
Abstract
Ubiquitous yet unique, lipid droplets are intracellular organelles that are increasingly being recognized for their versatility beyond energy storage. Advances uncovering the intricacies of their biogenesis and the diversity of their physiological and pathological roles have yielded new insights into lipid droplet biology. Despite these insights, the mechanisms governing the biogenesis and functions of lipid droplets remain incompletely understood. Moreover, the causal relationship between the biogenesis and function of lipid droplets and human diseases is poorly resolved. Here, we provide an update on the current understanding of the biogenesis and functions of lipid droplets in health and disease, highlighting a key role for lipid droplet biogenesis in alleviating cellular stresses. We also discuss therapeutic strategies of targeting lipid droplet biogenesis, growth or degradation that could be applied in the future to common diseases, such as cancer, hepatic steatosis and viral infection.
Key points
-
The primary roles of lipid droplet biogenesis are to alleviate cellular stress and maintain energy homeostasis.
-
Excess accumulation of lipid droplets and mutations in genes associated with lipid droplets are implicated in a plethora of pathologies.
-
Although they are beneficial to cell physiology under most conditions, lipid droplets might also exacerbate cytotoxicity and disease progression in certain disease settings.
-
Therapies targeting lipid droplet biogenesis, growth and degradation might be promising avenues for treating cancer, viral infection and hepatic steatosis.
Similar content being viewed by others
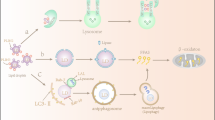


Introduction
Delimited by a surfactant phospholipid monolayer, lipid droplets are evolutionarily conserved organelles that enable the secure storage of hydrophobic neutral lipids in the hydrophilic cytosol. Although lipid droplets were initially observed in the late 1890s1,2, it was not until a century later, following the discovery of perilipin (PLIN) and oleosin3,4, that the molecular detail of lipid droplets received attention. Investigations over the past three decades have established lipid droplets as dynamic cellular organelles that affect many aspects of cell physiology5,6,7,8. In addition to energy storage, lipid droplets are crucial for relieving cellular stresses, including lipotoxic stress, endoplasmic reticulum (ER) stress, oxidation and starvation. Accordingly, their dysregulation or accumulation is associated with many disease conditions. Here, we review the latest advances in the current understanding of lipid droplet biogenesis and functions in health and disease.
Lipid droplet biogenesis
Based on their subcellular localization in eukaryotes, three types of lipid droplets have been described: cytoplasmic, nuclear and luminal (Fig. 1). In this section, we review the latest mechanistic insights into their biogenesis.

(1) Cytoplasmic lipid droplet biogenesis involves the synthesis, nucleation, cytoplasmic budding and growth of neutral lipids from the endoplasmic reticulum (ER). DGAT1 and DGAT2 catalyse the formation of triacylglycerols (TAG; see inset for the biosynthesis pathway), which, once accumulated beyond 2.8–10.0 mol%, form lens structures at sites putatively defined by proteins, such as seipin and its interacting partners. Regulated by monolayer tension asymmetry, budding towards the cytosol might then occur spontaneously or be promoted by both proteins and lipids, including seipin, FIT2 or diacylglycerol (DAG). Ostwald ripening, coalescence, membrane bridges and both ERTOLD and CYTOLD proteins might then contribute to the growth of cytoplasmic lipid droplets. (2) De novo nuclear lipid droplet biogenesis is similar to cytoplasmic lipid droplet formation but occurs at the inner nuclear membrane. While the role of seipin remains controversial, increased phosphatidic acid levels are hypothesized to be pivotal. (3) In lipoprotein-secreting cells, nuclear lipid droplet biogenesis might also occur via the engulfment of luminal lipid droplets. During ER stress, degradation of ApoB but maintenance of MTP levels (top right) enable ApoB-free luminal lipid droplet accumulation in the ER lumen before entry into the nucleus through type I nucleoplasmic reticulum breakage. (4) Luminal lipid droplet biogenesis remains poorly characterized and confounded with VLDL biogenesis (lipidated ApoB represents nascent VLDL particles). Luminal lipid droplets are characterized as ApoB-free precursors. MTP might connect cytoplasmic lipid droplets with the formation of luminal lipid droplets and might also mediate the lipid transfer between luminal lipid droplets and lipidated ApoB. AGPAT, 1-acylglycerol-3-phosphate acyltransferase; ApoB, apolipoprotein B-100; CIDE, cell death-inducing DFFA-like effector; CYTOLD, cytoplasm to lipid droplet targeting; DGAT, diacylglycerol transferase; ERTOLD, ER to lipid droplet targeting; FIT2, fat storage-inducing transmembrane protein 2; G-3-P, glycerol-3-phosphate; GPAT, glycerol-3-phosphate acyltransferase; LPA, lysophosphatidic acid; MTP, microsomal lipid transfer protein; PA, phosphatidic acid; PAP, phosphatidic acid phosphatase; Pi, inorganic phosphate; PLIN3, perilipin 3.
Cytoplasmic lipid droplets
Often used to refer generally to all lipid droplets, cytoplasmic lipid droplets are by far the most well characterized form of these increasingly recognized, diverse organelles. Many models have been proposed for their biogenesis9,10,11,12,13,14,15,16,17,18,19,20,21,22, with the most empirically supported version involving the synthesis, nucleation and cytoplasmic budding and growth of neutral lipids from the ER5,7,8 (section 1 of Fig. 1).
Triacylglycerols and sterol esters are the most abundant neutral lipids in eukaryotes23. Triacylglycerols are synthesized from glycerol-3-phosphate through a series of enzymatic reactions starting with the rate limiting enzyme, glycerol-3-phosphate acyltransferase (GPAT) (section 1 of Fig. 1). In mammals, triacylglycerols are synthesized by the esterification of diacylglycerol through diacylglycerol acyltransferases (DGAT1 and DGAT2)24,25,26. Cholesteryl esters are synthesized by acyl-CoA cholesterol O-acyltransferases 1 and 2 (ACAT1 and ACAT2), which are also known as sterol O-acyltransferase 1 and 2 (SOAT1 and SOAT2)27,28,29. The molecular structures of most of these acyltransferases have been resolved30,31,32,33,34, revealing similar catalysing mechanisms between the closely related DGAT1, ACAT1 and ACAT2 enzymes. In the budding yeast, Saccharomyces cerevisiae, Dga1 and the luminally oriented Lro1 (refs. 20,35,36), as well as Are1 and Are2 (ref. 37), catalyse the esterification of triacylglycerol and sterol esters. Crucially, although genetic deletion in yeast (ARE1Δ ARE2Δ LRO1Δ DGA1Δ)38,39 or mice (Dgat1Dgat2−/−)40 ablates lipid droplet content, individual expression of Dga1, Lro1 and Are2 (listed in decreasing order of efficacy) is sufficient for the restoration of lipid droplet formation41. This finding demonstrates the necessity of neutral lipid synthesis for cytoplasmic lipid droplet biogenesis, which thus far remains the only obligatory process in cytoplasmic lipid droplet formation8,42,43.
Once the newly synthesized neutral lipids accumulate beyond a critical concentration (2.8–10.0 mol% triacylglycerol)44,45,46, a lens between the ER bilayers might form via the de-mixing phenomenon of phase separation23,47, experimentally observed in yeast as 30–60 nm diameter structures48. This lens formation occurs when interaction of neutral lipids with themselves becomes thermodynamically more favourable than interaction with their membrane components. In addition, lens formation might occur spontaneously47,49 or be influenced by membrane tension50, curvature51,52 and/or local lipid and protein composition53,54. The molecular details of neutral lipid nucleation and lens formation are unknown; however, seipin and its yeast orthologue Sei1 (also known as Fld1) seem to have a prominent role in this process55,56. Namely, Sei1 forms a decameric cage-like structure that might facilitate the phase separation of triacylglycerol57,58. Moreover, seipin59 and proteins that interact with it, such as lipid droplet assembly factor 1 (LDAF1)60, NEM1 (ref. 61), multiple C2 and transmembrane domain containing 2 and its yeast homologue, Pex30 (refs. 62,63,64), help define the sites at which the formation of cytoplasmic lipid droplets is initiated. Other proteins, such as PLIN3 (refs. 15,18), as well as fat storage-inducing transmembrane protein 2 (FIT2) and its ER tubule-forming binding partners, receptor expression-enhancing protein 5, reticulon 4 and the cytoskeletal septin 7, also contribute to initiating the formation of cytoplasmic lipid droplets65. In addition to proteins, lipids such as phosphatidic acid and diacylglycerol might also have a crucial role in regulating triacylglycerol nucleation and lens formation8.
Following nucleation, neutral lipids might either be resorbed or bud into spherical lipid droplets by overcoming the deformation energy barrier of their encapsulating leaflets23,47. In yeast, cytoplasmic lipid droplets remain functionally connected to the ER66,67, whereas both fully detached and ER-connected cytoplasmic lipid droplets have been observed in mammals59,68. Explained by the principles of dewetting (spreading propensity of a liquid determined by tensions between interacting phases), budding can result from a gradual increase in the contact angle between emerging lipid droplets and the ER bilayer47,69. Theoretically, cytoplasmic lipid droplet budding can occur spontaneously without assistance from curvature-inducing agents or proteins70,71, and its direction might be regulated by an asymmetry in monolayer tension, with cytoplasmic lipid droplet budding occurring due to reduced tension towards the cytosol72. Empirically, however, both proteins and lipids have been implicated in the promotion of cytoplasmic lipid droplet budding73,74. In particular, seipin and PEX30 have been shown to organize budding-permissive ER domains, potentially through alterations in phosphatidylcholine, phosphatidylinositol and diacylglycerol62. Furthermore, FIT2 might determine budding direction via its luminally localized phosphatase activity, which depletes phospholipids to form diacylglycerol48,51,75,76.
To mature into their full storage potential, budded cytoplasmic lipid droplets might then grow via several biophysical and/or protein-assisted mechanisms. One key mechanism involves a process called Ostwald ripening, which promotes the transfer of neutral lipids from smaller lipid droplets to larger, more stable lipid droplets23,48,70. This transfer is assisted by the lipid droplet-associated, cell death-inducing DFFA-like effector (CIDE) family of proteins (CIDEA, CIDEB and CIDEC), which can form pores that connect and mediate the directional transfer of contents from small to large lipid droplets77 (section 1 of Fig. 1). Distinct from lipid droplet growth mediated by CIDE proteins, rapid coalescence of lipid droplets could also take place in the presence of inadequate levels of phosphatidylcholine78 or an accumulation of phosphatidic acid54. Membrane bridges connecting cytoplasmic lipid droplets to the ER represent another means of cytoplasmic lipid droplet growth. Here, activation of ARF1–COPI complexes might trigger the formation of membrane bridges79, allowing certain enzymes that synthesize triacylglycerol, including GPAT4, to relocate from the ER to lipid droplets80. Besides CIDE proteins and GPAT4, many other proteins can reach and associate with lipid droplets through the ER to lipid droplet (ERTOLD) or cytosol to lipid droplet (CYTOLD) pathways and contribute to the growth of lipid droplets81,82,83 (section 1 of Fig. 1). Finally, lipid transfer proteins, such as oxysterol binding protein related protein 5, might deliver phospholipids from the ER to lipid droplets, thereby promoting lipid droplet growth84.
Nuclear lipid droplets
Originally considered cytoplasmic lipid droplet entrapments in the nucleus, nuclear lipid droplets are now emerging as true organelles with their own distinct set of features (sections 2 and 3 of Fig. 1). Indeed, although their identification was first limited to hepatic cells and tissues85,86,87,88, their characterization in various species (Arabidopsis thaliana, S. cerevisiae and Caenorhabditis elegans, as well as mammalian prostatic epithelial and osteosarcoma cell lines) and conditions (seipin knockdown)89,90,91,92,93,94,95,96 has suggested that nuclear lipid droplets might not be as scarce and physiologically irrelevant as once inferred. In fact, depending on the cell type in which they reside, two main mechanisms of nuclear lipid droplet biogenesis have been identified: first, de novo generation and budding at the inner nuclear membrane, and second, engulfment of luminal lipid droplets via the type I nucleoplasmic reticulum.
