
Abstract
Modern photovoltaic devices are often based on a heterojunction structure where two components with different optoelectronic properties are interfaced. The properties of each side of the junction can be tuned by either utilizing different materials (for example, donor/acceptor) or doping (for example, p–n junction) or even varying their dimensionality (for example, 3D/2D). Here we demonstrate the concept of phase heterojunction (PHJ) solar cells by utilizing two polymorphs of the same material. We demonstrate the approach by forming γ-CsPbI3/β-CsPbI3 perovskite PHJ solar cells. We find that all of the photovoltaic parameters of the PHJ device significantly surpass those of each of the single-phase devices, resulting in a maximum power conversion efficiency of 20.1%. These improvements originate from the efficient passivation of the β-CsPbI3 by the larger bandgap γ-CsPbI3, the increase in the built-in potential of the PHJ devices enabled by the energetic alignment between the two phases and the enhanced absorption of light by the PHJ structure. The approach demonstrated here offers new possibilities for the development of photovoltaic devices based on polymorphic materials.
Similar content being viewed by others


Main
Most photovoltaic technologies rely on the use of a junction to enable their function as an efficient solar cell1,2,3,4,5. The fundamental concept behind this approach is independent of how the junction is realized and is based on the combination of two components with different optoelectronic properties. The simplest realization of a heterojunction is the combination of two different materials. For example, organic solar cells rely on the formation of a junction between donor and acceptor organic materials in a ‘bulk heterojunction’, thus facilitating charge separation at the heterointerface (Fig. 1a)2,6,7,8,9. The well-established Si solar cells are based on a different type of junction, one that is formed by p- and n-doping the Si crystal on either side of the homojunction (Fig. 1b)10. However, in CdTe (ref. 11), CIGS (ref. 12) and GaAs (ref. 13) solar cells, a p–n junction can also be formed as a heterojunction (Fig. 1c). Finally, another noteworthy example is the use of junctions of varying dimensionality, such as a 3D/2D junction (Fig. 1d) in perovskite solar cells14, leading to improvements in their efficiency and stability.

a–e, Donor–acceptor heterojunction (a), p–n homojunction (b), p–n heterojunction (c), 3D/2D dimensional heterojunction (d) and phase heterojunction (e).
Polymorphism is observed in many classes of crystalline materials15,16,17,18,19. This phenomenon is based on the ability of a material to exist in several crystalline phases, with different structural arrangements of atoms or molecules15,18. Both organic and inorganic materials may exhibit polymorphism, which has been shown to strongly impact their optoelectronic properties20. It is thus interesting to explore the possibility of combining two polymorphs of the same material to form a phase heterojunction (PHJ).
A promising candidate for the demonstration of this concept is the material class of metal halide perovskites (MHPs). MHPs exhibit favourable optoelectronic properties, which have led to their successful integration in high-performance photovoltaic devices21,22,23,24,25. Moreover, MHPs exhibit multiple polymorphs based on their composition. For example, the inorganic CsPbI3 perovskite exhibits four different polymorphs26, three of which are photoactive and have been exploited for the fabrication of photovoltaic devices27. α-CsPbI3 can be formed by spin-coating a 1:1 CsI:PbI2 solution in N,N-dimethylformamide (DMF) and heating to 335 °C (ref. 28). With cooling to room temperature, the perovskite would not directly turn to the yellow phase (δ-CsPbI3). Instead, CsPbI3 tends to form metastable polymorphs, where the high-symmetry cubic phase (α-CsPbI3) initially distorts to a tetragonal phase (β-CsPbI3), followed by a further change to the orthorhombic phase (γ-CsPbI3) (ref. 27). Notably, the optical bandgaps of the two metastable phases, β-CsPbI3 and γ-CsPbI3, are clearly different (β-CsPbI3 ≈ 1.68 eV, γ-CsPbI3 ≈ 1.74 eV)29. Additionally, their lattice constants also change as the relative orientation of the [PbI6]4− octahedra is altered30. With the development of a stabilizing strategy, all three of the photoactive polymorphs can be maintained at room temperature. α-CsPbI3 or γ-CsPbI3 solar cells were reported to achieve a power conversion efficiency (PCE) of ~17% (refs. 31,32). β-CsPbI3 solar cells result in higher PCEs than α- and γ-CsPbI3-based devices, with high short circuit current densities (JSC) of over 20 mA cm−2 owing to their narrower bandgap29,33,34,35,36. The PCEs of β-CsPbI3-based solar cells have surpassed 20% and 18% in standard and inverted structure, respectively36,37.
To realize the PHJ concept, we chose to combine the β- and the γ-phases of CsPbI3. We find that compared with single-phase (SP) solar cell devices, the PHJ devices exhibit a simultaneous increase in all of the photovoltaic parameters, reaching a maximum fill factor of 84.17% and power conversion efficiency of 20.17%. Microscopic and spectroscopic characterization revealed that this increase in performance is a result of effective defect passivation, beneficial energetic alignment and enhanced light absorption in the PHJ structures.
Fabrication of the PHJ and structural characterization
Our choice of the β-CsPbI3 and γ-CsPbI3 phases for the demonstration of the PHJ concept was motivated by their relatively low-temperature processing, unlike the α-CsPbI3 that requires annealing at >300 °C (ref. 28). Considering that solution processing of all polymorphs of CsPbI3 requires the use of the same type of polar solvent38, we utilized a hybrid deposition approach that combines both solution processing and thermal evaporation. Specifically, we first deposited the narrower bandgap β-CsPbI3 by a single step solution deposition method29, followed by vapour deposition of the γ-CsPbI3 (Fig. 2a). The β-CsPbI3 was fabricated following the dimethylammonium iodide (DMAI) method. DMAI is an effective volatile additive that allows manipulation of the crystallization process of β-CsPbI3 by forming an intermediate phase22. The additive is then sublimated during the annealing process. The γ-CsPbI3 was fabricated by a co-evaporation process we developed in a previous study39. In this process, a small amount of 2-phenylethylammonium iodide is co-evaporated alongside the perovskite precursors and serves as the additive and stabilizer to maintain the γ-CsPbI3. Throughout our study, we kept the thickness of the solution-processed β-CsPbI3 fixed at 370 nm and varied the thickness of the vapour-deposited γ-CsPbI3 from 2 nm to 200 nm, labeled in the following as PHJ2 to PHJ200.

