
Abstract
In principle, humans can produce an antibody response to any non-self-antigen molecule in the appropriate context. This flexibility is achieved by the presence of a large repertoire of naive antibodies, the diversity of which is expanded by somatic hypermutation following antigen exposure1. The diversity of the naive antibody repertoire in humans is estimated to be at least 1012 unique antibodies2. Because the number of peripheral blood B cells in a healthy adult human is on the order of 5 × 109, the circulating B cell population samples only a small fraction of this diversity. Full-scale analyses of human antibody repertoires have been prohibitively difficult, primarily owing to their massive size. The amount of information encoded by all of the rearranged antibody and T cell receptor genes in one person—the ‘genome’ of the adaptive immune system—exceeds the size of the human genome by more than four orders of magnitude. Furthermore, because much of the B lymphocyte population is localized in organs or tissues that cannot be comprehensively sampled from living subjects, human repertoire studies have focused on circulating B cells3. Here we examine the circulating B cell populations of ten human subjects and present what is, to our knowledge, the largest single collection of adaptive immune receptor sequences described to date, comprising almost 3 billion antibody heavy-chain sequences. This dataset enables genetic study of the baseline human antibody repertoire at an unprecedented depth and granularity, which reveals largely unique repertoires for each individual studied, a subpopulation of universally shared antibody clonotypes, and an exceptional overall diversity of the antibody repertoire.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