In both mammalian U2OS cells (a human osteosarcoma cell line) and yeast, nuclear lipid droplet biogenesis is suggested to occur de novo. Increased phosphatidic acid levels at the inner nuclear membrane have a pivotal role, and various mechanisms have been hypothesized. In U2OS cells, the absence of seipin, which normally binds phosphatidic acid and inhibits upstream GPAT3 and GPAT4 (refs. 97,98), might enable free phosphatidic acid diffusion to the inner nuclear membrane, where upregulation of the diacylglycerol-producing enzyme (Lipin-1) might promote triacylglycerol synthesis96. In yeast, conditions that increase the levels of phosphatidic acid, such as inhibition of the phosphatidic acid to CDP diacylglycerol-converting enzyme, Cds1, or its transcriptional upregulators, Ino2 and Ino4 (ref. 54), are considered essential for nuclear lipid droplet biogenesis90. However, the role of seipin remains controversial. Seipin is directly implicated in the promotion of yeast nuclear lipid droplet biogenesis through its formation of membrane bridges that link the inner nuclear membrane and nuclear lipid droplets90. However, a suppressive role for seipin has been demonstrated; similarly to U2OS cells, yeast deficient in seipin or its binding partner Ldb16 contains increased levels of nuclear lipid droplets99,100. Despite these discrepancies, the presence of enzymes that synthesize neutral lipids (yeast Pah1 and Cds1, and mammalian Lipin-1, GPAT3, GPAT4, DGAT1 and DGAT2) at the inner nuclear membrane90,92,96 suggests that their nuclear lipid droplet biogenesis should still mimic cytoplasmic lipid droplet formation.
In hepatocytes, nuclear lipid droplet biogenesis might occur via the engulfment of luminal lipid droplets (see next section ‘Luminal lipid droplets’). Here, instead of secreting VLDLs containing both lipidated apolipoprotein B-100 (ApoB) and ApoB-free luminal lipid droplets during ER stress, ApoB-free luminal lipid droplets accumulate in the ER lumen before eventually entering the nucleus through the type I nucleoplasmic reticulum101. This process relies on both degradation of the scaffolding protein, ApoB102, and maintenance of the lipid transferring chaperone, microsomal triglyceride transfer protein (MTP)103 (section 4 of Fig. 1). Their concurrent imbalance in hepatocytes then enables the extension of type I nucleoplasmic reticulum through the expression of promyelocytic leukaemia protein isoform II101,104. Being deficient in lamin A, lamin C and lamin B1 (ref. 105), the type I nucleoplasmic reticulum is then vulnerable to breakage upon contact with large ApoB-free luminal lipid droplets, enabling them to enter the nucleus.
Luminal lipid droplets
Luminal lipid droplets are the least characterized members of the eukaryotic lipid droplet family, but they are known to have similar origins to cytoplasmic lipid droplets and are precursors in nuclear lipid droplet biogenesis (section 4 of Fig. 1). Due to their topological and ultrastructural resemblance to lipoproteins20,106, luminal lipid droplet biogenesis is often confounded with the established VLDL biogenesis pathway, in which triacylglycerols are transferred to partially lipidated ApoB-free precursors via a two-step process that is facilitated by MTP107. Distinct from VLDL particles, luminal lipid droplets are characterized as lipid droplets that have low levels of triacylglycerol and lack ApoB, PLIN2 and albumin; instead, they contain ApoE, triacylglycerol hydrolase, carboxylesterase 1 and MTP108. Minimal molecular insights into the biogenesis of luminal lipid droplets are currently available, but MTP might have a crucial role in facilitating the transfer of triacylglycerol from cytoplasmic lipid droplets to luminal lipid droplets and possibly between luminal lipid droplets and nascent VLDL particles95,108. It also remains to be unequivocally determined if luminal equivalents of cytoplasmic lipid droplets actually exist, and if so, what role (or roles) they have in lipid droplet biology. Key cytoplasmic lipid droplet proteins, LRO1, FIT2 and seipin, have been demonstrated to have active sites (synthesizing triacylglycerol (LRO1) or diacylglycerol (FIT2)) or mutations associated with diseases (N88S and S90L seipin) in the ER lumen20,48,54,75,76,109. These findings suggest that the biogenesis of cytoplasmic lipid droplets and luminal lipid droplets might share some common components and/or mechanisms. Moreover, the accessibility of high affinity cytoplasmic lipid droplet proteins106 and milk-producing lipid droplets110,111 to the ER lumen further support the idea that true luminal lipid droplet biogenesis occurs. Thus, akin to the initial dismissal of both cytoplasmic lipid droplets and nuclear lipid droplets, further interrogation of luminal lipid droplets is warranted and might yield exciting new insights.
Lipid droplet biogenesis as a response to cellular stress
Although the most recognized function of cytoplasmic lipid droplets is to serve as energy storage organelles, many lines of evidence suggest that the primary goal of lipid droplet biogenesis is to alleviate cellular lipotoxic stress, as well as stresses associated with disturbed ER homeostasis, oxidation and starvation6,112,113,114 (Fig. 2). Substrates of triacylglycerol and cholesterol ester synthesis (fatty acids, fatty acyl CoA, diacylglycerol and cholesterol) are bioactive lipids that can disrupt normal cell function when in excess115,116,117,118,119. Oleate, the most commonly used lipid for inducing lipid droplet formation, is highly toxic to both yeast and mammalian cells when the ability to synthesize neutral lipids is compromised116,120. However, oleate is far less toxic than palmitate in wild-type cells as it can easily be converted to triacylglycerol for storage in lipid droplets116. In fact, unsaturated fatty acids, including oleate, can rescue palmitate-induced cytotoxicity by promoting the incorporation of palmitate into triacylglycerols116. These toxicity-lowering effects of triacylglycerol synthesis and lipid droplet biogenesis have also been demonstrated in numerous models, including the fission yeast Schizosaccharomyces pombe112, mouse adipocytes121, mouse endothelial cells122, mouse cardiomyocytes123 and fly brain124.

Cellular stress, including endoplasmic reticulum (ER), oxidation and starvation might trigger increased lipogenesis, autophagy, lysosomal phospholipid turnover or the activity of phospholipases. In turn, increased flux of free fatty acids and other lipid intermediates to the ER might induce lipotoxicity. Lipid droplet biogenesis, which is initiated by increased neutral lipids at the ER, might then be promoted to sequester toxic free fatty acids. Through their various roles, including maintaining energy and membrane homeostasis and enabling protein storage and degradation, lipid droplets might further support stress alleviation.
In addition to lipotoxicity, other forms of stress can also trigger lipid droplet biogenesis6,113,114. When nutrients are limited, yeast cells enter a stationary growth phase and accumulate lipid droplets37,112. This accumulation is partly due to inositol deficiency, which slows down the synthesis of phospholipids but promotes that of diacylglycerol and triacylglycerol125. In mammalian cells, long-term starvation or mTORC1 inhibition by TORIN1 triggers autophagy, which then increases fatty acid release via membrane degradation. DGAT1 converts these fatty acids into triacylglycerol, which promotes lipid droplet biogenesis and prevents lipotoxic damage to mitochondria126,127. In a study published in 2022, mTORC1 inhibition or nutrient deprivation were demonstrated to increase the delivery of endosomal cargo to lysosomes and promote triacylglycerol accumulation through lysosome-dependent but autophagy-independent hydrolysis of phospholipids128. Increased phospholipase expression and activity might also account for the hydrolysis of phospholipids under these conditions128. Mechanistic differences aside, cells seem to adapt to starvation by increasing membrane turnover and the flux of fatty acids and other lipid intermediates to the ER for triacylglycerol synthesis and lipid droplet biogenesis.
Similar to starvation, conditions of ER stress increase the synthesis of neutral lipids and lipid droplet formation in the budding yeast113. ER stress pathways can also promote lipogenesis and increase triacylglycerol synthesis in mammals129. In addition to lipogenesis, phospholipid remodelling and turnover could also contribute to triacylglycerol synthesis and lipid droplet formation under these conditions. For instance, ER stress activates the mitogen-activated protein kinases, which can phosphorylate and activate phospholipases such as cPLA2α to promote lipid droplet biogenesis130,131,132.
Oxidative stress arising from reactive oxygen species (ROS) is often associated with increased lipid droplet biogenesis114. The underlying mechanism remains to be elucidated but might involve the activation of lipogenesis mediated by sterol regulatory element-binding protein (SREBP), as well as phospholipid turnover and remodelling133,134. Upon oxidative stress, phospholipases might release polyunsaturated fatty acids (PUFAs) from membranes so that they can be stored in lipid droplets, where PUFAs are less susceptible to peroxidation134. Whether lysosomal digestion of phospholipids contributes to increased levels of fatty acids and lipid droplet biogenesis under ER or oxidative stress is an interesting topic for future investigation128.
In summary, it seems that most, if not all, forms of stress factors can induce membrane degradation and/or lipogenesis, thereby increasing the flux of free fatty acids to the ER. Triacylglycerol synthesis and lipid droplet biogenesis are then induced to reduce the level of toxic polar lipids (Fig. 2). Once formed, lipid droplets can also relieve cellular stress in other ways, such as by facilitating protein storage and turnover and shielding PUFAs from ROS damage114,135. By alleviating cellular stress, lipid droplets help maintain membrane homeostasis, a crucial function for normal cell physiology, as well as the survival of viruses and some forms of cancer (see sections ‘Bacterial and viral infection’ and ‘Cancer’). Finally, while many stress conditions might increase the release and/or synthesis of free fatty acids, the long-term presence of excessive free fatty acids, especially palmitate, can also cause ER and oxidative stress, thereby potentially creating a vicious cycle and driving lipid droplet accumulation further129,136.
Lipid droplets in health and disease
From alleviating cellular stresses and facilitating energy regulation to providing membrane lipid precursors, acting as hubs in lipid trafficking and docking sites for protein storage and degradation, lipid droplets serve many vital homeostatic and operational functions. Not surprisingly, both their dysregulated accumulation in cells and mutations in genes associated with lipid droplets (Table 1) have been linked with disease (Box 1). In this section, we review the latest understanding of the multifactorial roles of lipid droplets in disease.
Obesity
Through their regulation of lipid droplet size, several proteins associated with lipid droplets have been linked with the pathogenesis of diet-induced obesity. For instance, the expression of CIDEA, CIDEC and PLIN1 were found to be upregulated in the visceral adipose tissue of adult women with obesity137. Likewise, single nucleotide polymorphisms (rs1154588, rs4796955, rs8092502 and rs7230480) in the gene encoding CIDEA were identified as risk factors for obesity in Han Chinese and Swedish populations138,139. Null mutations in mice or cellular knockdown of CIDEA140, CIDEB141, CIDEC (also known as fat-specific protein 27, FSP27)142 or PLIN1 (refs. 143,144) have also demonstrated a clear pro-obesogenic role for these proteins. Mechanistically, promotion of a large unilocular lipid droplet phenotype through interaction and activation of CIDEC (residues 38–217) with PLIN1 (residues 291–318)145 might be responsible for the obesogenic effect. Interestingly, while both CIDEC-knockout mice and PLIN1-knockout mice exhibit drastically reduced levels of white adipose tissue and elevated lipolysis, only CIDEC-knockout mice remain insulin-sensitive, potentially through their increased fragmentation of, and mitochondrial access to, lipid droplets146. Contrary to the interaction between CIDEC and PLIN1, the interaction of CIDEA and CIDEC with CLSTN3β promotes lipid droplet multilocularity through disrupting lipid transfer between lipid droplets147. This finding suggests a role for other factors that interact with CIDE proteins in the regulation of lipid droplet size in obesity.
In addition to posing its own health challenges, many of the effects of obesity stem from the morbidities that are associated with obesity, such as type 2 diabetes mellitus (T2DM), non-alcoholic fatty liver disease (NAFLD), cardiovascular diseases, respiratory diseases and some cancers. Although these associations are firmly established on an epidemiological level, the causal and temporal relationships between obesity and its putative pathogenic factors, insulin resistance and hyperinsulinaemia, remain unclear148. Indeed, although insulin resistance was initially presumed to increase in parallel with adiposity149, ectopic lipid accumulation and lipotoxicity resulting from limited subcutaneous (hyperplastic) adipose tissue expansion (Supplementary Box 1) have now emerged as being key contributors to the onset of metabolic diseases.
The influence of ectopic lipid accumulation is best exemplified by the characterization of lipid distribution in the athlete’s paradox, metabolically healthy obesity and lipodystrophy. In the athlete’s paradox, elevated intramyocellular lipid storage, which is normally negatively associated with insulin resistance, is observed in insulin-sensitive athletes150. An increased number of lipid droplets in the intramyofibrillar region of type I fibres in trained individuals might be responsible for this paradox, whereas increased lipid droplet size in the subsarcolemmal region of type II fibres is characteristic of T2DM150. Moreover, in metabolically healthy obesity, where individuals have a reduced risk of cardiometabolic abnormalities in relation to their BMI, reduced mean adipocyte size, inflammatory markers and ectopic (visceral and liver) adiposity are observed151. These observations are accompanied by increased subcutaneous gluteofemoral and leg adiposity, increased capacity for hyperplastic adipose tissue expansion and preserved insulin sensitivity152. Finally, severe insulin resistance is observed in lipodystrophy (see next section ‘Lipodystrophy’), a disorder characterized by insufficient rather than excess adipose tissue.