a, Schematic illustration of the fabrication procedure of the β-CsPbI3/γ-CsPbI3 PHJ. The devices are based on the structure glass/indium tin oxide (ITO)/[2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid (MeO-2PACz)/β-CsPbI3/γ-CsPbI3/phenyl-C61-butyric acid methyl ester (PCBM)/bathocuproine (BCP)/Ag. b, Surface SEM images of β-CsPbI3, PHJ10, PHJ20, PHJ50, PHJ100, PHJ200 and γ-CsPbI3 are shown from left to right. c, Cross-sectional SEM of PHJ100. d, XRD of β-CsPbI3, γ-CsPbI3 and PHJ layers with varying thicknesses. The range 14.4° to 28.4° is not shown in order to highlight the perovskite diffraction peaks. The complete XRD patterns are shown in Supplementary Fig. 3b. e, XRD of PHJ100 measured at different incidence angles.
To characterize the microstructure of the deposited layers, their surface was imaged using scanning electron microscopy (SEM). The solution-processed β-CsPbI3 shows a large and uniform grain size with an average diameter of ~300 nm. The microstructures of PHJ10 and PHJ20 are similar, however, with some additional small domains visible at the grain boundaries and on the surface of the β-CsPbI3 grains. Increasing the thickness of γ-CsPbI3 further leads to the formation of grains with comparable sizes to those of the underlying β-CsPbI3, but with orderly striped edges on the surfaces of the grain, particularly evident for the PHJ100 films. This is probably associated with the highly oriented growth of γ-CsPbI3 along the (00l) planes, as shown in our previous work and Supplementary Fig. 1a39. With the thickness of the top γ-CsPbI3 increasing to 200 nm, the surface morphology becomes more disordered with a myriad of grains with different shapes and sizes. A comparison to a 200 nm-thick γ-CsPbI3 film deposited on the glass reveals that such a film exhibits far smaller grains than the β-CsPbI3 or the PHJ films with thicknesses <200 nm. This suggests that the large grains of the underlying β-CsPbI3 impact on the growth of the γ-CsPbI3 perovskite, such that the grain size of the latter is increased when deposited on a perovskite layer. This is further confirmed by SEM images of γ-CsPbI3 of varying thicknesses deposited on glass substrates (Supplementary Fig. 1b–f). Even the thinnest 10 nm-thick γ-CsPbI3 samples exhibit a compact film with small grains, which are hard to distinguish due to the lack of contrast at the boundaries. While the grains become slightly larger with increasing thickness of γ-CsPbI3, they remain much smaller than those observed for the PHJ samples, confirming that the growth is influenced by the underlying β-CsPbI3 microstructure.
To investigate the composition of the small clusters observed on top of the large grains of the PHJ samples, we performed energy-dispersive x-ray spectroscopy elemental mapping on the PHJ100 sample (Supplementary Fig. 2a–d). The experiment revealed that the distribution of Pb, Cs and I is fully homogeneous across the layer, suggesting that these small clusters are CsPbI3 domains formed during the evaporation process.
Cross-sectional SEM imaging was performed to examine the horizontal microstructure of the PHJ100 film (Fig. 2c). The cross section shows that the β-CsPbI3 layer exhibits large vertical grains with few boundaries in the direction of transport. Meanwhile, the 100 nm-thick γ-CsPbI3 perovskite layer is comprised of similarly sized grains, with a very clear boundary between the two layers. For comparison, the cross-sectional SEM images of single β-CsPbI3 and γ-CsPbI3 layers are shown in Supplementary Fig. 1a,b, respectively. As has been already observed from surface images, the growth of γ-CsPbI3 on top of the β-CsPbI3 leads to far larger grain sizes than when deposited on glass, suggesting that the microstructure of the γ-CsPbI3 is templated by that of the underlying β-CsPbI3 layer.
To determine the crystal structure of the perovskite films, we used x-ray diffraction (XRD) techniques, including grazing-incidence XRD. We indexed the XRD patterns to β-CsPbI3 and γ-CsPbI3 (Supplementary Fig. 3a)26,40,41. The obvious difference in the peak positions between the β and γ phases can be observed and is especially evident for the ~28° peak (Fig. 2d). The vapour-deposited γ-CsPbI3 grows along the (00l) planes, leading to strong diffraction peaks of the (002) and (004) reflections, which appear at lower 2θ angles than the diffraction peaks of the β-CsPbI3, which correspond to the (110) and (220) reflections. With increasing thickness of the PHJs, contributions from both phases can be observed, especially noticeable in the PHJ100 and PHJ200 films. The full XRD patterns can be seen in Supplementary Fig. 3b.
To further confirm the structure of the γ-CsPbI3/β-CsPbI3 PHJ, grazing-incidence XRD experiments were performed on the PHJ100 sample by varying the incidence angle Ω from 0.4° to 5° (Fig. 2e). The lower incidence angles are more surface sensitive, and the XRD patterns (measured around ~28°) show a stronger contribution of γ-CsPbI3 (004) diffraction peak, with a smaller contribution of the β-CsPbI3 (220) peak. Increasing Ω to 5° makes the measurement more bulk sensitive, leading to a far stronger signal from the β-CsPbI3 (220) peak. These measurements confirm the formation of a γ-CsPbI3/β-CsPbI3 PHJ that retains the phase of each of the junction layers.
