected and spun at 300g for 10 min. Next, the supernatant was collected and spun at 2,000g for 10 min. The supernatant after this second centrifugation was collected and used as conditioned medium. For vesicle isolation, the conditioned medium was subsequently ultracentrifuged at 10,000g for 30 min and at 100,000g for 70 min. The vesicle pellet at this stage was washed with PBS, spun at 100,000g for 70 min and resuspended again in PBS for in vitro uptake experiments.
ImageStream analysis
Image stream analyses were carried out on an ImageStream Mark X II Imaging Flow Cytometer (Amnis Merck). The acquired data were analysed using IDEA software (Amnis Merck).
Electron microscopy
Experiments were performed on glass bottom dishes with a numbered grid (MatTek) to enable subsequent location of the same cell imaged by confocal microscopy. After confocal imaging, cells were fixed in 8% formaldehyde in 0.1 M phosphate buffer (pH 7.4) added in equal quantities to cell medium for 15 min and then further fixed in 2.5% glutaraldehyde and 4% formaldehyde in 0.1 M phosphate buffer (pH 7.4) for 1 h and then processed using the National Center for Microscopy and Imaging Research protocol41. For transmission electron microscopy, 70-nm serial sections were cut using a UC6 ultramicrotome (Leica Microsystems) and collected on formvar-coated slot grids. No post-staining was required owing to the density of metal deposited using the NCMIR protocol. Images were acquired using a 120-kV Tecnai G2 Spirit transmission electron microscope (FEI Company Thermo Fisher Scientific) and an Orius CCD camera (Gatan).
RNA sequencing sample preparation
Bulk RNA sequencing
CD45−Ter119− (CD45−) cells were sorted from single-cell suspensions of metastatic lungs stained with anti-mouse CD45 and Ter119 antibodies and DAPI. RNA isolation was performed using the MagMax-96 Total RNA Isolation Kit (Thermo Fisher Scientific), which enables high-quality RNA extraction from samples with low cell numbers (<10,000 cells). RNA quality for each sample was assessed using the Agilent RNA 6000 Pico Kit (Agilent Technologies). RNA was amplified and analysed at the Barts and London Genome Centre.
Single-cell RNA sequencing
CD45−Ter119− cells were sorted from single-cell suspensions of metastatic lungs stained with anti-mouse CD45 and Ter119 antibodies and DAPI. Library generation for 10x Genomics analysis were performed following the Chromium Single Cell 3′ Reagents Kits (10x Genomics) and sequenced on an Hiseq4000 (Illumina), to achieve an average of 50,000 reads per cell.
Determination of intracellular ROS levels
Single-cell suspensions from mouse lungs were incubated with mouse FcR blocking reagent for 5 min on ice and subsequently incubated with CellROX Deep Red Reagent (Thermo Fisher Scientific) for 30 min at 37 °C following the manufacturer’s recommendations. Next, cells were washed twice with MACS buffer, stained with DAPI and analysed by flow cytometry.
Quantitative proteomic analysis of Ly6G cells
Neutrophils were sorted by FACS from single-cell suspensions of metastatic lungs stained with a conjugated anti-mouse Ly6G–APC antibody (three samples from independent sorts). Ly6G cells from the metastatic niche (mCherry+) and the distal lung (mCherry−) were digested into peptides using a previously described protocol42 and analysed by data-independent acquisition mass spectrometry43 on a Orbitrap Fusion Lumos instrument (Thermo Fisher Scientific). A hybrid spectral library was generated using the search engine Pulsar in Spectronaut Professional+ (v.11.0.15038, Biognosys) by combing data-dependent acquisition runs obtained from a pooled sample of Ly6G cells, and the data-independent acquisition data. Data analysis and differential protein expression was performed using Spectronaut Professional+. A detailed description of sample processing, data acquisition and processing can be provided on request from the corresponding authors.