Lipodystrophy
Lipodystrophy is a heterogeneous disorder defined by the selective loss of adipose tissue. Historically, it was regarded as infrequent with estimates of ~1 in 1–10 million people153, but it is now estimated to be as prevalent as ~1 in 7,000 people in cohort-based health record studies154. Despite representing the adipose tissue antithesis of obesity, lipodystrophy manifests with severe obesity-associated metabolic complications155. This superficial paradox occurs due to overwhelmed adipose tissue stores, where drastically reduced adipose tissue capacity and expandability culminate in lipotoxicity156. Mechanistically, reduced adipose tissue capacity involves impaired adipocyte differentiation or premature adipocyte death resulting in increased circulating levels of fatty acids, ectopic lipid accumulation and/or hyperphagia and reduced energy expenditure linked to low leptin levels157. In turn, these sequelae might promote systemic insulin resistance and subsequent cardiometabolic dysfunction157.
Depending on its aetiology (genetic or acquired) and the extent of adipose tissue loss (general or partial), lipodystrophy can be categorized into four broad groups: congenital generalized lipodystrophy, acquired general lipodystrophy, familial partial lipodystrophy and acquired partial lipodystrophy. The acquired forms can originate from autoimmune disorders, result in progressive loss of adipose tissue and are three to four times more common in female than in male individuals158, whereas the generalized forms are predominantly inherited via mutations in genes involved in lipid droplet biogenesis or biology.
Currently, four molecularly distinct subtypes are described for congenital generalized lipodystrophy, and there are six genetic subtypes of familial partial lipodystrophy. In congenital generalized lipodystrophy type 1, mutations in the gene encoding the lysophosphatidic acid to phosphatidic acid converting enzyme, AGPAT2, lead to the ablation of metabolically active adipose tissue and preservation of mechanical adipose tissue in humans159. Although AGPAT2 is upregulated 30-fold during adipocyte differentiation and is involved in the regulation of early stages of adipogenesis160,161, the redundancy of AGPAT isoforms might be responsible for the maintenance of mechanical adipose tissue depots162. By contrast, mutations in BSCL2, the gene encoding seipin, result in the absence of all forms of adipose tissue in congenital generalized lipodystrophy type 2. Specifically, global or adipose-specific Bscl2-knockout mice exhibit loss of white and brown adipose tissue, adipocyte hypertrophy, inflammation and insulin resistance, as well as a range of complications163,164. Both seipin and AGPAT2 are essential in the biogenesis and growth of lipid droplets and are associated with phosphatidic acid97,98,165; however, exactly how they regulate both lipid droplet biogenesis and adipogenesis remains to be elucidated (Supplementary Box 2).
In congenital generalized lipodystrophy type 3 and type 4, mutations in CAV1 (encoding caveolin 1) and PTRF (encoding cavin 1), respectively, enable retention of mechanical adipose tissue and bone marrow adipose tissue despite loss of metabolically active adipose tissue162. Caveolae maintain the integrity of lipid droplets166; caveolin 1 is a major component of caveolae and cavin 1 is essential for caveolae biogenesis. Therefore, the pathogenesis of these proteins is also linked, albeit indirectly, to the regulation of lipid droplets. With the exception of familial partial lipodystrophy type 1 and type 2, the link between lipid droplets and lipodystrophy also extends to most types of familial partial lipodystrophies, where mutations in the genes encoding PPARγ, PLIN1, LIPE and CIDEC lead to familial partial lipodystrophy types 3, 4, 5 and 6, respectively (Table 1).
Non-alcoholic fatty liver disease
Defined by an accumulation of lipid droplets in the liver, NAFLD is a chronic heterogeneous disease with a global prevalence of 32.4%167. As NAFLD can occur as a cause, consequence and exacerbator of metabolic dysfunction168,169, a reclassification of NAFLD to metabolic associated fatty liver disease (MAFLD) has been proposed in the past few years170. Originating as simple steatosis, NAFLD can progress to steatohepatitis with fibrosis, cirrhosis, liver failure and potentially hepatocellular carcinoma171. This progression has two proposed means of pathogenesis: the two-hit hypothesis and the multiple-hit hypothesis. In the two-hit hypothesis, hepatic lipid droplet accumulation (first hit) potentiates subsequent oxidative stress and inflammation (second hit)172. In the multiple-hit hypothesis, several factors (such as the aforementioned first and second hits, insulin resistance, mitochondrial dysfunction, gut microbiota, genetic factors and epigenetic factors) act in parallel to initiate and progress hepatic steatosis173,174.
At the molecular level, initial accumulation of lipid droplets in the liver might occur as an adaptive response to imbalanced fatty acid supply and removal. During fasting, adipose tissue lipolysis (Supplementary Box 3) almost exclusively mediates fatty acid flux to the liver175. During postprandial periods, chylomicron incorporation of dietary fats or de novo lipogenesis of dietary glucose mediate this flux176. Although NAFLD can occur in lean individuals177, and occurs independently of comorbidities178, it is often accompanied by selective hepatic insulin resistance, in which considerably elevated levels of de novo lipogenesis might further contribute to NAFLD179. Depending on physiological needs, fatty acids entering hepatocytes can be partitioned for β-oxidation or esterification into neutral lipids180. Following neutral lipid synthesis and lens formation (see section ‘Cytoplasmic lipid droplets’), cytoplasmic lipid droplet biogenesis might either proceed with budding towards the cytosol or remain embedded in the ER bilayer. In the latter state, the unfolded protein response might be triggered, leading to ER stress, increased production of ROS and exacerbation of lipid droplet accumulation in the liver181. Thus, lipid droplet biogenesis in the liver is initially protective; however, long-term and excessive lipid droplet accumulation can be detrimental to hepatocyte function. For instance, mice fed a high-fat diet for 12 weeks exhibit accumulation of enlarged lipid droplets in the liver and sequestration of proteins from other cellular compartments, thereby disrupting the function of other organelles, in particular the Golgi182.
In addition to dysregulated lipid droplet metabolism, several genetic factors associated with lipid droplets have been implicated in the pathogenesis of NAFLD. The most prominent risk factor is the single nucleotide polymorphism I148M in patatin-like phospholipase domain-containing protein 3 (PNPLA3), which was independently identified in two genome-wide association studies183,184. Whilst PNPLA3 depletion does not result in hepatic steatosis185,186, PNPLA3 I148M (rs738409) promotes NAFLD through three distinct yet cumulative functions. Namely, it ablates PNPLA3-mediated triacylglycerol hydrolase activity187,188, leading to lipid droplet accumulation. It also evades ubiquitination and autophagic degradation189,190, resulting in increased levels of PNPLA3 I148M on accumulated lipid droplets. PNPLA3 I148M on the lipid droplet surface might then competitively bind to and sequester αβ-hydrolase domain-containing 5 (ABHD5; also known as CGI-58) away from PNPLA2 (also known as ATGL)191,192, preventing subsequent lipolysis and lipophagy of large and small lipid droplets, respectively193. In addition, PNPLA3 I148M also exacerbates hepatic ballooning and fibrosis194, potentially through the pro-inflammatory attenuation of its retinol palmitate lipase activity195,196.
Two variants of 17β-hydroxysteroid dehydrogenase 13 (17β-HSD13), rs72613567:TA and rs6834314, which have retinol dehydrogenase activity197, have been suggested to protect against PNPLA3 I148M-induced hepatic fibrosis198,199. Initially identified in a proteomics screen200, 17β-HSD13 is a hepatocyte-specific protein that is localized to lipid droplets and is involved in the promotion of hepatic lipogenesis and NAFLD pathogenesis. The mechanism by which 17β-HSD13 and mutated forms of this protein regulate steatosis and lipid droplet dynamics remains unclear; however, a study linking 17β-HSD13 with lipolysis regulation revealed new insights. Specifically, wild-type 17β-HSD13 physically interacts with PNPLA2 on the surface of lipid droplets, but this interaction is reduced by protein kinase A-mediated phosphorylation of 17β-HSD13 at serine 33, thereby enabling PNPLA2–ABHD5 binding and subsequent hepatic lipolysis201.
Mutations in the gene encoding CIDEB, another hepatocyte-specific protein localized to lipid droplets, have also been linked to NAFLD. Although >20 somatic CIDEB mutations have been identified in chronic liver disease202, rare germline mutations in CIDEB offer substantial protection against liver disease203. The role of CIDEB in NAFLD regulation remains poorly characterized; however, CIDEB is known to facilitate lipid droplet growth and lipid exchange from small to large lipid droplets in hepatocytes204. It also promotes VLDL lipidation and the biogenesis of its transport vesicles205,206. Moreover, CIDEB also acts as the linchpin of SREBP activation through selectively promoting SREBP and SCAP loading into COPII-coated vesicles207.
Finally, numerous other proteins and genetic variants, including TM6SF2 E167K208, MBOAT7 rs641738 (ref. 209), ATG7 rs143545741 (ref. 210), rare coding variants in ABCB4 (ref. 211), APOB212 and SLC30A10 (ref. 213), PLIN2 S251P214 as well as PLIN1 and PLIN3 (ref. 215), are associated with NAFLD pathogenesis or protection. However, the mechanisms of action of these variants remain largely elusive.
Pancreatic steatosis: T2DM, pancreatitis, pancreatic cancer
Increasingly recognized as being as clinically important as NAFLD, pancreatic fat accumulation comprises either adipocyte infiltration in the exocrine pancreas (predominant route) or ectopic accumulation of triacylglycerol-rich lipid droplets in the endocrine pancreas216. Despite being identified almost a century ago217, methods to accurately and efficiently detect and quantify pancreatic fat are lacking, and there is no consensus on the classification for its pathology. Unsurprisingly, characterization of its true global prevalence is limited, with the best, albeit biased, estimates suggesting 16–35%218,219. Although minimal pancreatic fat is normal in healthy individuals and fatty acids serve as potentiators of glucose-stimulated insulin secretion220, excess fat accumulation in the pancreas, most broadly termed pancreatic steatosis221, is now recognized as detrimental for both its endocrine and exocrine roles. Indeed, whilst increased pancreatic fat content has long been correlated with obesity and T2DM222,223,224,225, pancreatic steatosis is also emerging as a potential primary cause of chronic pancreatitis226, and is associated with pancreatic ductal adenocarcinoma227.
Pancreatic steatosis is affected by several risk factors, including age, sex, the metabolic syndrome, NAFLD (67.9% concurrence rate) and lifestyle factors such as alcohol, tobacco and cannabis consumption226,228. As such, pancreatic steatosis might have a causative role in the pathogenesis of many diseases of the pancreas. In T2DM, which accounts for ~90% of the 537 million cases of diabetes mellitus worldwide229, pancreatic steatosis might disturb glucose metabolism and impair insulin secretion. Specifically, high levels of liver-originating fetuin A coupled with local fatty acid release might activate pro-inflammatory toll-like receptor 4, altering both the secretome230 and the differentiation potential of pancreatic adipocytes231. A vicious twin cycle involving blunted insulin suppression of hepatic glucose production and compensatory hyperinsulinaemia might then ensue, leading to hyperglycaemia and β-cell dysfunction115. Bidirectionally linked with T2DM228, the pathogenesis of chronic pancreatitis likewise involves inflammation derived from pancreatic steatosis in the form of toll-like receptor-dependent signalling. Moreover, in type 3c diabetes mellitus, where diabetes mellitus of the exocrine pancreas might develop after acute pancreatitis, cells and secreted hormones of pancreatic islets have been suggested to be impaired by inflammation, fibrosis and sclerosis232. Finally, increased cytokine production arising from pancreatic steatosis might also predispose human pancreatic cells to malignant transformation233.