Optical properties of the PHJ
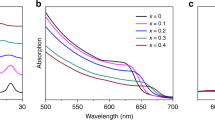
To investigate the optical properties of the perovskite films, the absorption and photoluminescence (PL) properties were characterized. Figure 3a displays the ultraviolet–visible (UV–vis) absorption spectra of the SP and PHJ films. The absorption edges of the γ- and β-CsPbI3 are ~710 nm and ~740 nm, respectively, which is consistent with previous reports in the literature29,32,34,36,41. These measurements further confirm that the phases of the CsPbI3 films deposited by solution and evaporation indeed correspond to the β and γ phases. To precisely evaluate the bandgap of each of the two phases, Tauc plots (Supplementary Fig. 4) were extracted from the absorbance data. The bandgap of γ- and β-CsPbI3 were found to be ~1.74 eV and ~1.69 eV, respectively. Their two slightly different bandgaps result in the absorbance spectra of the PHJ samples displaying features originating from both phases, especially evident in the PHJ200 spectrum that exhibits a strong absorption feature around 680 nm.

a–d, UV–vis (a), PL (b), PLQY (c) and PL lifetime (d) measured on SP γ-CsPbI3 and β-CsPbI3 and PHJ with varying layer thickness. The box plot in c displays the mean, median line, 25~75% box limits with 1.5× interquartile range whiskers. To obtain the PLQY data displayed in c, 7, 4, 5, 7, 5, 7, 5 measurements were performed for β-CsPbI3, PHJ10, PHJ20, PHJ50, PHJ100, PHJ200 and γ-CsPbI3, respectively.
Steady-state PL measurements are shown in Fig. 3b. The PL peak positions of γ- and β-CsPbI3 are at ~710 nm and ~740 nm, respectively, which is consistent with their absorption edges. The PL spectra of the PHJ samples are centred around 740 nm, consistent with light emission only from the low-bandgap β-CsPbI3. It is curious that no emission from the γ-CsPbI3 is observed, even for the PHJ samples with the thickest γ-CsPbI3 layers. There are two things to consider when examining the PL behaviour. First, the bandgap of the γ-CsPbI3 is larger than that of the β-CsPbI3 which would enable energy transfer from the γ-CsPbI3 to the β-CsPbI3, thus suppressing emission from the γ-CsPbI3. Secondly, the energetic alignment between the two phases may lead to charge transfer. Indeed, based on UV photoemission spectroscopy (UPS) depth profiling measurements (Supplementary Fig. 5), the valence bands of the β-CsPbI3 and γ-CsPbI3 phases are aligned at the heterointerface, which suggests that the conduction band of the β-CsPbI3 is slightly lower than that of the γ-CsPbI3. Upon photoexcitation, this would result in electron extraction towards the β-CsPbI3, while the holes remain in the γ-CsPbI3, thus serving as an additional mechanism that prevents radiative recombination in the γ-CsPbI3.
Monitoring the intensity of the PL and the corresponding evolution of the PL quantum yield (PLQY) (Fig. 3c) reveals an interesting trend. With the introduction of 10 nm of γ-CsPbI3 on top of the β-CsPbI3, the PLQY increases by a factor of two, suggesting a thin layer of γ-CsPbI3 can effectively passivate trap states at the surface of the β-CsPbI3, thus suppressing non-radiative recombination. However, with the further increase in the thickness of the γ-CsPbI3 layer, the PLQY gradually decreases up to the value of the SP γ-CsPbI3. We believe that this trend is a result of electron transfer from the γ-CsPbI3 to the β-CsPbI3 phase across the heterointerface, which opens an additional relaxation pathway via an exciton-electron annihilation in the β-CsPbI3. As the thickness of the 𝛾-CsPbI3 increases, a larger number of electrons formed upon a photoexcitation of the γ-CsPbI3 can be transferred to the β-CsPbI3, thus progressively lowering the PLQY of the β-CsPbI3.
Figure 3d displays the time-resolved photoluminescence decay curves of SP and PHJ films. Considering that the curves exhibit a non-monoexponential decay, the lifetime was evaluated using τave, with the details of the fitting process and the resulting parameters summarized in Supplementary Fig. 6 and Supplementary Table 1. We observe that the τave of β-CsPbI3 is 9.5 ns, which is consistent with other reported values in the literature34,37. However, γ-CsPbI3 exhibits a much longer τave up to 125 ns. With the introduction and increasing thickness of γ-CsPbI3 in the PHJ, the τave increases from 9.56 ns of β-CsPbI3 to 104 ns of PHJ100. However, the τave of PHJ200 decreases to 77 ns. These results are consistent with our previous observation that γ-CsPbI3 can effectively passivate the underlying β-CsPbI3.
Photovoltaic performance of SP and PHJ solar cells
To investigate the photovoltaic performance of the SP and PHJ layers, they were incorporated into photovoltaic devices with an inverted architecture following the structure: glass/indium tin oxide (ITO)/[2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid (MeO-2PACz)/β-CsPbI3/γ-CsPbI3/phenyl-C61-butyric acid methyl ester (PCBM)/bathocuproine (BCP)/Ag (Fig. 2a). Additionally, SP devices with the optimal thickness (370 nm for β-CsPbI3 and 500 nm for γ-CsPbI3) were fabricated. The photovoltaic performance parameters of the devices are summarized in Fig. 4a–d. Comparing the performance of the SP solar cells reveals that the open-circuit voltage (VOC) of the β-CsPbI3 SP devices is approximately 0.1 V lower than that of the γ-CsPbI3 SP solar cells, a much higher difference than the 0.05 eV change in their bandgaps. This suggests that additional non-radiative losses are present in the β-CsPbI3 solar cells, in agreement with the short PL lifetime of the corresponding thin films and with the previous reports of a high density of surface defects in the case of β-CsPbI3 solar cells29. On the other hand, the short circuit current density (JSC) of the β-CsPbI3 devices is substantially higher than the JSC of γ-CsPbI3 devices. This is consistent with the lower bandgap of β-CsPbI3, which enables absorption across a larger spectral region, thus resulting in a higher photocurrent. Both SP devices result in a similar range of fill factors (FF), with slightly higher values in the case of the γ-CsPbI3 solar cells. The opposing trends of the VOC and JSC between the two SP devices lead to a similar power conversion efficiency (PCE) in both cases of up to 15%.