Bioinformatics analysis
Bulk RNA sequencing
The sequencing was performed on biological triplicates for each condition, generating approximately 35 million 76-bp paired-end reads. The RSEM package44 (v.1.2.29) and Bowtie2 were used to align reads to the mouse mm10 transcriptome, taken from the known-gene reference table available from University of California Santa Cruz (https://genome.ucsc.edu/). For RSEM, all parameters were run as default except “–forward-prob” which was set to 0.5. Differential-expression analysis was carried out with DESeq2 package45 (v.1.12.4) in R v.3.3.1 (https://www.r-project.org/). Genes were considered to be differentially expressed if the adjusted P was less than 0.05. Differentially expressed genes were taken forward and their pathway and process enrichments were analysed using Metacore (https://portal.genego.com). Hypergeometric test was used to determine statistical enriched pathways and processes and the associated P-value was corrected using the Benjamini–Hochberg method. GSEA (v.2.2.3)46,47 was carried out using ranked gene lists using the Wald statistic and the gene sets of C2 canonical pathways and C5 biological processes. All parameters were kept as default except for enrichment statistic (classic) and maximum size, which was changed to 5,000. Gene signatures with FDR q-value equal to or less than 0.05 were considered statistically significant. A weighted Kolmogorov–Smirnov-like statistic was derived and the associated P-value was corrected with the Benjamini–Hochberg method.
Single-cell RNA sequencing
Raw reads were initially processed by the Cell Ranger v.2.1.1 pipeline, which deconvolved reads to their cell of origin using the UMI tags, aligned these to the mm10 transcriptome using STAR (v.2.5.1b) and reported cell-specific gene expression count estimates. All subsequent analyses were performed in R v.3.4.1 using the cellrangerRkit, monocle and pheatmap packages. Genes were considered to be ‘expressed’ if the estimated (log10) count was at least 0.1. Primary filtering was then performed by removing from consideration: genes expressed in fewer than 20 cells; cells expressing fewer than 50 genes; cells for which the total yield (that is, sum of expression across all genes) was more than two standard deviations from the mean across all cells in that sample; and cells for which mitochondrial genes made up greater than 10% of all expressed genes. PCA decomposition was performed and, after consideration of the eigenvalue ‘elbow-plots’, the first 25 components were used to construct t-SNE plots for both samples. Niche cells expressing Epcam were subdivided into those also expressing Cdh1 and those not expressing Cdh1. Other genes expressed in at least 50% of cells in a given group were said to be co-expressed and the set of genes co-expressed in one or more groups was presented as a heat map, with the columns (cells) clustered using the standard Euclidean hierarchical method.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The RNA-sequencing datasets have been deposited in the Gene Expression Omnibus with accession number GSE117930; the single-cell RNA-sequencing datasets have been deposited with accession number GSE131508. The proteomic datasets have been deposited in the Proteomics Identifications Database with accession number PXD010597.
Change history
13 November 2019
An Amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Hanahan, D. & Coussens, L. M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013).
Barash, S., Wang, W. & Shi, Y. Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem. Biophys. Res. Commun. 294, 835–842 (2002).
Flinterman, M. et al. Delivery of therapeutic proteins as secretable TAT fusion products. Mol. Ther. 17, 334–342 (2009).
Shaner, N. C., Steinbach, P. A. & Tsien, R. Y. A guide to choosing fluorescent proteins. Nat. Methods 2, 905–909 (2005).
del Pozo Martin, Y. et al. Mesenchymal cancer cell-stroma crosstalk promotes niche activation, epithelial reversion, and metastatic colonization. Cell Rep. 13, 2456–2469 (2015).
Peinado, H. et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017).
Wculek, S. K. & Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015).
Coffelt, S. B., Wellenstein, M. D. & de Visser, K. E. Neutrophils in cancer: neutral no more. Nat. Rev. Cancer 16, 431–446 (2016).
Singhal, S. et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 30, 120–135 (2016).