Neurodegeneration
Lipid droplets were originally observed over a century ago alongside amyloid-β plaques and hyperphosphorylated tau tangles in Alzheimer disease234. As the most common neurodegenerative disorder, Alzheimer disease is expected to reach a global prevalence of >150 million cases in 2050 (ref. 235). Since this initial observation, lipid droplets have been identified in numerous brain cell types, including neurons, astrocytes, oligodendrocytes, microglia and ependymal cells. Although only microglia and ependymal cells generally accumulate lipid droplets under non-pathological conditions236,237, lipid droplets in the brain commonly form in response to inflammation, oxidative stresses and ageing133,238. Indeed, increased ROS production, hypoxia and metabolic stress lead to the formation of lipid droplets in astrocytes as a neuronal detoxification mechanism239,240. Specifically, as neurons have a limited capacity to store and catabolize lipid droplets due to weak antioxidant defences241,242, peroxidated lipids generated via ROS are sequestered by apolipoproteins (ApoE and ApoD) and are stored in glial lipid droplets239,243. The APOE4 allele, the strongest genetic risk factor for both early-onset and late-onset Alzheimer disease244, has reduced lipid transfer capacity, causing impaired formation of glial lipid droplets239,243. Several other genetic risk factors for Alzheimer disease also affect the formation of neuroprotective glial lipid droplets245. In Parkinson disease, lipid droplet accumulation might also have a protective role, as enhanced cytotoxicity is observed in the absence of lipid droplet formation in yeast models expressing human α-synuclein246. Interestingly, the pathological, α-synuclein protein binds to the surface of lipid droplets, which promotes their accumulation by preventing their lipolysis247,248. This binding ability is either enhanced (E46K and A53T)247,249 or reduced (A30P and G51D)250,251 in mutated forms of α-synuclein that are involved in Parkinson disease. Finally, a large number of proteins involved in lipid droplet biogenesis or biology have been implicated in motor neuron diseases, including amyotrophic lateral sclerosis (associated with seipin and hVAPB)252, and hereditary spastic paraplegia (associated with seipin, DDHD2, atlastin-1 and spastin)253. However, their mechanistic details are yet to be elucidated.
Bacterial and viral infection
Lipid droplets serve as dynamic platforms for fighting against bacterial infection254. Although bacteria-derived lipopolysaccharide has long been known to increase lipid droplet formation in mammalian cells, it remains unclear whether lipid droplets function to support or undermine bacterial infection. In a bacterial killing assay of Escherichia coli, purified lipid droplet proteins reduced bacterial growth254. Lipid droplet proteins purified from lipopolysaccharide-treated cells have enhanced antibacterial activity, suggesting that mammalian lipid droplets might have an intrinsic antibacterial function that is enhanced by infection. Mechanistically, lipopolysaccharide treatment increases levels of PLIN2 and concentrates multiple host defence proteins and antimicrobial peptides on lipid droplets254. Lipopolysaccharide treatment also downregulates PLIN5, a key protein for establishing contacts between lipid droplets and mitochondria. This downregulation of PLIN5 uncouples lipid droplets from mitochondria and reduces fatty acid metabolism and oxidative phosphorylation. Thus, mammalian lipid droplets can organize and concentrate immune proteins to attack the invading pathogen while also modulating cell metabolism to adapt to infection.
By contrast, many lines of evidence indicate a supportive role for lipid droplets in the replication and proliferation of positive-stranded RNA viruses, such as flaviviruses (for example, hepatitis C virus and dengue virus) and SARS-CoV-2 (refs. 255,256). Accumulation of cytoplasmic lipid droplets is commonly observed in cells infected by positive-stranded RNA viruses, and lipid droplets can serve as stations for virus replication and assembly. Coronaviruses, including SARS-CoV-2, induce the formation of a replication organelle predominantly composed of double membrane vesicles, which protects their genomes from detection and destruction by host cells. SARS-CoV-2-induced double membrane vesicles are connected to the ER by thin membrane connectors, which enable lipids (but not bulky proteins) to access double membrane vesicles256. Three transmembrane proteins of SARS-CoV-2 have critical roles in generating double membrane vesicles and connectors: non-structural proteins 3 and 4 (NSP3 and NSP4) for double membrane vesicles and NSP6 for the connectors. Notably, ~40% of double-stranded RNA and NSP6-positive viral replication areas are associated with lipid droplets255. NSP6 is a key factor in connecting lipid droplets with the replication organelle; possibly through interacting with and recruiting double FYVE domain-containing protein 1 (DFCP1), which regulates lipid droplet biogenesis and growth and the replication of SARS-CoV-2 (refs. 257,258,259,260). Functionally, lipid droplets are consumed during the formation of double membrane vesicles and this consumption depends on NSP6 and DFCP1 (refs. 256,261). Depleting DFCP1 and PLIN2, or inhibiting DGAT1 with xanthohumol, can suppress the replication of SARS-CoV-2 (refs. 256,261). Together, these data suggest that lipid droplets might help sustain SARS-CoV-2 replication by providing fatty acids for the synthesis of phospholipids.
Other viruses also engage with and exploit lipid droplets in multiple ways. The capsid (core) proteins of hepatitis C virus and dengue virus, and some NSPs of hepatitis C virus are found on the lipid droplet surface262. Similar to many other lipid droplet surface proteins, these viral proteins often have amphipathic helices. Host factors such as PLIN2, RAB18 and DGAT1 might also affect viral protein targeting to lipid droplets. Disrupting the association of these viral proteins with lipid droplets reduced viral replication and production in various cell models262. The replication compartments of enteroviruses can form contact sites with lipid droplets263. The viral proteins of enteroviruses can also physically interact with and activate the host’s lipolytic machinery, enabling fatty acid mobilization from lipid droplets to fuel the growth of replication compartments. Inhibiting this lipolytic pathway or the formation of membrane contact sites between lipid droplets and replication compartments disrupts replication compartment biogenesis and enterovirus replication. Together, these findings suggest that proteins associated with lipid droplets, including DGATs, DFCP1, RAB18 and PLIN2, are promising antiviral targets.
Cancer
Accumulation of lipid droplets has been observed in many cancer cell lines and neoplastic tissues264. Lipid droplets can provide building blocks for membrane lipid synthesis during cancer cell growth; however, cumulative evidence suggests that lipid droplets promote the proliferation, migration and survival of cancer cells through at least two other mechanisms. Firstly, through alleviation of cellular stresses and secondly, via provision of substrates for ATP production through β-oxidation265,266. The common kidney cancer, clear-cell renal cell carcinoma (ccRCC) is unique among all cancers through its accumulation of a large number of lipid droplets, which gives rise to its ‘clear cell’ phenotype265. Another hallmark of ccRCC is the constitutive activation of hypoxia-inducible factors (HIF), especially HIF2α, which drives metabolic alterations that enhance cell proliferation265. The expression of PLIN2, but not other PLINs, is specifically upregulated in ccRCC in a HIF2α-dependent manner265. PLIN2 is both necessary and sufficient for promoting lipid droplet accumulation in ccRCC cells. Importantly, PLIN2 activity and the accumulation of lipid droplets are required for the tumour-promoting function of HIF2α. Mechanistically, upregulated PLIN2 expression and enhanced lipid droplet formation reduce cytotoxic ER stress, thereby promoting cancer cell survival.
In addition to relieving ER stress, lipid droplets also have a major role in combating oxidative stress and its related cytotoxicity and cell death134. Conditions known to increase levels of ROS, such as hypoxia or chemical pro-oxidant treatment, can induce the formation of lipid droplets. ROS preferentially attack the double bonds in PUFAs, resulting in lipid peroxidation that disrupts normal cell function and causes cell death under certain conditions. For instance, ferroptosis is driven by iron-dependent peroxidation of membrane lipids, especially PUFAs267. Lipid droplets protect against ROS damage because upon oxidative stress, phospholipases can release PUFAs from membranes to be stored in lipid droplets, which are less vulnerable to peroxidation than cellular membranes134. When the amount of PUFAs overwhelms the buffering capacity of lipid droplets, cancer cells (which are acidic) readily undergo ferroptosis268. Moreover, reducing lipid droplet formation by inhibiting DGAT1 or PLIN2 increases ROS toxicity, impairs cell proliferation and enhances oxidative cell death and ferroptosis268,269. Therefore, lipid droplets can promote cancer cell survival under multiple oxidative conditions, including haematogenous dissemination in iron-rich blood, hypoxia and radiotherapy270.
As well as alleviating cellular stresses, lipid droplets also act as potent energy supplies for cancer cell invasion and metastasis266,271. Acidosis, like hypoxia, is a hallmark of the tumour microenvironment. Acidosis induces lipid droplet formation in cancer cells through CD36-mediated free fatty acid uptake and DGAT-mediated triacylglycerol synthesis266. In acidosis-adapted cancer cells, lipid droplets enhance anoikis resistance, invasiveness and metastatic spreading by providing free fatty acids for energy production through fatty acid oxidation266. The provision of free fatty acids seems to be a common mechanism by which lipid droplets promote metastasis. Pancreatic ductal adenocarcinoma cells, for instance, accumulate lipid droplets through oncogenic KRAS suppression of hormone sensitive lipase expression in primary tumours271. These stored lipid droplets are then catabolized by lipases during metastatic cell invasion, resulting in increased ATP generation and fatty acid oxidation, which then drives the highly energy-intensive process of metastatic cell invasion.
Together, these data suggest that DGAT1, PLIN2 and possibly hormone-sensitive lipase could be promising targets for anticancer treatment. A diet rich in PUFAs could also be considered as a complementary approach to pharmacological interventions against cancer268. However, further work is needed to validate these therapeutic targets and strategies.
Cardiovascular diseases
There are two major aspects of lipid droplet physiology in the heart: lipid droplets in cardiomyocytes and lipid droplets in macrophage foam cells272. Cardiomyocytes of adult mammals have high energy demands and derive ~70% of their energy from the oxidation of long-chain fatty acids (LCFAs) under fasting conditions273. Notably, the majority of LCFAs oxidized by cardiac mitochondria originate from the triacylglycerol and lipid droplet pool274. Through the action of lipases, especially PNPLA2, cardiomyocyte lipid droplets not only provide LCFAs for mitochondrial oxidation, but also ligands for PPARα activation275. PPARα and PPARγ coactivator 1α (or PPARγ coactivator 1β) work together to promote mitochondrial biogenesis and oxidative phosphorylation, which is essential for normal cardiac function. Triacylglycerol synthesis and lipid droplet biogenesis also have a cardioprotective role, as they reduce lipotoxicity through their transient storage of excess LCFAs273,276,277. Despite these physiological functions, accumulation of lipid droplets in heart cells might also contribute to cardiomyopathy and heart failure in both common genetic diseases (hyperlipidaemia in severe obesity and diabetes mellitus) and rare genetic diseases (neutral lipid storage disease with myopathy). The turnover rate of cardiac triacylglycerol and lipid droplets is often increased in the hearts of people with chronic diabetes mellitus, contributing to elevated oxidation of LCFAs, cytotoxicity and cell death272,278. Thus, while the biogenesis of lipid droplets can positively sustain cardiac function under normal physiological conditions, excessive accumulation and enlargement of cardiac lipid droplets under pathological settings might promote disease progression. The molecular and biochemical features underlying the functional differences between these ‘physiological’ and ‘pathological’ lipid droplets remain to be elucidated272.
The accumulation of cholesterol ester-rich lipid droplets in macrophage foam cells is a prominent feature of atherosclerosis. Foam cells arise from monocytes, which differentiate into macrophages during atherogenesis to take up large amounts of oxidized LDL279. Cholesterol released from oxidized LDL is then converted to cholesteryl esters by ACAT1 (ref. 280). The synthesis of cholesteryl esters and the formation of lipid droplets are cytoprotective as ACAT1 deletion from macrophages in LDL receptor-deficient mice increased macrophage lipotoxicity and atherogenesis281. Consistently, treatment with the ACAT inhibitor, pactimibe, in a clinical trial did not limit atherosclerosis, and instead promoted atherogenesis282. Nevertheless, lipid droplets might have different roles at different stages of an atherosclerotic lesion. In advanced lesions, lipid droplets and cholesteryl esters in foam cells might also contribute to innate immune responses and inflammation283.
Therapeutic strategies
Lipid droplets serve different roles in different diseases and might even have opposite functions during different stages of the same disease (that is, protection in initial stages and exacerbation upon disease progression). A thorough understanding of lipid droplet biogenesis, growth and degradation, especially in the context of disease, might thus inform new therapeutic targets and strategies. In some disease conditions, such as certain cancers and viral infections, lipid droplets promote cell growth and viral replication256,265,266. Here, disrupting lipid droplet biogenesis could be an effective treatment strategy. For instance, small-molecule inhibitors or small interfering RNAs that target DGAT1, which is highly expressed in glioblastoma tissues in which lipid droplets accumulate, promoted the death of glioblastoma cell lines in vitro and suppressed glioblastoma growth in a mouse model in vivo by increasing oxidative stress269. Similar results were also obtained in human melanoma cell lines and a mouse model of human melanoma284, as well as human prostate cancer cell lines and a mouse model of prostate cancer285. Therefore, DGAT1 inhibitors could be tested for use in other cancers, especially those that involve lipid droplet accumulation. Cholesteryl ester-rich lipid droplets also accumulate in advanced prostate cancers in humans and ACAT1 inhibitors can reduce cancer proliferation and aggressiveness in mouse xenograft models with negligible toxicity286. PLIN2 promotes lipid storage and tumour growth in ccRCC xenografts by reducing ER stress265. Disrupting PLIN2 function considerably reduces cell viability in ccRCC cell lines and xenograft tumours265. Lipid droplets also contribute to double membrane vesicle formation and viral replication during SARS-CoV-2 infection256. Inhibiting DGAT1 with xanthohumol or depleting DFCP1 or PLIN2 by genetic manipulation can suppress SARS-CoV-2 replication256,261. Therefore, targeting proteins involved in lipid droplet biogenesis might offer new avenues for combating viral infections.