a–g, Distribution of VOC (a), JSC (b), FF (c), PCE parameters (d), EQE spectra (e), J–V characteristics (f) and hysteresis index (g) of SP and PHJ solar cells with varying thicknesses. h, Comparison of PCEs of standard and inverted architecture CsPbI3 solar cells (data taken from refs. 28,29,31,33,36,37,43,44,45,46,47,48,49). The curves drawn on top of the data are presented as guides to the eye. The box plots in a–d and g display the mean, median line, 25~75% box limits with 1.5× interquartile range whiskers. The number of samples of each column of panels a, b, c, d and g is 64, 7, 8, 23, 30, 46, 120, 39, 74, respectively. Twenty-one data points from the same batch for ‘β-CsPbI3’ are also used in Supplementary Fig. 12 as ‘370 nm’. From ‘β-CsPbI3’, ‘γ-CsPbI3’, ‘PHJ100’ in d, 18 data points of the same batch are also provided in Supplementary Fig. 14.
The PHJ devices show a gradual increase in all of the photovoltaic parameters, reaching an optimum at PHJ100. PHJ100 devices exhibit a high VOC of up to 1.15 V, a JSC reaching up to 20 mA cm−2 and a high FF of 82%. The simultaneous increase in all the photovoltaic performance parameters leads to a substantial increase in the PCE of PHJ devices, surpassing 19% in the case of PHJ100 (Supplementary Fig. 7 provides for maximum power point tracking). Considering that the PL studies indicated that thin layers of γ-CsPbI3 may passivate surface defects of the underlying β-CsPbI3, we fabricated devices in which ultra-thin layers of γ-CsPbI3 were evaporated (2 nm and 5 nm). Because passivation is a surface effect, such thin layers make it possible to identify to which degree passivation is responsible for the increase in the photovoltaic performance. We observe that the evaporation of 2 nm of γ-CsPbI3 results in an increase in VOC and FF, thus increasing the average PCE of the β-CsPbI3 SP devices from 13.2% to 14.3%. This moderate increase is consistent with surface passivation and is similar to what is observed when other surface passivation methods are applied (Supplementary Fig. 8). This result and the fact that the VOC and FF continue to increase up to PHJ100 suggests that while passivation of the surface defects of the β-CsPbI3 contributes to the increase in photovoltaic performance of the PHJ, it is not the sole factor leading to an enhancement in performance. Examining the energy band diagram reveals that the resultant energetic alignment increases the built-in potential of the device. Considering that all of the devices are fabricated with the same extraction layers, and that these extraction layers are always pinned a certain energetic distance below/above the conduction/valence band (Supplementary Fig. 9, left), the extraction of electrons from the higher-lying conduction band of the γ-CsPbI3 results in an increase in the built-in potential (VBI), and consequently the VOC and FF. Such a strategy has been previously proposed by device simulations in which higher-bandgap layers were introduced near the extraction layers, thus resulting in an increase in the VBI (ref. 42). Importantly, this highlights that with the structure β-CsPbI3/γ-CsPbI3, the observed increase in VOC and FF would only occur for inverted-architecture devices, because introducing the PHJ in a standard device architecture would lead to the opposite effect in which the VBI is reduced (Supplementary Fig. 9, right). Indeed, when we fabricated devices with the structure glass/ITO/TiO2/PVK/Spiro-OMeTAD/Au (PVK = β-CsPbI3 or β-CsPbI3/γ-CsPbI3), the PHJ devices showed a far lower performance, with a particularly strong reduction in the VOC and FF (Supplementary Fig. 10). To utilize the PHJ concept in standard architecture devices would thus require reversing the order of layers, that is, γ-CsPbI3/β-CsPbI3; however, this is not yet possible using existing fabrication protocols because the deposition of the β-CsPbI3 occurs from solution and would thus dissolve the underlying γ-CsPbI3. In the future, we plan to develop protocols to deposit β-CsPbI3 by thermal evaporation, which would enable the formation of PHJs in any order and thus introduce them into standard architecture devices as well.