Blomberg, O. S., Spagnuolo, L. & de Visser, K. E. Immune regulation of metastasis: mechanistic insights and therapeutic opportunities. Dis. Model. Mech. 11, dmm036236 (2018).
Kessenbrock, K., Plaks, V. & Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010).
Kowanetz, M. et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl Acad. Sci. USA 107, 21248–21255 (2010).
Qian, B.-Z. et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011).
Acharyya, S. et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178 (2012).
Oskarsson, T. et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874 (2011).
Erez, N. Cancer: Opening LOX to metastasis. Nature 522, 41–42 (2015).
Onnis, B., Fer, N., Rapisarda, A., Perez, V. S. & Melillo, G. Autocrine production of IL-11 mediates tumorigenicity in hypoxic cancer cells. J. Clin. Invest. 123, 1615–1629 (2013).
Malanchi, I. et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89 (2012).
Su, F., Overholtzer, M., Besser, D. & Levine, A. J. WISP-1 attenuates p53-mediated apoptosis in response to DNA damage through activation of the Akt kinase. Genes Dev. 16, 46–57 (2002).
Costa, A. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463–479 (2018).
Karnoub, A. E. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563 (2007).
Hosaka, K. et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl Acad. Sci. USA 113, E5618–E5627 (2016).
Murgai, M. et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 23, 1176–1190 (2017).
Treutlein, B. et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014).
Kim, C. F. B. et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835 (2005).
Lee, J.-H. et al. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4–NFATc1–thrombospondin-1 axis. Cell 156, 440–455 (2014).
Zacharias, W. J. et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255 (2018).
Chapman, H. A. et al. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Invest. 121, 2855–2862 (2011).
McQualter, J. L., Yuen, K., Williams, B. & Bertoncello, I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl Acad. Sci. USA 107, 1414–1419 (2010).
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. & Desai, T. J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123 (2018).
Liu, Y. et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell 30, 243–256 (2016).
Lee, J. W. et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 567, 249–252 (2019).
Guy, C. T., Cardiff, R. D. & Muller, W. J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12, 954–961 (1992).
Okabe, M., Ikawa, M., Kominami, K., Nakanishi, T. & Nishimune, Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 407, 313–319 (1997).
Rock, J. R. et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl Acad. Sci. USA 108, E1475–E1483 (2011).
Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001).
Rawlins, E. L. et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4, 525–534 (2009).
Lee, J.-H. et al. Anatomically and functionally distinct lung mesenchymal populations marked by Lgr5 and Lgr6. Cell 170, 1149–1163 (2017).
Théry, C., Amigorena, S., Raposo, G. & Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 30, 3.22.1–3.22.29 (2006).
Deerink, T. J. et al. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. National Center for Microscopy and Imaging Research https://ncmir.ucsd.edu/sbem-protocol (2010).
Heinze, I. et al. Species comparison of liver proteomes reveals links to naked mole-rat longevity and human aging. BMC Biol. 16, 82 (2018).
Bruderer, R. et al. Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteomics 16, 2296–2309 (2017).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003).
Acknowledgements
We thank E. Sahai, P. Scaffidi (The Francis Crick Institute) and V. Sanz-Moreno (Barts Cancer Institute) for scientific discussions, critical reading of the manuscript and sharing cell lines and mouse strains; M. Izquierdo (CSIC, Madrid) for sharing the CD63–GFP plasmid; E. Nye and the pathologists G. Stamp and E. Herbert from the Experimental Histopathology Unit at the Francis Crick Institute for histological processing and analysis support; J. Bee from the Biological Resources Unit at the Francis Crick Institute for technical support with mice and mouse tissues; R. Goldstone and A. Edwards from the Advanced Sequencing Facility at the Francis Crick Institute for technical support; M. Llorian-Sopena from the Bioinformatics and Biostatistics Unit at the Francis Crick Institute for helping with the RNA sequencing analysis; the Flow Cytometry Unit at the Francis Crick Institute, particularly S. Purewal and J. Cerveira, for invaluable technical help; the Cell Services Unit at the Francis Crick Institute; C. Moore (The Francis Crick Institute) for intra-tracheal injections; and I. Pshenichnaya, P. Humphreys, S. McCallum and Cambridge Stem Cell Institute core facilities for technical assistance. We acknowledge support from the FLI Core Facility Proteomics, which is a member of the Leibniz Association and is financially supported by the Federal Government of Germany and the State of Thuringia. This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001112), the UK Medical Research Council (FC001112), and the Wellcome Trust (FC001112) and the European Research Council grant (ERC CoG-H2020-725492); and by the Wellcome Trust—MRC Stem Cell Institute, which receives funding from the Sir Henry Dale Fellowship from Wellcome, the Royal Society (107633/Z/15/Z) and the European Research Council Starting Grant (679411).