NAFLD and non-alcoholic steatohepatitis (NASH) are characterized by lipid droplet accumulation. However, increased lipid droplet abundance in hepatocytes does not necessarily indicate cellular dysfunction178. To date, no pharmacological therapies have been approved for the treatment of NAFLD and NASH287. Data from the past few years suggest that dysregulated lipolysis (for example, NAFLD-linked mutations in PNPLA3 and 17β-HSD13 affect lipolysis) or lipid droplet growth (for example, NAFLD-linked mutations in CIDEB affect lipid droplet growth) can contribute to the development of NAFLD and NASH178,288,289,290 (see also the section ‘Non-alcoholic fatty liver disease’). Accordingly, both small-molecule inhibitors and RNA interference-based therapies against 17β-HSD13 have entered clinical trials for treating NAFLD and NASH291. A small-molecule inhibitor of DGAT2 also reduced hepatic steatosis in both mice and humans with few adverse effects in preclinical studies and in two phase I clinical studies289. An antisense oligonucleotide inhibitor of DGAT2 (IONIS-DGAT2Rx) also statistically significantly reduced liver lipid content in a phase II study292. Moreover, when combined with an acetyl-CoA carboxylase 1 and 2 inhibitor, clesacostat (PF-05221304), the DGAT2 inhibitor, ervogastat (PF-06865571) reduced liver lipid content, with a favourable safety and tolerability profile293. The FDA has granted Fast Track designation to this combination therapy for the treatment of NASH with liver fibrosis. CIDEB seems to be a key driver of liver diseases in humans and loss-of-function variants in CIDEB are highly protective203. Therefore, new small interfering RNA-based or small-molecule-based therapies targeting CIDEB might also be developed. Finally, it should be noted that there are limitations in targeting lipid droplets for disease treatment or prevention. For instance, inhibiting DGATs might harm cancer cells, but this strategy could also adversely affect healthy cells throughout the body.
Conclusions
The current understanding of lipid droplets has markedly progressed since their initial identification over a century ago. Now well-recognized as dynamic organelles, lipid droplets are increasingly appreciated for their diversity and crucial cellular roles. Despite this progress, several fundamental features regarding their biogenesis and functions remain incompletely resolved. Investigation to provide deeper insights into the formation of nuclear, luminal and potentially other subcellular lipid droplets is warranted and might uncover new mechanistic details about cytoplasmic lipid droplets. In particular, little is known about the biogenesis of luminal lipid droplets, which might have a critical role in the formation and secretion of lipoproteins in the plasma and therefore have profound implications for many metabolic disorders.
Further elucidation of the exact roles of lipid droplets in numerous disease conditions is also essential. Whereas the initial generation of lipid droplets is usually protective, long-term accumulation and uncontrolled enlargement of lipid droplets in chronic disease can exacerbate disease progression. It is thus necessary that the unique protein and lipid fingerprint of physiological and pathological lipid droplets is dissected to enable greater insights into their transition from being beneficial to being detrimental in different disease settings. How lipid droplets interact with other organelles in health and disease states, and whether such contacts contribute to disease progression, also remain to be determined. Technological breakthroughs providing sensitive and specific methods for accurate detection and visualization of lipids in their natural environments will no doubt prove pivotal in facilitating these advances in our understanding. In turn, the development of lipid droplet-based therapeutic strategies will be greatly enhanced by improving our understanding of the biogenesis and function of lipid droplets.
References
Altman, R. Die Elementarorganismen und Ihre Beziehungen zu den Zellen [German] (Viet, 1890).
Wilson, E. B. The Cell in Development and Inheritance (Macmillan, 1896).
Greenberg, A. S. et al. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 266, 11341–11346 (1991).
Huang, A. H. C. Oil bodies and oleosins in seeds. Annu. Rev. Plant Physiol. Plant Mol. Biol. 43, 177–200 (1992).
Walther, T. C., Chung, J. & Farese, R. V. Lipid droplet biogenesis. Annu. Rev. Cell Dev. Biol. 33, 491–510 (2017).
Henne, W. M., Reese, M. L. & Goodman, J. M. The assembly of lipid droplets and their roles in challenged cells. EMBO J. 37, e98947 (2018).
Olzmann, J. A. & Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 20, 137–155 (2019).
Gao, M., Huang, X., Song, B.-L. & Yang, H. The biogenesis of lipid droplets: lipids take center stage. Prog. Lipid Res. 75, 100989 (2019).
Wanner, G., Formanek, H. & Theimer, R. R. The ontogeny of lipid bodies (spherosomes) in plant cells: ultrastructural evidence. Planta 151, 109–123 (1981).
Murphy, D. J. & Vance, J. Mechanisms of lipid-body formation. Trends Biochem. Sci. 24, 109–115 (1999).
Zweytick, D., Athenstaedt, K. & Daum, G. Intracellular lipid particles of eukaryotic cells. Biochim. Biophys. Acta 1469, 101–120 (2000).
Robenek, M. J. et al. Lipids partition caveolin-1 from ER membranes into lipid droplets: updating the model of lipid droplet biogenesis. FASEB J. 18, 866–868 (2004).
Wältermann, M. et al. Mechanism of lipid-body formation in prokaryotes: how bacteria fatten up. Mol. Microbiol. 55, 750–763 (2005).
Andersson, L. et al. PLD1 and ERK2 regulate cytosolic lipid droplet formation. J. Cell Sci. 119, 2246–2257 (2006).
Wolins, N. E., Brasaemle, D. L. & Bickel, P. E. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 580, 5484–5491 (2006).
Ploegh, H. L. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 448, 435–438 (2007).
Ohsaki, Y. et al. Biogenesis of cytoplasmic lipid droplets: from the lipid ester globule in the membrane to the visible structure. Biochim. Biophys. Acta 1791, 399–407 (2009).
Skinner, J. R. et al. Diacylglycerol enrichment of endoplasmic reticulum or lipid droplets recruits perilipin 3/TIP47 during lipid storage and mobilization. J. Biol. Chem. 284, 30941–30948 (2009).
Walther, T. C. & Farese, R. V. The life of lipid droplets. Biochim. Biophys. Acta 1791, 459–466 (2009).
Choudhary, V., Jacquier, N. & Schneiter, R. The topology of the triacylglycerol synthesizing enzyme Lro1 indicates that neutral lipids can be produced within the luminal compartment of the endoplasmatic reticulum: implications for the biogenesis of lipid droplets. Commun. Integr. Biol. 4, 781–784 (2011).
Brasaemle, D. L. & Wolins, N. E. Packaging of fat: an evolving model of lipid droplet assembly and expansion. J. Biol. Chem. 287, 2273–2279 (2012).
Pol, A., Gross, S. P. & Parton, R. G. Review: biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J. Cell Biol. 204, 635–646 (2014).
Thiam, A. R. & Ikonen, E. Lipid droplet nucleation. Trends Cell Biol. 31, 108–118 (2021).
Cases, S. et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl Acad. Sci. USA 95, 13018–13023 (1998).
Cases, S. et al. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 276, 38870–38876 (2001).
Lardizabal, K. D. et al. DGAT2 is a new diacylglycerol acyltransferase gene family. J. Biol. Chem. 276, 38862–38869 (2001).
Chang, C. C., Huh, H. Y., Cadigan, K. M. & Chang, T. Y. Molecular cloning and functional expression of human acyl-coenzyme A: cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 268, 20747–20755 (1993).
Anderson, R. A. et al. Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 273, 26747–26754 (1998).
Cases, S. et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J. Biol. Chem. 273, 26755–26764 (1998).
Wang, L. et al. Structure and mechanism of human diacylglycerol O-acyltransferase 1. Nature 581, 329–332 (2020).
Sui, X. et al. Structure and catalytic mechanism of a human triacylglycerol-synthesis enzyme. Nature 581, 323–328 (2020).
Guan, C. et al. Structural insights into the inhibition mechanism of human sterol O-acyltransferase 1 by a competitive inhibitor. Nat. Commun. 11, 2478 (2020).
Long, T., Sun, Y., Hassan, A., Qi, X. & Li, X. Structure of nevanimibe-bound tetrameric human ACAT1. Nature 581, 339–343 (2020).
Long, T., Liu, Y. & Li, X. Molecular structures of human ACAT2 disclose mechanism for selective inhibition. Structure 29, 1410–1418.e4 (2021).
Oelkers, P. et al. A lecithin cholesterol acyltransferase-like gene mediates diacylglycerol esterification in yeast. J. Biol. Chem. 275, 15609–15612 (2000).
Oelkers, P., Cromley, D., Padamsee, M., Billheimer, J. T. & Sturley, S. L. The DGA1 gene determines a second triglyceride synthetic pathway in yeast. J. Biol. Chem. 277, 8877–8881 (2002).
Yang, H. et al. Sterol esterification in yeast: a two-gene process. Science 272, 1353–1356 (1996).
Sandager, L. et al. Storage lipid synthesis is non-essential in yeast. J. Biol. Chem. 277, 6478–6482 (2002).
Sorger, D., Athenstaedt, K., Hrastnik, C. & Daum, G. A yeast strain lacking lipid particles bears a defect in ergosterol formation. J. Biol. Chem. 279, 31190–31196 (2004).
Harris, C. A. et al. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J. Lipid Res. 52, 657–667 (2011).
Petschnigg, J. et al. Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J. Biol. Chem. 284, 30981–30993 (2009).
Guo, Y. et al. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453, 657–661 (2008).
Sturley, S. L. & Hussain, M. M. Lipid droplet formation on opposing sides of the endoplasmic reticulum. J. Lipid Res. 53, 1800–1810 (2012).
Hamilton, J. A. & Small, D. M. Solubilization and localization of triolein in phosphatidylcholine bilayers: a 13C NMR study. Proc. Natl Acad. Sci. USA 78, 6878–6882 (1981).
Khandelia, H., Duelund, L., Pakkanen, K. I. & Ipsen, J. H. Triglyceride blisters in lipid bilayers: implications for lipid droplet biogenesis and the mobile lipid signal in cancer cell membranes. PLoS ONE 5, e12811 (2010).
Duelund, L. et al. Composition, structure and properties of POPC-triolein mixtures. Evidence of triglyceride domains in phospholipid bilayers. Biochim. Biophys. Acta 1828, 1909–1917 (2013).
Thiam, A. R. & Forêt, L. The physics of lipid droplet nucleation, growth and budding. Biochim. Biophys. Acta 1861, 715–722 (2016).
Choudhary, V., Ojha, N., Golden, A. & Prinz, W. A. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J. Cell Biol. 211, 261–271 (2015).
Zoni, V. et al. Pre-existing bilayer stresses modulate triglyceride accumulation in the ER versus lipid droplets. eLife 10, e62886 (2021).
Ben M’barek, K. et al. ER membrane phospholipids and surface tension control cellular lipid droplet formation. Dev. Cell 41, 591–604.e7 (2017).
Choudhary, V. et al. Architecture of lipid droplets in endoplasmic reticulum is determined by phospholipid intrinsic curvature. Curr. Biol. CB 28, 915–926.e9 (2018).
Santinho, A. et al. Membrane curvature catalyzes lipid droplet assembly. Curr. Biol. CB 30, 2481–2494.e6 (2020).
Adeyo, O. et al. The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J. Cell Biol. 192, 1043–1055 (2011).
Fei, W. et al. A role for phosphatidic acid in the formation of ‘supersized’ lipid droplets. PLoS Genet. 7, e1002201 (2011).
Szymanski, K. M. et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl Acad. Sci. USA 104, 20890–20895 (2007).
Fei, W. et al. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J. Cell Biol. 180, 473–482 (2008).
Klug, Y. A. et al. Mechanism of lipid droplet formation by the yeast Sei1/Ldb16 seipin complex. Nat. Commun. 12, 5892 (2021).
Arlt, H. et al. Seipin forms a flexible cage at lipid droplet formation sites. Nat. Struct. Mol. Biol. 29, 194–202 (2022).