The increase in the JSC observed when thicker layers of γ-CsPbI3 are used is associated with the increased absorption of the PHJ devices as compared to the SP ones. This is evident from comparing the external quantum efficiency (EQE) spectra of the devices (Fig. 4e), which confirm the far higher contribution to the photocurrent in the range 600 nm to 740 nm of the PHJ devices as compared with either of the SP solar cells. We highlight that only the γ-CsPbI3 device exhibits a different onset of the EQE spectra due to its larger bandgap. All of the PHJ devices show the same onset as that of the β-CsPbI3-only devices, with an increase in the 600–740 nm range due to the additional absorption by the γ-CsPbI3 layer. Correspondingly, the predicted JSC from the EQE curves is increased in the PHJ devices and is in a good agreement with the measured JSC (Supplementary Table 2). To confirm that this increase originates from the PHJ structure, we performed optical simulations, the results of which are shown in Supplementary Fig. 11. The excellent agreement in the spectral features of the simulated and experimentally measured EQE spectra confirms that the increased photocurrent originates from the increased light absorption enabled by the addition of the γ-CsPbI3. The high photocurrents also confirm that the small offset in the conduction band position across the PHJ (0.05 eV) does not hinder efficient electron transport from the β-CsPbI3 to the γ-CsPbI3 and its extraction at the PCBM layer. Importantly, the same improvement in the photovoltaic performance cannot be achieved by simply increasing the thickness of the SP photovoltaic devices. We found that the optimal thickness for the β-CsPbI3 SP device is 370 nm (Supplementary Fig. 12) and attempting to make the active layer thicker results in a loss of performance. The maximum thickness that can be achieved (420 nm) is limited by the solubility of the perovskite precursors, thus not allowing to reach the optimum thickness of the PHJ devices (that is, 470 nm). However, considering that the 420 nm devices show an inferior performance to that of the 370 nm, it is clear that a further increase would not lead to a better performance. This decrease in performance is probably caused by the poorer microstructure of the thicker β-CsPbI3 devices. Similarly, in our previous work, we found the optimal thickness of γ-CsPbI3 SP devices to be 500 nm (ref. 39), which is close to the 470 nm thickness of the PHJ100 devices, yet the SP γ-CsPbI3 devices show a lower photovoltaic performance, due to its larger bandgap and consequently lower JSC, highlighting that the PHJ structure is beneficial over each of the SP devices on their own.
The observation that the PHJ200 devices show a slightly reduced performance in comparison with the PHJ100 is probably associated with the poorer microstructure of the 200 nm-thick γ-CsPbI3 layers (Fig. 2b). As described above, the surface morphology becomes more disordered with a myriad of grains with a range of shapes and sizes, suggesting that the templating effect by the underlying β-CsPbI3 becomes weaker as the γ-CsPbI3 becomes very thick. Such a disordered microstructure may lead to additional defects, evidenced by a reduction in the PL lifetime (Fig. 3d) and a reduced VOC of the PHJ200 devices.
Current density–voltage (J–V) characteristics of representative devices of each kind are displayed in Fig. 4f, and their photovoltaic parameters are summarized in Table 1. It is noticeable that the β-CsPbI3 devices exhibit non-negligible hysteresis, which is significantly suppressed in both the γ-CsPbI3 SP and PHJ devices. To quantify this, we defined the hysteresis index, (PCErev − PCEfor) / PCErev, which is displayed in Fig. 4g for the devices whose performance is shown in Fig. 4a–d. Consistently, the hysteresis index is lower in the PHJ devices than in the β-CsPbI3 solar cells. In Fig. 4h, we summarize the PCE evolution of CsPbI3-based solar cells since their first introduction in 2014 (refs. 28,29,31,33,36,37,43,44,45,46,47,48,49). The performance of the inverted architecture-based PHJ100 is comparable with the standard-architecture CsPbI3 solar cells, thus demonstrating the enormous potential of the phase heterojunction solar cell concept.
To further investigate the origin of the improved photovoltaic performance, we focused on the characterization of the SP β-CsPbI3 solar cell and the PHJ100 device. Light intensity-dependent VOC measurements (Fig. 5a) show that the PHJ100 solar cell exhibits an overall lower trap-assisted recombination, as the ideality factor (n) measured for the reference β-CsPbI3 device (n =1.703) is reduced to n =1.163 for the PHJ devices. While it is not trivial to ascribe charge recombination mechanisms from light intensity-dependent VOC and ideality factor measurements in perovskite solar cells50, these measurements are supported by direct characterization of the sub-bandgap trap density in the β-CsPbI3 and PHJ100 films by photothermal deflection spectroscopy (PDS). The PDS spectra (Fig. 5b) show that the PHJ100 structure results in a notably reduced energetic disorder, corresponding to an Urbach energy of 19.80 meV, in comparison with 33.27 meV for the SP β-CsPbI3 thin films. The reduction in the trap density of the PHJ100 structure results in a suppression of non-radiative recombination losses as can be directly visualized by measuring the solar cells as light-emitting diodes. The electroluminescence quantum yield (ELQY) of the PHJ100 devices is approximately one order of magnitude higher than that of the SP β-CsPbI3 solar cells (Fig. 5c).

a, Light intensity-dependent VOC showing ideality factors (n) of 1.703 and 1.163 for SP β-CsPbI3 and PHJ100, respectively. b, PDS spectra showing the Urbach energies (EU) of 33.27 meV and 19.80 meV for SP β-CsPbI3 and PHJ100, respectively. c, ELQY. d, Temperature-dependent conductivity showing ion migration activation energies (Ea) of 0.79 eV and 1.66 eV and the transition temperatures of 260 K and 273 K for SP β-CsPbI3 and PHJ100, respectively. The inset in d shows the conductivity device with a lateral structure consisting of two Au (50 nm) electrodes with a length of 111 mm and spacing gap of 0.2 mm deposited on the surface of the perovskite film. e, Storage stability of PCE. Encapsulated devices were stored in nitrogen atmosphere at 25 °C and under dim light conditions. f, Performance evolution of encapsulated SP β-CsPbI3 and PHJ100 solar cells under continuous illumination. Up to 550 h stability was tested under 40% relative humidity (RH), 25 °C, constant xenon lamp-simulated solar illumination (100 mW cm−2) without a UV filter. The fitting lines in a, b and d were obtained by linear fitting of data points. f displays the mean, with 1.5× interquartile range whiskers.
To explore the origin of the reduced hysteresis in the PHJ devices, we characterized the temperature-dependent conductivity of the SP β-CsPbI3 and PHJ100 structures integrated in lateral devices, shown in the inset of Fig. 5d. The measurements allow extraction of the value of the activation energy (Ea) for ion migration by using the Nernst–Einstein equation, \(\sigma \left( T \right) = \frac{{\sigma _0}}{T}\mathrm{e}^{ - \frac{{E_\mathrm{a}}}{{kT}}}\), where σ, k and T are conductivity, Boltzmann constant and temperature, respectively. Such measurements have been previously utilized to characterize ion migration in perovskite solar cells51,52. We find that the Ea of the SP β-CsPbI3 (0.79 eV) is significantly increased in the case of PHJ100 (Ea = 1.66 eV). Moreover, the threshold temperature at which ion conduction starts to dominate the total current is increased from 260 K to 273 K. Both of these factors point towards a suppressed ion migration in the PHJ structures, consistent with the significantly reduced hysteresis observed in Fig. 4g.