Author information
Authors and Affiliations
Contributions
L.O. designed and performed most of the experiments, analysed and interpreted the data and contributed to the manuscript preparation. E.N. assisted with data collection, performed all the 3D-scaffold co-culture experiments, the in vivo WISP1 experiments and the scRNA sequencing, and interpreted and analysed the data and contributed to the manuscript preparation. I.K. performed the RT–qPCR analysis, some of the tissue immunofluorescence staining and analysed the data. A.M. and J.-H.L. performed some of the tissue immunofluorescence staining and all the lung organoid experiments, and interpreted and analysed the data. V.B. performed some of the tissue immunofluorescence staining. P.C. and S.H. performed bioinformatics analysis. I.H., J.K. and A.O. performed the proteomics and analysed the data. E.G.-G. helped with the collection of Ly6G+ cells for proteomics. G.M. performed the 3D-scaffold co-culture to analyse CD104+ cells. A.W. and L.C. performed the electron microscopy experiments. E.H. and V.S. provided human samples. L.O., E.N., I.K., V.B. and J.-H.L. critically reviewed the manuscript. J.-H.L. supervised the lung organoid experiments. I.M. designed and supervised the study, interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information Nature thanks Marie-Liesse Asselin-Labat, Thomas Tüting and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Fig. 1 The mCherry-niche system in vitro.
a, sLP–mCherry design. b, Fluorescence images of labelling-4T1 cells after thawing. Scale bar, 10 μm. c, Representative FACS plot of labelling-4T1 cells. d, In vitro cultures of the indicated cell types with LCM: culture scheme and representative fluorescence images of HC11 (mouse mammary epithelial cells) and hNLF (human normal lung fibroblasts) with LCM (scale bar, 10 μm). e, FACS plots of 4T1, HC11, RAW264.7 (mouse macrophages), hNLF and mouse breast CAFs cultured with LCM. f, FACS analysis of 293T cells cultured with LCM, at different time points after LCM removal (black dots); white dots show the theoretical decrease considering the cell proliferation rate only (the amount of 293T cells labelled with mCherry after 24 h incubation with LCM was set to 100%). g, Representative fluorescence image of 4T1-CD63–GFP cells cultured with LCM. Scale bars: main panels, 5 μm; enlarged region, 1 μm. h, Representative correlative light and electron microscopy of labelling-4T1 cells showing re-uptake of sLP–mCherry (n = 5 different cells analysed). Top left, bright-field image overlaid with mCherry immunofluorescence (∼700 nm optical section). Bottom left, electron microscopy of the same cell (∼70-nm section thickness). Centre, best approximation of immunofluorescence–bright-field–electron microscopy overlay (scale bar, 5 μm). Right, electron microscopy of the outlined regions (centre, labelled a–c) (black arrows point at vesicular structures containing mCherry; scale bar, 1 μm). i, j, Analysis of in vitro labelling potential of soluble fraction and extracellular vesicles isolated from LCM by FACS. i, Schematic representation of LCM fractionation. j, HC11 cells cultured with either LCM, soluble fraction after depletion of extracellular vesicles (soluble) or purified extracellular vesicles. k, ImageStream analysis of mCherry+ extracellular vesicles in LCM (16% of total extracellular vesicles are mCherry+). Data are representative of three (b), ten (c) or two (d–g, j, k) independent experiments.