Salo, V. T. et al. Seipin facilitates triglyceride flow to lipid droplet and counteracts droplet ripening via endoplasmic reticulum contact. Dev. Cell 50, 478–493.e9 (2019).
Chung, J. et al. LDAF1 and seipin form a lipid droplet assembly complex. Dev. Cell 51, 551–563.e7 (2019).
Choudhary, V., El Atab, O., Mizzon, G., Prinz, W. A. & Schneiter, R. Seipin and Nem1 establish discrete ER subdomains to initiate yeast lipid droplet biogenesis. J. Cell Biol. 219, e201910177 (2020).
Wang, S. et al. Seipin and the membrane-shaping protein Pex30 cooperate in organelle budding from the endoplasmic reticulum. Nat. Commun. 9, 2939 (2018).
Joshi, A. S. et al. Lipid droplet and peroxisome biogenesis occur at the same ER subdomains. Nat. Commun. 9, 2940 (2018).
Joshi, A. S., Ragusa, J. V., Prinz, W. A. & Cohen, S. Multiple C2 domain-containing transmembrane proteins promote lipid droplet biogenesis and growth at specialized endoplasmic reticulum subdomains. Mol. Biol. Cell 32, 1147–1157 (2021).
Chen, F. et al. FIT2 organizes lipid droplet biogenesis with ER tubule-forming proteins and septins. J. Cell Biol. 220, e201907183 (2021).
Jacquier, N. et al. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Sci. 124, 2424–2437 (2011).
Grippa, A. et al. The seipin complex Fld1/Ldb16 stabilizes ER-lipid droplet contact sites. J. Cell Biol. 211, 829–844 (2015).
Valm, A. M. et al. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167 (2017).
Thiam, A. R., Farese, R. V. & Walther, T. C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 14, 775–786 (2013).
Deslandes, F., Thiam, A. R. & Forêt, L. Lipid droplets can spontaneously bud off from a symmetric bilayer. Biophys. J. 113, 15–18 (2017).
Hegaard, F. V., Klenow, M. B. & Simonsen, A. C. Lens nucleation and droplet budding in a membrane model for lipid droplet biogenesis. Langmuir 38, 9247–9256 (2022).
Chorlay, A. & Thiam, A. R. An asymmetry in monolayer tension regulates lipid droplet budding direction. Biophys. J. 114, 631–640 (2018).
Chorlay, A. et al. Membrane asymmetry imposes directionality on lipid droplet emergence from the ER. Dev. Cell 50, 25–42.e7 (2019).
Zoni, V. et al. To bud or not to bud: a perspective on molecular simulations of lipid droplet budding. Front. Mol. Biosci. 6, 124 (2019).
Hayes, M. J. et al. Fat storage-inducing transmembrane (FIT or FITM) proteins are related to lipid phosphatase/phosphotransferase enzymes. Microb. Cell 5, 88–103 (2018).
Becuwe, M. et al. FIT2 is an acyl-coenzyme A diphosphatase crucial for endoplasmic reticulum homeostasis. J. Cell Biol. 219, e202006111 (2020).
Gong, J. et al. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J. Cell Biol. 195, 953–963 (2011).
Krahmer, N. et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP: phosphocholine cytidylyltransferase. Cell Metab. 14, 504–515 (2011).
Wilfling, F., Haas, J. T., Walther, T. C. & Farese, R. V. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 29, 39–45 (2014).
Wilfling, F. et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399 (2013).
Prévost, C. et al. Mechanism and determinants of amphipathic helix-containing protein targeting to lipid droplets. Dev. Cell 44, 73–86.e4 (2018).
Olarte, M.-J., Swanson, J. M. J., Walther, T. C. & Farese, R. V. The CYTOLD and ERTOLD pathways for lipid droplet-protein targeting. Trends Biochem. Sci. 47, 39–51 (2022).
Song, J. et al. Identification of two pathways mediating protein targeting from ER to lipid droplets. Nat. Cell Biol. 24, 1364–1377 (2022).
Du, X. et al. ORP5 localizes to ER–lipid droplet contacts and regulates the level of PI(4)P on lipid droplets. J. Cell Biol. 219, e201905162 (2020).
Leduc, E. H. & Wilson, J. W. An electron microscope study of intranuclear inclusions in mouse liver and hepatoma. J. Biophys. Biochem. Cytol. 6, 427–430 (1959).
Thoenes, W. Fett im nucleolus [German]. J. Ultrastruct. Res. 10, 194–206 (1964).
Karasaki, S. The fine structure of proliferating cells in preneoplastic rat livers during azo-dye carcinogenesis. J. Cell Biol. 40, 322–335 (1969).
Karasaki, S. Passage of cytoplasmic lipid into interphase nuclei in preneoplastic rat liver. J. Ultrastruct. Res. 42, 463–478 (1973).
Taurino, M. et al. SEIPIN proteins mediate lipid droplet biogenesis to promote pollen transmission and reduce seed dormancy. Plant. Physiol. 176, 1531–1546 (2018).
Romanauska, A. & Köhler, A. The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets. Cell 174, 700–715.e18 (2018).
Romanauska, A. & Köhler, A. Reprogrammed lipid metabolism protects inner nuclear membrane against unsaturated fat. Dev. Cell 56, 2562–2578.e3 (2021).
Lee, J., Salsman, J., Foster, J., Dellaire, G. & Ridgway, N. D. Lipid-associated PML structures assemble nuclear lipid droplets containing CCTα and Lipin1. Life Sci. Alliance 3, e202000751 (2020).
Morales, A. et al. Loss of ephrin B2 receptor (EPHB2) sets lipid rheostat by regulating proteins DGAT1 and ATGL inducing lipid droplet storage in prostate cancer cells. Lab. Invest. 101, 921–934 (2021).
Mosquera, J. V., Bacher, M. C. & Priess, J. R. Nuclear lipid droplets and nuclear damage in Caenorhabditis elegans. PLoS Genet. 17, e1009602 (2021).
Sołtysik, K., Ohsaki, Y., Tatematsu, T., Cheng, J. & Fujimoto, T. Nuclear lipid droplets derive from a lipoprotein precursor and regulate phosphatidylcholine synthesis. Nat. Commun. 10, 473 (2019).
Sołtysik, K. et al. Nuclear lipid droplets form in the inner nuclear membrane in a seipin-independent manner. J. Cell Biol. 220, e202005026 (2021).
Pagac, M. et al. SEIPIN regulates lipid droplet expansion and adipocyte development by modulating the activity of glycerol-3-phosphate acyltransferase. Cell Rep. 17, 1546–1559 (2016).
Yan, R. et al. Human SEIPIN binds anionic phospholipids. Dev. Cell 47, 248–256.e4 (2018).
Cartwright, B. R. et al. Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology. Mol. Biol. Cell 26, 726–739 (2015).
Wolinski, H. et al. Seipin is involved in the regulation of phosphatidic acid metabolism at a subdomain of the nuclear envelope in yeast. Biochim. Biophys. Acta 1851, 1450–1464 (2015).
Sołtysik, K., Ohsaki, Y. & Fujimoto, T. Duo in a mystical realm – nuclear lipid droplets and the inner nuclear membrane. Contact 2, 251525641989696 (2019).
Ginsberg, H. N. & Fisher, E. A. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J. Lipid Res. 50, S162–S166 (2009).
Raabe, M. et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J. Clin. Invest. 103, 1287–1298 (1999).
Ohsaki, Y. et al. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 212, 29–38 (2016).
Chen, N. Y. et al. Fibroblasts lacking nuclear lamins do not have nuclear blebs or protrusions but nevertheless have frequent nuclear membrane ruptures. Proc. Natl Acad. Sci. USA 115, 10100–10105 (2018).
Mishra, S. et al. Mature lipid droplets are accessible to ER luminal proteins. J. Cell Sci. 129, 3803–3815 (2016).
Tiwari, S. & Siddiqi, S. A. Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 32, 1079–1086 (2012).
Wang, H., Gilham, D. & Lehner, R. Proteomic and lipid characterization of apolipoprotein B-free luminal lipid droplets from mouse liver microsomes. J. Biol. Chem. 282, 33218–33226 (2007).
Kim, S., Swanson, J. M. J. & Voth, G. A. Computational studies of lipid droplets. J. Phys. Chem. B 126, 2145–2154 (2022).
Ladinsky, M. S., Mardones, G. A., Orlicky, D. J., Howell, K. E. & McManaman, J. L. Electron tomography revels that milk lipids originate from endoplasmic reticulum domains with novel structural features. J. Mammary Gland. Biol. Neoplasia 24, 293–304 (2019).
Monks, J., Ladinsky, M. S. & McManaman, J. L. Organellar contacts of milk lipid droplets. Contact https://doi.org/10.1177/2515256419897226 (2020).
Zhang, Q. et al. Schizosaccharomyces pombe cells deficient in triacylglycerols synthesis undergo apoptosis upon entry into the stationary phase. J. Biol. Chem. 278, 47145–47155 (2003).
Fei, W., Wang, H., Fu, X., Bielby, C. & Yang, H. Conditions of endoplasmic reticulum stress stimulate lipid droplet formation in Saccharomyces cerevisiae. Biochem. J. 424, 61–67 (2009).
Welte, M. A. & Gould, A. P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta 1862, 1260–1272 (2017).
Lee, Y. et al. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc. Natl Acad. Sci. USA 91, 10878–10882 (1994).
Listenberger, L. L. et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl Acad. Sci. USA 100, 3077–3082 (2003).
Piccolis, M. et al. Probing the global cellular responses to lipotoxicity caused by saturated fatty acids. Mol. Cell 74, 32–44.e8 (2019).
Song, Y., Liu, J., Zhao, K., Gao, L. & Zhao, J. Cholesterol-induced toxicity: an integrated view of the role of cholesterol in multiple diseases. Cell Metab. 33, 1911–1925 (2021).
Yoon, H., Shaw, J. L., Haigis, M. C. & Greka, A. Lipid metabolism in sickness and in health: emerging regulators of lipotoxicity. Mol. Cell 81, 3708–3730 (2021).
Garbarino, J. et al. Sterol and diacylglycerol acyltransferase deficiency triggers fatty acid-mediated cell death. J. Biol. Chem. 284, 30994–31005 (2009).
Chitraju, C. et al. Triglyceride synthesis by DGAT1 protects adipocytes from lipid-induced ER stress during lipolysis. Cell Metab. 26, 407–418.e3 (2017).
Kuo, A., Lee, M. Y. & Sessa, W. C. Lipid droplet biogenesis and function in the endothelium. Circ. Res. 120, 1289–1297 (2017).
Liu, L. et al. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J. Biol. Chem. 284, 36312–36323 (2009).
Long, M. et al. DGAT1 activity synchronises with mitophagy to protect cells from metabolic rewiring by iron depletion. EMBO J. 41, e109390 (2022).
Kwiatek, J. M., Han, G.-S. & Carman, G. M. Phosphatidate-mediated regulation of lipid synthesis at the nuclear/endoplasmic reticulum membrane. Biochim. Biophys. Acta 1865, 158434 (2020).
Rambold, A. S., Cohen, S. & Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692 (2015).
Nguyen, T. B. et al. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev. Cell 42, 9–21.e5 (2017).
Hosios, A. M. et al. mTORC1 regulates a lysosome-dependent adaptive shift in intracellular lipid species. Nat. Metab. 4, 1792–1811 (2022).
Han, J. & Kaufman, R. J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 57, 1329–1338 (2016).
Lin, L.-L. et al. cPLA2 is phosphorylated and activated by MAP kinase. Cell 72, 269–278 (1993).
Gubern, A. et al. Group IVA phospholipase A2 is necessary for the biogenesis of lipid droplets. J. Biol. Chem. 283, 27369–27382 (2008).
Darling, N. J. & Cook, S. J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 1843, 2150–2163 (2014).
Liu, L. et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190 (2015).
Bailey, A. P. et al. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 163, 340–353 (2015).
Roberts, M. A. & Olzmann, J. A. Protein quality control and lipid droplet metabolism. Annu. Rev. Cell Dev. Biol. 36, 115–139 (2020).
Listenberger, L. L., Ory, D. S. & Schaffer, J. E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 276, 14890–14895 (2001).
Park, C. Y. et al. Dysregulation of lipid droplet protein expression in adipose tissues and association with metabolic risk factors in adult females with obesity and type 2 diabetes. J. Nutr. 153, 691–702 (2023).
Dahlman, I. et al. The CIDEA gene V115F polymorphism is associated with obesity in Swedish subjects. Diabetes 54, 3032–3034 (2005).
Wu, J. et al. The genetic contribution of CIDEA polymorphisms, haplotypes and loci interaction to obesity in a Han Chinese population. Mol. Biol. Rep. 40, 5691–5699 (2013).