To evaluate the stability of the PHJ devices, we monitored their performance upon storage under dim light conditions (Fig. 5e). The PHJ100 devices showed an improvement over the first 50 days, reaching a maximum performance of 20.17% (Supplementary Fig. 13). A gradual increase in the photovoltaic performance with storage time has been reported in the literature with several different mechanisms proposed53,54. While the elucidation of the exact mechanisms at play in our case is beyond the scope of this study, we consistently observed that stored PHJ devices outperform those measured directly after fabrication (Supplementary Fig. 14). The performance remains stable up to 100 days and is only slightly reduced after 180 days. Importantly, throughout the stability study, the PHJ devices consistently exhibited a superior performance to the β-CsPbI3 solar cells. Moreover, we monitored the evolution of the PCE of the devices upon continuous illumination under 1 sun conditions (Fig. 5f). Both types of devices maintained their performance for approximately 400 h, followed by a significant decrease in performance. The fact that both devices exhibit a similar lifetime suggests that their degradation is linked to the stability of the β-CsPbI3, and the fact that over the first 400 h, the performance of the PHJ device remained superior to the SP counterpart suggests that the PHJ structure retained its stability for this duration. To further confirm that the PHJ structure is maintained upon illumination, we fabricated PHJ layers with different thicknesses of γ-CsPbI3 and exposed them to 1 sun illumination for 100 h. Probing the optical absorption of the aged PHJ films reveals that it remained unchanged as compared with that of the freshly prepared PHJs, in particular the PHJ100 and PHJ200 structures, clearly showing the contributions of both the β-CsPbI3 and γ-CsPbI3 phases in their absorption spectra (Supplementary Fig. 15). Furthermore, examining the XRD patterns of PHJ100 samples exposed to either 1 sun illumination for 100 h or thermally annealed at 85 °C for 15 h shows clear contributions from both the β-CsPbI3 and γ-CsPbI3 phases, just as is observed for the freshly prepared samples (Supplementary Fig. 16). While there is no doubt that further improvements to device stability are necessary, our initial results are promising because they suggest that the PHJ structure itself is maintained upon prolonged illumination. Future development of degradation-mitigation strategies will certainly enhance the device stability further; however, they go beyond the scope of the current study in which the PHJ concept is demonstrated.
Conclusions
To summarize, in this work, we propose a concept for photovoltaics exploiting the phenomenon of polymorphism of crystalline materials. We demonstrate this concept by fabricating γ-CsPbI3/β-CsPbI3 PHJ solar cells, which show superior performance to either of the SP devices. Our findings offer a promising approach towards highly efficient photovoltaic devices that might in the future be realized also via other classes of polymorphic materials such as, for example, organic materials or other perovskite compositions.
Methods
Indium tin oxide (ITO)-coated glass substrates were bought from PsiOTech Ltd. (2-(3,6-Dimethoxy-9H-carbazol-9-yl)ethyl)phosphonic acid (MeO-2PACz) was purchased from TCI. Phenyl-C61-butyric acid methyl ester (PC61BM) (99.5%) and bathocuproine (BCP, 99.99%, trace metals basis) were obtained from Solenne BV and Sigma-Aldrich, respectively. TiCl (99.9%) were obtained from Alfa Aesar. Spiro-OMeTAD (99%, HPLC), 4-tert-butylpyridine (TBP, 98%) and Li-TFSI were obtained from Sigma-Aldrich. All solvents were purchased from Sigma-Aldrich. For solution-processed β-CsPbI3, PbI2 (99.999%, trace metals basis), CsI (99.999%, trace metals basis) were obtained from TCI. The PbI2·xDMAI was prepared by using PbI2 (1.2 g) dissolved in 2.5 ml anhydrous dimethylformamide (DMF) at 80 °C under active stirring for 30 min in air atmosphere. Immediately after, 10 ml of hydroiodic acid was added to the solution with stirring for 24 h at 80 °C. The precipitate was filtered, then centrifuged and washed several times with copious diethyl ether and ethyl alcohol to remove the residual solvent. The collected powder was dried in a vacuum oven at 70 °C for 24 h. For evaporated γ-CsPbI3, PbI2 (99.999%, trace metals basis), CsI (99.999%, trace metals basis) and 2-phenylethylammonium iodide (98%) were obtained from Sigma-Aldrich. All materials were used as received without purification.