Extended Data Fig. 2 The mCherry-niche system in vivo.
a, b, Distance of labelled cells within metastases. a, Representative fluorescence images (lines measure the maximum distance of labelled cells (mCherry+) from labelling-4T1 cells (mCherry+GFP+); scale bar, 50 μm). b, Quantification of labelling distance in micro-metastases (n = 11) and macro-metastases (n = 4). c, Correlation between the percentage of mCherry-labelled niche cells and the percentage of cancer cells in metastatic lungs analysed by FACS. Left, analysis of lungs with a small number of cancer cells (n = 14 mice). Right, analysis with all cancer cell frequencies (n = 31 mice). Statistical analysis by Pearson correlation. d–f, CD45+ cell frequency on live cells in distal lung, mCherry+ niche and not-injected naive lungs by FACS. d, BALB/c mice injected with labelling-4T1 cells (n = 5 mice per group). e, BALB/c mice injected with labelling-HC11 cells (n = 4 mice). f, RAG1-knockout mice injected with labelling-4T1 cells (n = 10 mice). Statistical analysis by paired two-tailed t-test. Data are represented as mean ± s.e.m.
Extended Data Fig. 3 mCherry+-niche neutrophils increase ROS production.
a, b, CD11b+ (a) and Ly6G+ (b) cell frequencies among live cells in distal lung and mCherry+ niche by FACS (n = 9 mice per group). c, Enriched processes by MetaCore analysis and GSEA based on proteomic data by comparing mCherry+-niche (n = 3) and distal lung (n = 3) neutrophils; dominant mCherry+-niche proteins were obtained by using WebGestalt (http://www.webgestalt.org/option.php). d, PCA of proteins found in unlabelled or mCherry+-niche neutrophils (n = 3, each with 10 mice, small circles; large circles represent the average of the triplicates). e, f, Representative FACS plot (e) and scatter plot (f) of intrinsic ROS in Ly6G+ cells (n = 6 mice). g, GFP signal quantification of 3D co-culture with GFP+ MMTV–PyMT cancer cells and MACS-sorted Ly6G+ cells from either naive or metastatic lungs with or without the ROS inhibitor TEMPO (n = 3, each with 3 technical replicates). Data are normalized to cancer cell growth (statistical analysis on biological replicates). h, Representative cancer cell growth on the scaffold (from 14 independent experiments): integrated density of the GFP signal was measured on the scaffold using ImageJ and the corresponding fluorescent image of GFP+ cancer cell growth (scale bar, 400 μm). Statistical analysis by paired two-tailed t-test (a, b, f), hypergeometric test with Benjamini–Hochberg correction (c, Metacore), weighted Kolmogorov–Smirnov-like statistic with Benjamini–Hochberg correction (c, GSEA) and two-way ANOVA (g). Data are presented as mean ± s.d. (f) and mean ± s.e.m. (g).
Extended Data Fig. 4 RNA sequencing of non-immune mCherry+-niche cells.
a, b, GSEA of upregulated genes in mCherry+-niche cells. a, Percentage of correlating processes related to the indicated activity. b, Specific signalling pathways (indicated by the ∗ in a) at early or late time point. c, MetaCore analysis of genes differentially expressed in RNA-seq data, comparing early (n = 3) or late (n = 3) mCherry+ samples versus the respective mCherry− samples (see Fig. 3a, b). Statistical analysis by hypergeometric test with Benjamini–Hochberg correction.