Zhou, Z. et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat. Genet. 35, 49–56 (2003).
Li, J. Z. et al. Cideb regulates diet-induced obesity, liver steatosis, and insulin sensitivity by controlling lipogenesis and fatty acid oxidation. Diabetes 56, 2523–2532 (2007).
Nishino, N. et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Invest. 118, 2808–2821 (2008).
Martinez-Botas, J. et al. Absence of perilipin results in leanness and reverses obesity in Leprdb/db mice. Nat. Genet. 26, 474–479 (2000).
Tansey, J. T. et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl Acad. Sci. USA 98, 6494–6499 (2001).
Sun, Z. et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 4, 1594 (2013).
Puri, V. & Czech, M. P. Lipid droplets: FSP27 knockout enhances their sizzle. J. Clin. Invest. 118, 2693–2696 (2008).
Qian, K. et al. CLSTN3β enforces adipocyte multilocularity to facilitate lipid utilization. Nature 613, 160–168 (2023).
James, D. E., Stöckli, J. & Birnbaum, M. J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 22, 751–771 (2021).
Virtue, S. & Vidal-Puig, A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 6, e237 (2008).
Daemen, S. et al. Distinct lipid droplet characteristics and distribution unmask the apparent contradiction of the athlete’s paradox. Mol. Metab. 17, 71–81 (2018).
Blüher, M. Metabolically healthy obesity. Endocr. Rev. 41, bnaa004 (2020).
Stefan, N. Metabolically healthy and unhealthy normal weight and obesity. Endocrinol. Metab. Seoul. Korea 35, 487–493 (2020).
Garg, A. Acquired and inherited lipodystrophies. N. Engl. J. Med. 350, 1220–1234 (2004).
Gonzaga-Jauregui, C. et al. Clinical and molecular prevalence of lipodystrophy in an unascertained large clinical care cohort. Diabetes 69, 249–258 (2020).
Lim, K., Haider, A., Adams, C., Sleigh, A. & Savage, D. B. Lipodistrophy: a paradigm for understanding the consequences of ‘overloading’ adipose tissue. Physiol. Rev. 101, 907–993 (2021).
Zammouri, J. et al. Molecular and cellular bases of lipodystrophy syndromes. Front. Endocrinol. 12, 803189 (2021).
Fourman, L. T. & Grinspoon, S. K. Approach to the patient with lipodystrophy. J. Clin. Endocrinol. Metab. 107, 1714–1726 (2022).
Patni, N. & Garg, A. Lipodystrophy for the diabetologist – what to look for. Curr. Diab. Rep. 22, 461–470 (2022).
Simha, V. & Garg, A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J. Clin. Endocrinol. Metab. 88, 5433–5437 (2003).
Gale, S. E. et al. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J. Biol. Chem. 281, 11082–11089 (2006).
Subauste, A. R. et al. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes 61, 2922–2931 (2012).
Patni, N. & Garg, A. Congenital generalized lipodystrophies – new insights into metabolic dysfunction. Nat. Rev. Endocrinol. 11, 522–534 (2015).
Cui, X. et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum. Mol. Genet. 20, 3022–3030 (2011).
Liu, L. et al. Adipose-specific knockout of Seipin/Bscl2 results in progressive lipodystrophy. Diabetes 63, 2320–2331 (2014).
Mak, H. Y. et al. AGPAT2 interaction with CDP-diacylglycerol synthases promotes the flux of fatty acids through the CDP-diacylglycerol pathway. Nat. Commun. 12, 6877 (2021).
Blouin, C. M. et al. Lipid droplet analysis in caveolin-deficient adipocytes: alterations in surface phospholipid composition and maturation defects. J. Lipid Res. 51, 945–956 (2010).
Riazi, K., Swain, M. G., Congly, S. E., Kaplan, G. G. & Shaheen, A.-A. Race and ethnicity in non-alcoholic fatty liver disease (NAFLD): a narrative review. Nutrients 14, 4556 (2022).
Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2, 901–910 (2014).
Targher, G., Tilg, H. & Byrne, C. D. Non-alcoholic fatty liver disease: a multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol. Hepatol. 6, 578–588 (2021).
Eslam, M., Sanyal, A. J. & George, J., International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1 (2020).
Sheka, A. C. et al. Nonalcoholic steatohepatitis: a review. JAMA 323, 1175–1183 (2020).
Day, C. P. & James, O. F. W. Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–845 (1998).
Buzzetti, E., Pinzani, M. & Tsochatzis, E. A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048 (2016).
Tilg, H., Adolph, T. E. & Moschen, A. R. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: revisited after a decade. Hepatology 73, 833–842 (2021).
Barrows, B. R. & Parks, E. J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 91, 1446–1452 (2006).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 (2005).
Das, K. & Chowdhury, A. Lean NASH: distinctiveness and clinical implication. Hepatol. Int. 7, 806–813 (2013).
Mashek, D. G. Hepatic lipid droplets: a balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 50, 101115 (2021).
Smith, G. I. et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Invest. 130, 1453–1460 (2020).
Hodson, L. & Gunn, P. J. The regulation of hepatic fatty acid synthesis and partitioning: the effect of nutritional state. Nat. Rev. Endocrinol. 15, 689–700 (2019).
Scorletti, E. & Carr, R. M. A new perspective on NAFLD: focusing on lipid droplets. J. Hepatol. 76, 934–945 (2022).
Krahmer, N. et al. Organellar proteomics and phospho-proteomics reveal subcellular reorganization in diet-induced hepatic steatosis. Dev. Cell 47, 205–221.e7 (2018).
Romeo, S. et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465 (2008).
Yuan, X. et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am. J. Hum. Genet. 83, 520–528 (2008).
Chen, W., Chang, B., Li, L. & Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 52, 1134–1142 (2010).
Basantani, M. K. et al. Pnpla3/adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 52, 318–329 (2011).
He, S. et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 285, 6706–6715 (2010).
Huang, Y., Cohen, J. C. & Hobbs, H. H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 286, 37085–37093 (2011).
BasuRay, S., Smagris, E., Cohen, J. C. & Hobbs, H. H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 66, 1111–1124 (2017).
BasuRay, S., Wang, Y., Smagris, E., Cohen, J. C. & Hobbs, H. H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl Acad. Sci. USA 116, 9521–9526 (2019).
Wang, Y., Kory, N., BasuRay, S., Cohen, J. C. & Hobbs, H. H. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology 69, 2427–2441 (2019).
Yang, A., Mottillo, E. P., Mladenovic-Lucas, L., Zhou, L. & Granneman, J. G. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat. Metab. 1, 560–569 (2019).
Schott, M. B. et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 218, 3320–3335 (2019).
Kabbani, M. et al. Human hepatocyte PNPLA3-148M exacerbates rapid non-alcoholic fatty liver disease development in chimeric mice. Cell Rep. 40, 111321 (2022).
Pirazzi, C. et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 23, 4077–4085 (2014).
Bruschi, F. V., Tardelli, M., Claudel, T. & Trauner, M. PNPLA3 expression and its impact on the liver: current perspectives. Hepatic Med. Evid. Res. 9, 55–66 (2017).
Ma, Y. et al. 17-beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology 69, 1504–1519 (2019).
Abul-Husn, N. S. et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378, 1096–1106 (2018).
Seko, Y. et al. Attenuated effect of PNPLA3 on hepatic fibrosis by HSD17B13 in Japanese patients with non-alcoholic fatty liver disease. Liver Int. 40, 1686–1692 (2020).
Su, W. et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl Acad. Sci. USA 111, 11437–11442 (2014).
Su, W. et al. Phosphorylation of 17β-hydroxysteroid dehydrogenase 13 at serine 33 attenuates nonalcoholic fatty liver disease in mice. Nat. Commun. 13, 6577 (2022).
Ng, S. W. K. et al. Convergent somatic mutations in metabolism genes in chronic liver disease. Nature 598, 473–478 (2021).
Verweij, N. et al. Germline mutations in CIDEB and protection against liver disease. N. Engl. J. Med. 387, 332–344 (2022).
Xu, W. et al. Differential roles of cell death-inducing DNA fragmentation factor-α-like effector (CIDE) proteins in promoting lipid droplet fusion and growth in subpopulations of hepatocytes. J. Biol. Chem. 291, 4282–4293 (2016).
Ye, J. et al. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 9, 177–190 (2009).
Tiwari, S., Siddiqi, S. & Siddiqi, S. A. CideB protein is required for the biogenesis of very low density lipoprotein (VLDL) transport vesicle. J. Biol. Chem. 288, 5157–5165 (2013).
Su, L. et al. Cideb controls sterol-regulated ER export of SREBP/SCAP by promoting cargo loading at ER exit sites. EMBO J. 38, e100156 (2019).
Kozlitina, J. et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46, 352–356 (2014).
Mancina, R. M. et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150, 1219–1230.e6 (2016).
Baselli, G. A. et al. Rare ATG7 genetic variants predispose patients to severe fatty liver disease. J. Hepatol. 77, 596–606 (2022).
de Vree, J. M. et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc. Natl Acad. Sci. USA 95, 282–287 (1998).
Tanoli, T., Yue, P., Yablonskiy, D. & Schonfeld, G. Fatty liver in familial hypobetalipoproteinemia: roles of the APOB defects, intra-abdominal adipose tissue, and insulin sensitivity. J. Lipid Res. 45, 941–947 (2004).
Ward, L. D. et al. GWAS of serum ALT and AST reveals an association of SLC30A10 Thr95Ile with hypermanganesemia symptoms. Nat. Commun. 12, 4571 (2021).
Faulkner, C. S., White, C. M., Shah, V. H. & Jophlin, L. L. A single nucleotide polymorphism of PLIN2 is associated with nonalcoholic steatohepatitis and causes phenotypic changes in hepatocyte lipid droplets: a pilot study. Biochim. Biophys. Acta 1865, 158637 (2020).
Najt, C. P., Devarajan, M. & Mashek, D. G. Perilipins at a glance. J. Cell Sci. 135, jcs259501 (2022).
Wagner, R. et al. Metabolic implications of pancreatic fat accumulation. Nat. Rev. Endocrinol. 18, 43–54 (2022).
Schaefer, J. H. The normal weight of the pancreas in the adult human being: a biometric study. Anat. Rec. 32, 119–132 (1926).
Wong, V. W.-S. et al. Fatty pancreas, insulin resistance, and β-cell function: a population study using fat-water magnetic resonance imaging. Am. J. Gastroenterol. 109, 589–597 (2014).
Singh, R. G., Yoon, H. D., Poppitt, S. D., Plank, L. D. & Petrov, M. S. Ectopic fat accumulation in the pancreas and its biomarkers: a systematic review and meta-analysis. Diabetes Metab. Res. Rev. 33, e2918 (2017).
Prentki, M., Corkey, B. E. & Madiraju, S. R. M. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 63, 10–20 (2020).
Smits, M. M. & van Geenen, E. J. M. The clinical significance of pancreatic steatosis. Nat. Rev. Gastroenterol. Hepatol. 8, 169–177 (2011).
Ogilvie, R. F. The islands of Langerhans in 19 cases of obesity. J. Pathol. Bacteriol. 37, 473–481 (1933).
Olsen, T. S. Lipomatosis of the pancreas in autopsy material and its relation to age and overweight. Acta Pathol. Microbiol. Scand. A 86A, 367–373 (1978).
Shimabukuro, M., Zhou, Y. T., Levi, M. & Unger, R. H. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc. Natl Acad. Sci. USA 95, 2498–2502 (1998).
Unger, R. H., Zhou, Y.-T. & Orci, L. Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc. Natl Acad. Sci. USA 96, 2327–2332 (1999).
Ramkissoon, R. & Gardner, T. B. Pancreatic steatosis: an emerging clinical entity. Am. J. Gastroenterol. 114, 1726–1734 (2019).
Takahashi, M. et al. Fatty pancreas: a possible risk factor for pancreatic cancer in animals and humans. Cancer Sci. 109, 3013–3023 (2018).
Petrov, M. S. & Taylor, R. Intra-pancreatic fat deposition: bringing hidden fat to the fore. Nat. Rev. Gastroenterol. Hepatol. 19, 153–168 (2022).
Ahmad, E., Lim, S., Lamptey, R., Webb, D. R. & Davies, M. J. Type 2 diabetes. Lancet 400, 1803–1820 (2022).
Gerst, F. et al. Metabolic crosstalk between fatty pancreas and fatty liver: effects on local inflammation and insulin secretion. Diabetologia 60, 2240–2251 (2017).