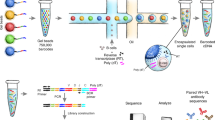

Data availability
Sequence data that support the findings in this study are available at the NCBI Sequencing Read Archive (www.ncbi.nlm.nih.gov/sra) under BioProject number PRJNA406949. Raw and processed datasets are available at www.github.com/briney/grp_paper.
References
Rajewsky, K. Clonal selection and learning in the antibody system. Nature 381, 751–758 (1996).
Alberts, B. et al. The Generation of Antibody Diversity (Garland Science, New York, 2002).
Boyd, S. D. & Crowe, J. E. Jr. Deep sequencing and human antibody repertoire analysis. Curr. Opin. Immunol. 40, 103–109 (2016).
Briney, B. & Burton, D. Massively scalable genetic analysis of antibody repertoires. Preprint at https://www.biorxiv.org/content/early/2018/10/19/447813 (2018).
Briney, B., Le, K., Zhu, J. & Burton, D. R. Clonify: unseeded antibody lineage assignment from next-generation sequencing data. Sci. Rep. 6, 23901 (2016).
Morbach, H., Eichhorn, E. M., Liese, J. G. & Girschick, H. J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 162, 271–279 (2010).
Morisita, M. Measuring of the dispersion of individuals and analysis of the distributional patterns. Mem. Fac. Sci. Kyushu Univ. Ser. E 2, 5–235 (1959).
Horn, H. S. Measurement of ‘overlap’ in comparative ecological studies. Am. Nat. 100, 419–424 (1966).
Setliff, I. et al. Multi-donor longitudinal antibody repertoire sequencing reveals the existence of public antibody clonotypes in HIV-1 infection. Cell Host Microbe 23, 845–854 (2018).
Chao, A. Estimating the population size for capture–recapture data with unequal catchability. Biometrics 43, 783–791 (1987).
Kaplinsky, J. & Arnaout, R. Robust estimates of overall immune-repertoire diversity from high-throughput measurements on samples. Nat. Commun. 7, 11881 (2016).
Chao, A. & Chiu, C.-H. Nonparametric Estimation and Comparison of Species Richness https://doi.org/10.1002/9780470015902.a0026329 (John Wiley & Sons, 2016).
Eren, M. I., Chao, A., Hwang, W.-H. & Colwell, R. K. Estimating the richness of a population when the maximum number of classes is fixed: a nonparametric solution to an archaeological problem. PLoS ONE 7, e34179 (2012).
DeKosky, B. J. et al. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat. Med. 21, 86–91 (2015).
Arnaout, R. et al. High-resolution description of antibody heavy-chain repertoires in humans. PLoS ONE 6, e22365 (2011).
Marcou, Q., Mora, T. & Walczak, A. M. High-throughput immune repertoire analysis with IGoR. Nat. Commun. 9, 561 (2018).
Morea, V., Tramontano, A., Rustici, M., Chothia, C. & Lesk, A. M. Conformations of the third hypervariable region in the VH domain of immunoglobulins. J. Mol. Biol. 275, 269–294 (1998).
Finn, J. A. et al. Improving loop modeling of the antibody complementarity-determining region 3 using knowledge-based restraints. PLoS ONE 11, e0154811 (2016).
Briney, B. S., Willis, J. R., Finn, J. A., McKinney, B. A. & Crowe, J. E. Jr. Tissue-specific expressed antibody variable gene repertoires. PLoS ONE 9, e100839 (2014).
van Dongen, J. J. M. et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 17, 2257–2317 (2003).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13, 31 (2012).
Meyerhans, A., Vartanian, J. P. & Wain-Hobson, S. DNA recombination during PCR. Nucleic Acids Res. 18, 1687–1691 (1990).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Rogers, T. F. et al. Zika virus activates de novo and cross-reactive memory B cell responses in dengue-experienced donors. Sci. Immunol. 2, eaan6809 (2017).
Acknowledgements
The authors thank all of the study subjects for their participation and the Genomic Services Laboratory at the HudsonAlpha Institute for Biotechnology for their sequencing expertise. This work was supported by the National Institute of Allergy and Infectious Diseases (Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery, UM1AI100663 (D.R.B.); Center for Viral Systems Biology, U19AI135995 (B.B.)), the International AIDS Vaccine Initiative (IAVI) through the Neutralizing Antibody Consortium SFP1849 (D.R.B.), and the Ragon Institute of MGH, MIT and Harvard (D.R.B.).
Author information
Authors and Affiliations
Contributions
B.B. and D.R.B. planned and designed the experiments. B.B., A.I. and C.J. performed experiments. B.B. analysed data. B.B. and D.R.B. wrote the manuscript. All authors contributed to manuscript revisions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Nearly full-length antibody gene amplification from biological and technical replicate samples.
a, Schematic of biological and technical replicate samples. Biological replicates (columns) are derived from distinct cell aliquots, so identical clonotypes or sequences found in multiple biological replicates must arise from different cells. Technical replicates (rows) were amplified using discrete RNA aliquots from a single-cell aliquot. b, Strategy for nearly full-length antibody heavy chains. Black arrows indicate primers. Primers in the cDNA synthesis step anneal to the heavy-chain constant region (CH) and add the first unique molecular identifier (UMI) and the Illumina read 1 primer annealing site. Primers in the second-strand synthesis step anneal to the framework 1 region of the variable gene and add a second UMI and the Illumina read 2 primer annealing site.
Extended Data Fig. 2 V and J frequency correlations of technical and biological replicates.
For each subject, the frequency of V and J combinations was compared for technical replicates (left panels) or biological replicates (right panels). The coefficient of determination (r2) is shown for each plot.
Extended Data Fig. 3 Nucleotide mutation frequencies.
a, The distribution of nucleotide mutations in sequences that encode IgM are shown. On the right, the number of unmutated sequences containing no mutations in the variable-gene segment is also plotted. b, The distribution of nucleotide mutations in sequences that encode IgG are shown. On the right, the mean mutation frequency for the IgG population of each subject is shown. Each line represents a single subject. For legibility, the legend is split between the two plots. Although only five subjects are shown in the legend of each plot, data from all ten subjects is present in each plot.
Extended Data Fig. 4 Cross-subject repertoire similarity.
Pairwise Morisita–Horn similarity comparisons between each subject and all other subjects. Similarity was computed using the frequency of V-gene, J-gene and CDRH3 length combinations. Each line represents the mean of 20 independent repertoire samplings (with replacement). The shading surrounding the mean line indicates the 95% confidence interval.
Extended Data Fig. 5 Collapsing sequences into clonotypes.
a, To demonstrate the effect of collapsing an expanded clonal lineage into clonotypes, we selected a previously reported lineage of Zika-specific monoclonal antibodies isolated from the plasmablast population of an acutely infected patient24. Of 119 sequences, 89 were unique at the nucleotide level. b, Sequences encoding the same V gene, J gene and an identical CDRH3 amino acid sequence were collapsed into clonotypes, and the sequence phylogeny was coloured by clonotype. A total of 119 sequences were collapsed into 18 clonotypes. c, Sequences were collapsed into clonotypes, allowing a single mismatch in the CDRH3 amino acid sequence, and the sequence phylogeny was coloured by clonotype. A total of 119 sequences were collapsed into 10 clonotypes. d, The clonotype fraction (number of clonotypes divided by the total number of filtered sequences), when collapsing clonotypes while allowing zero or one mismatch in the CDRH3 amino acid sequence for each subject in this study. e, Number of total clonotypes recovered when allowing zero or one mismatch in the CDRH3 amino acid sequence for each subject in this study.
Extended Data Fig. 6 Capture–recapture frequency.
a, Recapture frequency for each subject. Lines represent the mean of 10 random samplings (without replacement) for all subsample fractions except compete sampling (1.0). b, Mean recapture frequency for each subsample fraction.
Extended Data Fig. 7 Relative light-chain diversity estimation.
Using previously reported datasets of paired heavy and light antibody chains, clonotype diversity was estimated for heavy and light chains using both Chao 2 and Recon estimators. Estimates are shown in filled or unfilled points. Lines indicate the least-squares polynomial best fit (degree = 2) and is extrapolated to include both the lowest (1.17 × 108) and highest (9.06 × 108) number of UMI-corrected sequences from the 10 sequenced subjects.
Extended Data Fig. 8 Variance between inferred V(D)J recombination models.
a, Frequency of clonotype sharing between observed human subjects (black), synthetic datasets generated with IGoR’s default recombination model (red), synthetic datasets generated with subject-specific recombination models (blue) or synthetic datasets generated with a combined-subject recombination model (purple). b, Combined Kullback–Leibler divergence (KL divergence) between pairs of subject-specific models (blue), between subject-specific models and IGoR’s default model (red), or between subject-specific models and the combined-subject model (purple). c, Combined KL divergence between pairs of subject-specific models, separated by event type.
Supplementary information
Rights and permissions
About this article

Cite this article
Briney, B., Inderbitzin, A., Joyce, C. et al. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397 (2019). https://doi.org/10.1038/s41586-019-0879-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-0879-y
This article is cited by
-
Anchor Clustering for million-scale immune repertoire sequencing data
BMC Bioinformatics (2024)
-
Systematic evaluation of B-cell clonal family inference approaches
BMC Immunology (2024)
-
The dengue-specific immune response and antibody identification with machine learning
npj Vaccines (2024)
-
Adaptive immune receptor repertoire analysis
Nature Reviews Methods Primers (2024)
-
Immunogenetics in hematopathology and hematology: why a common language is important
Leukemia (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