Extended Data Fig. 5 WISP1 supports metastatic growth.
a, b, Representative immunofluorescence images of lung metastatic tissues (n = 2 mice) stained for GFP (green) to detect labelling-4T1 cells, WISP1 (red) and DAPI (blue), showing distal lung and metastatic areas (a; scale bar, 50 μm), and a representative image showing the enrichment of WISP1+ cells within lung metastasis including niche cells (white arrows) (b; scale bar, 50 μm). c–e, WISP1-blocking antibody treatment in vivo. c, Experimental design (IT, intratracheal injection; IP, intraperitoneal injection). d, Metastatic outcome measured as the percentage of lung area covered by metastases (quantification was performed on two lung levels 100 μm apart). e, Representative H&E staining (n = 5 mice per group; black arrows show metastatic foci). Scale bar, 500 μm. Two experiments with lower overall metastatic frequency are quantified in Fig. 3e. Statistical analysis by two-way ANOVA (d). Data are presented as mean ± s.e.m.
Extended Data Fig. 6 Lung pneumocytes react to cancer cells in human breast pulmonary metastases.
a–c, Histology of sections of human breast tumour lung metastases. a, Representative image of distal lung (scale bar, 100 μm). b, Image from the tumour–lung interface showing expression of the pneumocyte marker thyroid transcription factor 1 (TTF1) (scale bar, 50 μm). c, Representative histology of the metastatic border (scale bar, 100 μm). d–f, Alveolar cell proliferation in human breast tumour lung metastases analysed by immunofluorescence. Representative images from distal lung (d) and metastatic border (e) showing TTF1 (red), Ki67 (green) and DAPI (blue). Scale bars: all 100 μm, except e (far right), 50 μm. f, Quantification of alveolar proliferation. Box edges show 25th and 75th percentiles, the horizontal line shows the median and whiskers show the range of values. Statistical analysis by paired two-tailed t-test. Tissue sections from n = 4 independent patients were analysed.
Extended Data Fig. 7 Epithelial cells support cancer cell growth ex vivo.
a, GFP+ MMTV–PyMT cancer cell proliferation in 2D co-culture with MACS-sorted EPCAM+ and Ly6G+ cells stained with EdU and analysed by FACS (n = 3 independent experiments). Data are normalized to cancer cell proliferation. b–d, Three dimensional co-culture of GFP+ MMTV-PyMT cancer cells with MACS-sorted EPCAM+ and Ly6G+ cells. b, Co-culture scheme. c, Representative images from four independent experiments (day 4; scale bar, 400 μm). d, Quantification of GFP signal. Data are normalized to cancer cell growth (n = 4 independent experiments (dots), each with 3–4 technical replicates). Statistical analysis of biological replicates by one-sample two-tailed t-test (a) and two-way ANOVA (d). Data are represented as mean ± s.e.m.
Extended Data Fig. 8 scRNA-seq analysis reveals different sub-pools of stromal cells in the niche.
a, t-SNE plots of CD45− cells isolated from distal lung (n = 1,996) or mCherry+ niche (n = 1,473) after scRNA-seq analysis. Stromal cells are coloured on the basis of expression levels of the indicated genes. b, t-SNE niche plots from data in a; each plot shows (in red) the cells expressing the indicated stromal marker. c, MetaCore pathway enrichment analysis using the list of genes detected in at least 50% of the indicated marker-defined cells (n = 66 THY1+ cells, n = 175 PDGFRB1+ cells, n = 322 PDGFRA+ cells, n = 330 ACTA2+ cells, n = 25 LGR6+ cells). Statistical analysis by hypergeometric test with Benjamini–Hochberg correction.
Extended Data Fig. 9 mCherry+-niche epithelial cells are enriched for stem cell markers.