Perovskite film deposition
Substrates for film deposition were ultrasonically cleaned with 2% Hellmanex detergent, deionized water, acetone and isopropanol followed by 15 min oxygen plasma treatment. In a dry box (RH < 1 %), MeO-2PACz (3 mg ml−1 in ethanol) was spin-coated on the clean ITO substrates at 4,000 r.p.m. for 30 s and annealed at 100 °C for 10 min. For n–i–p devices, laser-patterned fluorine-doped tin oxide coated glass substrates (2.9 cm × 2.9 cm) were immersed into aqueous solution of TiCl (4.5 ml TiCl in 200 ml H2O) at 70 °C for 1 h, followed by annealing at 200 °C for 0.5 h to obtain a compact TiO2 electron transport layer. The TiO2 was then cleaned using an UV-ozone cleaner for 12 min. The CsPbI3 precursor solution was prepared by dissolving 802.5 mg PbI2·xDMAI, 210.7 mg PbI2 and 415.9 mg CsI in 2 ml DMF and dimethyl sulfoxide (v/v, 9:1) under active stirring for 12 h at 60 °C. The CsPbI3 solution was spin-coated at 1,000 r.p.m. for 10 s and 4,500 r.p.m. for 30 s. Then the samples were annealed at 180 °C for 15 min. Next, the samples were transferred into a vacuum chamber (CreaPhys GmbH) to deposit γ-CsPbI3 with the desired thickness following previously reported procedures39. Next, the samples were transferred into a nitrogen-filled glove box (GS), where PC61BM (20 mg ml−1 dissolved in chlorobenzene) was dynamically spin-coated at 2,000 r.p.m. for 30 s followed by a 3 min annealing at 100 °C. Finally, a BCP (0.5 mg ml−1 dissolved in isopropanol) hole-blocking layer was spin-coated at 4,000 r.p.m. for 30 s, followed by an 80 nm thermally evaporated Ag cathode (Mantis evaporator, base pressure of 10−7 mbar). For n–i–p devices, 6.0 μl of Spiro-OMeTAD solution prepared by dissolving 90 mg Spiro-OMeTAD in 1.0 ml chlorobenzene containing 36 μl TBP, and 22 μl of Li-TFSI was spin cast on the perovskite layer at 1,500 r.p.m. for 40 s. Then, the samples were transferred into a dry box for the oxidation of Spiro-OMeTAD for an entire night. Finally, a 100 nm-thick gold electrode was deposited by thermal evaporation to complete the cell. The as-prepared photovoltaic devices were sealed/encapsulated in a glovebox using an UV-hardened epoxy glue and a transparent clean encapsulation glass.
Photovoltaic device characterization
EQE spectra of the devices were recorded using a monochromatized light of a halogen lamp from 400 nm to 800 nm, and the reference spectra were calibrated using a National Institute of Standards and Technology (NIST)-traceable Si diode (Thorlabs). J–V characteristics of solar cells under a solar simulator (Abet Sun 3000 Class AAA solar simulator, AM 1.5 conditions) were recorded at room temperature in ambient conditions using a computer-controlled Keithley 2450 source meter unit. The incident light intensity was calibrated via a Si reference cell (NIST traceable, VLSI Standards Inc.) and tuned by measuring the spectral mismatch factor between the real solar spectrum, the spectral response of the reference cell and the perovskite devices. All devices were scanned from short circuit to forward bias (1.2 V) and reversed with a rate of 0.025 V s−1. No treatment was applied before measurements. The active area for all devices was 4.5 mm2 defined by thermal evaporation of the Ag electrodes.
Ion migration for activation energy characterization
The activation energy of ion migration was extracted from the temperature dependence of the conductivity of the CsPbI3 films. In short, we used a lateral structure device that consists of two Au (50 nm) electrodes with a length of 111 mm and a spacing gap of 0.2 mm deposited on the surface of CsPbI3 films with and without a hetero-phase structure. During the measurements, an electric field of 0.3 V μm−1 was applied to all the devices. The measurements were conducted using a homemade setup. To change the device temperature, the devices were placed in vacuum on a copper block, which was connected to a Peltier element (Peltron) controlled by a BelektroniG HAT Control device. For each conductivity, the current through the devices were stabilized for 5 min when the desired temperature was reached before the current measurement was performed. A semiconductor analyser (Keithley SMU2635A) was used for the current measurement. The measurement equipment was controlled with the software SweepMe! (https://sweep-me.net).
SEM
A SEM (Gemini 500, ZEISS) with an acceleration voltage of 3 kV under 5–6 × 10−4 mbar was utilized to obtain the surface and cross-sectional SEM images using the in-lens mode.
XRD
XRD patterns were measured in ambient air using a Bruker Advance D8 diffractometer equipped with a 1.6 kW Cu-Anode (λ = 1.54060 Å) and a LYNXEYE_XE_T 1D detector. The scans (2θ-Ω mode, 2θ = 10–40°, step size 0.01°, 0.1 s per step) were measured in a parallel beam geometry with a height-limiting slit of 0.2 mm. For grazing-incidence XRD, the parameters of scans are 2theta mode, 2θ = 13.5–15°, 28–29.6°, step size 0.01°, 0.5 s per step. The incidence angle (Ω) was fixed at 0.4°, 0.5°, 1°, 2°, 5°, respectively.
UV−vis absorption and PL measurements
The UV–vis absorbance spectra were recorded using a Shimadzu UV-3100 spectrometer. The PL measurements were performed using a CW laser (405 nm, 10 mW, Coherent) as the excitation source. The PL signal was collected using a near-infrared spectrometer (OceanOptics). The PLQY was measured in an integrating sphere (Labsphere Inc). All samples were encapsulated to prevent decomposition and to enable all measurements being carried out in ambient air at room temperature.
PDS
PDS measurements were performed following the procedure described in our previous work55,56,57. A quartz substrate with a perovskite film was mounted in the signal-enhancing liquid-filled (Fluorinert FC-770) quartz cuvette inside a N2-filled glovebox. Then, the samples were excited using a tunable, chopped, monochromatic light source (150 W xenon short arc lamp with a Cornerstone monochromator) and probed using a laser beam (635 nm diode laser, Thorlabs) propagating parallel to the surface of the sample. The heat generated through the absorption of light changes the refractive index of the Fluorinert liquid, resulting in the deflection of the laser beam. This deflection was measured using a position sensitive detector (Thorlabs, PDP90A) and a lock-in amplifier (Amatec SR7230) and is directly correlated to the absorption of the film.