Dasgupta, S. et al. NF-κB mediates lipid-induced fetuin-A expression in hepatocytes that impairs adipocyte function effecting insulin resistance. Biochem. J. 429, 451–462 (2010).
Richardson, A. & Park, W. G. Acute pancreatitis and diabetes mellitus: a review. Korean J. Intern. Med. 36, 15–24 (2021).
Roshani, R., McCarthy, F. & Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 345, 157–163 (2014).
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N. & Murtagh, F. R. An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 8, 429–431 (1995).
GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 7, e105–e125 (2022).
Ralhan, I., Chang, C.-L., Lippincott-Schwartz, J. & Ioannou, M. S. Lipid droplets in the nervous system. J. Cell Biol. 220, e202102136 (2021).
Islimye, E., Girard, V. & Gould, A. P. Functions of stress-induced lipid droplets in the nervous system. Front. Cell Dev. Biol. 10, 863907 (2022).
Farmer, B. C., Walsh, A. E., Kluemper, J. C. & Johnson, L. A. Lipid droplets in neurodegenerative disorders. Front. Neurosci. 14, 742 (2020).
Ioannou, M. S. et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177, 1522–1535.e14 (2019).
Smolič, T. et al. Astrocytes in stress accumulate lipid droplets. Glia 69, 1540–1562 (2021).
Schönfeld, P. & Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow. Metab. 33, 1493–1499 (2013).
Schönfeld, P. & Reiser, G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 109, 68–77 (2017).
Liu, L., MacKenzie, K. R., Putluri, N., Maletić-Savatić, M. & Bellen, H. J. The glia–neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 26, 719–737.e6 (2017).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C.-C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Moulton, M. J. et al. Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease-associated genes. Proc. Natl Acad. Sci. USA 118, e2112095118 (2021).
Fanning, S. et al. Lipidomic analysis of α-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol. Cell 73, 1001–1014.e8 (2019).
Cole, N. B. et al. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein α-synuclein. J. Biol. Chem. 277, 6344–6352 (2002).
Outeiro, T. F. & Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775 (2003).
Choi, W. et al. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 576, 363–368 (2004).
Jo, E., Fuller, N., Rand, R. P., St George-Hyslop, P. & Fraser, P. E. Defective membrane interactions of familial Parkinson’s disease mutant A30P α-synuclein 1. J. Mol. Biol. 315, 799–807 (2002).
Fares, M.-B. et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509 (2014).
Pennetta, G. & Welte, M. A. Emerging links between lipid droplets and motor neuron diseases. Dev. Cell 45, 427–432 (2018).
Tadepalle, N. & Rugarli, E. I. Lipid droplets in the pathogenesis of hereditary spastic paraplegia. Front. Mol. Biosci. 8, 673977 (2021).
Bosch, M. et al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science 370, eaay8085 (2020).
Dias, S. S. G. et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 16, e1009127 (2020).
Ricciardi, S. et al. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 606, 761–768 (2022).
Li, D. et al. The ER-localized protein DFCP1 modulates ER-lipid droplet contact formation. Cell Rep. 27, 343–358.e5 (2019).
Gao, G., Sheng, Y., Yang, H., Chua, B. T. & Xu, L. DFCP1 associates with lipid droplets. Cell Biol. Int. 43, 1492–1504 (2019).
Twu, W.-I. et al. Contribution of autophagy machinery factors to HCV and SARS-CoV-2 replication organelle formation. Cell Rep. 37, 110049 (2021).
Lukmantara, I. et al. PI(3)P and DFCP1 regulate the biogenesis of lipid droplets. Mol. Biol. Cell 33, ar131 (2022).
Yuan, S. et al. SARS-CoV-2 exploits host DGAT and ADRP for efficient replication. Cell Discov. 7, 100 (2021).
Pereira-Dutra, F. S., Teixeira, L., de Souza Costa, M. F. & Bozza, P. T. Fat, fight, and beyond: the multiple roles of lipid droplets in infections and inflammation. J. Leukoc. Biol. 106, 563–580 (2019).
Laufman, O., Perrino, J. & Andino, R. Viral generated inter-organelle contacts redirect lipid flux for genome replication. Cell 178, 275–289.e16 (2019).
Cruz, A. L. S. et al. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis. 11, 105 (2020).
Qiu, B. et al. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 5, 652–667 (2015).
Corbet, C. et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 11, 454 (2020).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Dierge, E. et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 33, 1701–1715.e5 (2021).
Cheng, X. et al. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 32, 229–242.e8 (2020).
Tirinato, L. et al. Lipid droplets and ferritin heavy chain: a devilish liaison in human cancer cell radioresistance. eLife 10, e72943 (2021).
Rozeveld, C. N., Johnson, K. M., Zhang, L. & Razidlo, G. L. KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase HSL. Cancer Res. 80, 4932–4945 (2020).
Goldberg, I. J. et al. Deciphering the role of lipid droplets in cardiovascular disease: a report from the 2017 National Heart, Lung, and Blood Institute Workshop. Circulation 138, 305–315 (2018).
Goldberg, I. J., Trent, C. M. & Schulze, P. C. Lipid metabolism and toxicity in the heart. Cell Metab. 15, 805–812 (2012).
Banke, N. H. et al. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARα. Circ. Res. 107, 233–241 (2010).
Haemmerle, G. et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 17, 1076–1085 (2011).
Liu, L. et al. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J. Clin. Invest. 117, 1679–1689 (2007).
Drevinge, C. et al. Perilipin 5 is protective in the ischemic heart. Int. J. Cardiol. 219, 446–454 (2016).
Anderson, E. J. et al. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898 (2009).
Glass, C. K. & Witztum, J. L. Atherosclerosis: the road ahead. Cell 104, 503–516 (2001).
Chang, T.-Y., Li, B.-L., Chang, C. C. Y. & Urano, Y. Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 297, E1–E9 (2009).
Fazio, S. et al. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J. Clin. Invest. 107, 163–171 (2001).
Nissen, S. E. et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N. Engl. J. Med. 354, 1253–1263 (2006).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721 (2013).
Wilcock, D. J. et al. Oxidative stress from DGAT1 oncoprotein inhibition in melanoma suppresses tumor growth when ROS defenses are also breached. Cell Rep. 39, 110995 (2022).
Nardi, F. et al. DGAT1 inhibitor suppresses prostate tumor growth and migration by regulating intracellular lipids and non-centrosomal MTOC protein GM130. Sci. Rep. 9, 3035 (2019).
Yue, S. et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 19, 393–406 (2014).
Ferguson, D. & Finck, B. N. Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 17, 484–495 (2021).
Xu, N. et al. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER-lipid droplet interface. J. Cell Biol. 198, 895–911 (2012).
Amin, N. B. et al. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci. Transl. Med. 11, eaav9701 (2019).
Gluchowski, N. L. et al. Hepatocyte deletion of triglyceride-synthesis enzyme Acyl CoA: diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology 70, 1972–1985 (2019).
Zhang, H.-B., Su, W., Xu, H., Zhang, X.-Y. & Guan, Y.-F. HSD17B13: a potential therapeutic target for NAFLD. Front. Mol. Biosci. 8, 824776 (2021).
Loomba, R. et al. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 5, 829–838 (2020).
Calle, R. A. et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 27, 1836–1848 (2021).
Haas, J. T. et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Invest. 122, 4680–4684 (2012).
Hong, Y. B. et al. DGAT2 mutation in a family with autosomal-dominant early-onset axonal Charcot-Marie-Tooth disease. Hum. Mutat. 37, 473–480 (2016).
Kantartzis, K. et al. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin. Sci. 116, 531–537 (2009).
Seco, C. Z. et al. A homozygous FITM2 mutation causes a deafness-dystonia syndrome with motor regression and signs of ichthyosis and sensory neuropathy. Dis. Model. Mech. 10, 105–118 (2016).
Shakir, A., Wadley, A. F., Purcarin, G. & Wierenga, K. J. The first case of deafness-dystonia syndrome due to compound heterozygous variants in FITM2. Clin. Case Rep. 6, 1815–1817 (2018).
Zhang, L., Miyaki, K., Nakayama, T. & Muramatsu, M. Cell death-inducing DNA fragmentation factor α-like effector A (CIDEA) gene V115F (G→T) polymorphism is associated with phenotypes of metabolic syndrome in Japanese men. Metabolism 57, 502–505 (2008).
Zhou, L. et al. Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 56, 95–107 (2012).
Whitfield, A. J., Barrett, P. H. R., van Bockxmeer, F. M. & Burnett, J. R. Lipid disorders and mutations in the APOB gene. Clin. Chem. 50, 1725–1732 (2004).
Burnett, J. R., Hooper, A. J. & Hegele, R. A. APOB-related familial hypobetalipoproteinemia. In GeneReviews (eds Adam, M. P. et al.) (University of Washington, 1993).
Huang, Y. & Mahley, R. W. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 72 Pt A, 3–12 (2014).
Luo, F., Oldoni, F. & Das, A. TM6SF2: a novel genetic player in nonalcoholic fatty liver and cardiovascular disease. Hepatol. Commun. 6, 448–460 (2022).
Prescher, M., Kroll, T. & Schmitt, L. ABCB4/MDR3 in health and disease – at the crossroads of biochemistry and medicine. Biol. Chem. 400, 1245–1259 (2019).
Schreiber, R., Xie, H. & Schweiger, M. Of mice and men: the physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta 1864, 880–899 (2019).
Lass, A. et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 3, 309–319 (2006).
Brown, A. L. & Brown, J. M. Critical roles for α/β hydrolase domain 5 (ABHD5)/comparative gene identification-58 (CGI-58) at the lipid droplet interface and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1233–1241 (2017).
Zhang, H. Lysosomal acid lipase and lipid metabolism: new mechanisms, new questions, and new therapies. Curr. Opin. Lipidol. 29, 218–223 (2018).
Magré, J. et al. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes 52, 1573–1578 (2003).
Agarwal, A. K. et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet. 31, 21–23 (2002).
Magré, J. et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 28, 365–370 (2001).
Fei, W., Du, X. & Yang, H. Seipin, adipogenesis and lipid droplets. Trends Endocrinol. Metab. 22, 204–210 (2011).
Kim, C. A. et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J. Clin. Endocrinol. Metab. 93, 1129–1134 (2008).
Hill, M. M. et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124 (2008).
Hayashi, Y. K. et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J. Clin. Invest. 119, 2623–2633 (2009).
Krimm, I. et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 10, 811–823 (2002).
Tontonoz, P., Hu, E., Devine, J., Beale, E. G. & Spiegelman, B. M. PPAR gamma 2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 15, 351–357 (1995).
Agarwal, A. K. & Garg, A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-γ gene in a patient with familial partial lipodystrophy. J. Clin. Endocrinol. Metab. 87, 408–411 (2002).
Granneman, J. G., Moore, H.-P. H., Krishnamoorthy, R. & Rathod, M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 284, 34538–34544 (2009).
Shen, W.-J., Patel, S., Miyoshi, H., Greenberg, A. S. & Kraemer, F. B. Functional interaction of hormone-sensitive lipase and perilipin in lipolysis. J. Lipid Res. 50, 2306–2313 (2009).
Kozusko, K. et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes 64, 299–310 (2015).
Rubio‐Cabezas, O. et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol. Med. 1, 280–287 (2009).
Albert, J. S. et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 370, 2307–2315 (2014).
Acknowledgements
Work in the laboratory of H.Y. is supported by grants 1141938, 1141939 and 1144726 from the National Health and Medical Research Council (NHMRC) of Australia, as well as grant DP210102576 from the Australian Research Council. H.Y. is a Level 3 Investigator of the NHMRC (2009852). We apologize to colleagues whose work is not cited due to space limitations.
Author information
Authors and Affiliations
Contributions
A.Z. and H.Y. contributed equally to all aspects of the article. X.D. contributed substantially to discussion of the content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Endocrinology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zadoorian, A., Du, X. & Yang, H. Lipid droplet biogenesis and functions in health and disease. Nat Rev Endocrinol 19, 443–459 (2023). https://doi.org/10.1038/s41574-023-00845-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41574-023-00845-0
This article is cited by
-
ASGR1 deficiency diverts lipids toward adipose tissue but results in liver damage during obesity
Cardiovascular Diabetology (2024)
-
Lipid droplets in pathogen infection and host immunity
Acta Pharmacologica Sinica (2024)
-
Lipid droplets and cellular lipid flux
Nature Cell Biology (2024)
-
Hairpin protein partitioning from the ER to lipid droplets involves major structural rearrangements
Nature Communications (2024)
-
Could Cytoplasmic Lipid Droplets be Linked to Inefficient Oxidative Phosphorylation in Cancer?
Current Tissue Microenvironment Reports (2024)