a, Representative FACS plots showing Lin− (CD45−CD31−Ter119−) cells in distal lung and mCherry+ niche from labelling-4T1-injected mice (quantification in Fig. 4i). b, c, Scatter plots showing FACS quantification of EPCAM+SCA1+ cell frequency on Lin− (CD45−CD31−Ter119−) cells in distal lung and mCherry+ niche with injection of labelling-RENCA (b; n = 5 mice) and labelling-CT26 (c; n = 4 mice). d–f, Scatter plot of CD49f+CD104+ cell frequency among Lin− (CD45−CD31−Ter119−) cells in distal lung and mCherry+ niche detected by FACS (d; n = 5 mice), representative FACS plots (e) and representative immunofluorescence image of FACS-sorted mCherry+-niche CD49f+CD104+ cells stained for E-cadherin (green) and with DAPI (blue) (f; scale bar, 20 μm). g–i, Three-dimensional co-culture of GFP+ MMTV–PyMT cancer cells with MACS-sorted EPCAM+ cells. g, Quantification of integrin β4 (CD104) expression on EPCAM+ cells. h, Number of CD104+ cells proximal to cancer cells (n = 4 from three independent sorts). i, Representative immunofluorescence image from the co-culture stained for CD104 (red), GFP+ cancer cells (green) and with DAPI (blue). Scale bar, 20 μm. Statistical analysis of biological replicates by paired two-tailed t-test (b–d, g). Data are presented as mean ± s.e.m.
Extended Data Fig. 10 Cancer cells change lung epithelial cell-lineage commitment ex vivo.
a, Representative immunofluorescence images of lung metastatic sections (n = 3 mice) co-stained for an airway marker (SCGB1A1 (top; white) or SOX2 (bottom; white)) and mCherry (red), and with DAPI (blue). Scale bar, 100 μm. b, c, Lung organoids from EPCAM+ FACS-sorted cells in co-culture with either lung stromal CD31+ cells or MLg fibroblasts, alone or in the presence of non-labelling 4T1-GFP cells from metastatic lungs in the lower chamber; quantification (b) and representative bright-field images (c; scale bar, 150 μm) of organoids. d, e, Lung organoids with Scgb1a1-CreERT2 lineage cells with or without 4T1-GFP: quantification (d) and representative bright-field images (e; scale bar, 150 μm). f, Representative staining of lineage cells in metastatic lungs from Scgb1a1-CreERT2 mice injected with MMTV–PyMT cancer cells. Scale bars: top left, 200 μm; other panels, 50 μm; top middle inset, 25 μm. Data are generated with sorted EPCAM+ (b) or club-lineage cells (d) and represented as cumulative percentage presented as mean ± s.d. of three co-cultures per sorting. Statistical analysis by two-tailed t-test on original non-cumulative values (b, d). Images are representative of three organoid cultures (c, e).
Supplementary information
Supplementary Information
FACS gating strategy examples, a list of antibodies used for FACS and immune-staining, a list of primers used for the qRT–PCRs (Fig. 4h) and information about the human pulmonary breast cancer metastases from four patients. In addition, supplementary data shows the sLP-mCherry nucleotide sequence (design shown in Ext.Data Fig.1a) that was cloned in a pRRL lentiviral backbone.
Supplementary Data
The entire list of differentially detected proteins (AVG log2 ratio) in mCherry-niche versus unlabelled Ly6G+ neutrophils. Differences were considered when changes were >0.58 and <−0.58.
Supplementary Data
Upregulated genes (>2 fold) in non-immune-mCherry-niche compared to unlabelled lung tissue at both 5 and 10 days post cancer cells seeding.
Source data
Rights and permissions
About this article

Cite this article
Ombrato, L., Nolan, E., Kurelac, I. et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 572, 603–608 (2019). https://doi.org/10.1038/s41586-019-1487-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1487-6
This article is cited by
-
Crosstalk between colorectal CSCs and immune cells in tumorigenesis, and strategies for targeting colorectal CSCs
Experimental Hematology & Oncology (2024)
-
Cell-cell communication characteristics in breast cancer metastasis
Cell Communication and Signaling (2024)
-
Lung endothelium exploits susceptible tumor cell states to instruct metastatic latency
Nature Cancer (2024)
-
Universal recording of immune cell interactions in vivo
Nature (2024)
-
Host-derived growth factors drive ERK phosphorylation and MCL1 expression to promote osteosarcoma cell survival during metastatic lung colonization
Cellular Oncology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