Time-correlated single photon counting
A time-correlated single photon counting setup containing a 375 nm laser diode head (Pico Quant LDHDC375), a PMA Hybrid Detector (PMA Hybrid 40), a TimeHarp platine (all PicoQuant) and a Monochromator SpectraPro HRS-300 (Princeton Instruments) was utilized. Perovskite films on quartz were excited with the 375 nm laser diode. The emission was collected by the PMA hybrid detector. The excitation fluence is around 7.5 nJ cm−2. The lifetimes were evaluated using reconvolution algorithms of FluoFit (PicoQuant).
Electroluminescence measurements
The devices were mounted in an integrating sphere (Labsphere Inc.). The current–voltage characteristics were recorded using a Keithley 2450 source meter unit. At the same time, a scientific grade spectrometer (Ocean Optics QE65PRO) was used to record the emission spectra, which were used to calculate the luminance. The optical system (spectrometer, integrating sphere and coupling optical fibre) was calibrated via a calibrated light source (Ocean Optics HL-2000-CAL).
UPS depth profiling
The samples were transferred to an ultra-high vacuum chamber (ESCALAB 250Xi) with a base pressure of 2 × 10−10 mbar for UPS depth profiling measurements. UPS measurements were performed using a double differentially pumped He gas discharge lamp emitting He I radiation (hν = 21.22 eV) with a pass energy of 2 eV and a bias of −5 V to ensure secondary electron onset detection. The UPS spectra are shown as a function of the binding energy with respect to the Fermi energy. The energy edge of the valence band features is used to determine the valence band level position with respect to the Fermi level. The conduction bands were estimated using the optical gaps of the γ-CsPbI3 and β-CsPbI3. Cluster etching was performed using large Ar clusters generated by the MAGCIS Dual Beam Ion Source (Thermo Scientific) with an energy of 4,000 eV.
Optical simulations
Optical simulations were performed using an in-house-developed optical simulation tool based on the transfer matrix method. The optical constants of perovskite layers were obtained by variable angle spectroscopic ellipsometry utilizing an EP4 imaging ellipsometer (Accurion GmbH). An effective isotropic optical model was applied to extract the complex refractive indices. The dispersion of both perovskite layer types was modelled by a superposition of four Tauc–Lorentz oscillators resulting in unbiased root mean square errors of <2.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated or analysed during this study are included in the published article and its supplementary information and source data files. Source data are provided with this paper.
References
Bach, U. et al. Solid-state dye-sensitized mesoporous TiO2 solar cells with high photon-to-electron conversion efficiencies. Nature 395, 583–585 (1998).
Peumans, P., Uchida, S. & Forrest, S. R. Efficient bulk heterojunction photovoltaic cells using small-molecular-weight organic thin films. Nature 425, 158–162 (2003).
Pattantyus-Abraham, A. G. et al. Depleted-heterojunction colloidal quantum dot solar cells. ACS Nano 4, 3374–3380 (2010).
Heo, J. H. et al. Efficient inorganic–organic hybrid heterojunction solar cells containing perovskite compound and polymeric hole conductors. Nat. Photonics 7, 486–491 (2013).
Liu, M., Johnston, M. B. & Snaith, H. J. Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 501, 395–398 (2013).
Yu, G., Gao, J., Hummelen, J. C., Wudl, F. & Heeger, A. J. Polymer photovoltaic cells: enhanced efficiencies via a network of internal donor-acceptor heterojunctions. Science 270, 1789–1791 (1995).
Wadsworth, A., Hamid, Z., Kosco, J., Gasparini, N. & McCulloch, I. The bulk heterojunction in organic photovoltaic, photodetector, and photocatalytic applications. Adv. Mater. 32, 2001763 (2020).
Yuan, J. et al. Effects of energetic disorder in bulk heterojunction organic solar cells. Energy Environ. Sci. 15, 2806–2818 (2022).
Chen, J. et al. Hole (donor) and electron (acceptor) transporting organic semiconductors for bulk-heterojunction solar cells. EnergyChem 2, 100042 (2020).
Shah, A., Torres, P., Tscharner, R., Wyrsch, N. & Keppner, H. Photovoltaic technology: the case for thin-film solar cells. Science 285, 692–698 (1999).
Möller, H. J. Semiconductors for Solar Cells (Artech House, 1993).
Heske, C. et al. Observation of intermixing at the buried CdS/Cu(In, Ga)Se2 thin film solar cell heterojunction. Appl. Phys. Lett. 74, 1451–1453 (1999).
Fedotov, Y. A., Zased, V. S. & Matson, É. A. in Physics of p–n Junctions and Semiconductor Devices (eds Ryvkin, S. M. & Shmartsev, Y. V.) 104–106 (Springer, 1971).
McGott, D. L. et al. 3D/2D passivation as a secret to success for polycrystalline thin-film solar cells. Joule 5, 1057–1073 (2021).
Bernstein, J. Polymorphism in Molecular Crystals 2e (Oxford University Press, 2020).
Gentili, D., Gazzano, M., Melucci, M., Jones, D. & Cavallini, M. Polymorphism as an additional functionality of materials for technological applications at surfaces and interfaces. Chem. Soc. Rev. 48, 2502–2517 (2019).
Caira, M. R. in Design of Organic Solids, Vol. 198 (ed Weber, E.) 163–208 (Springer-Verlag, 1998).
Bu, R., Li, H. & Zhang, C. Polymorphic transition in traditional energetic materials: influencing factors and effects on structure, property, and performance. Cryst. Growth Des. 20, 3561–3576 (2020).
Sood, S. & Gouma, P. Polymorphism in nanocrystalline binary metal oxides. Nanomater. Energy 2, 82–96 (2013).
Nogueira, B. A., Castiglioni, C. & Fausto, R. Color polymorphism in organic crystals. Commun. Chem. 3, 34 (2020).
Schmidt-Mende, L. et al. Roadmap on organic–inorganic hybrid perovskite semiconductors and devices. APL Mater. 9, 109202 (2021).
Min, H. et al. Perovskite solar cells with atomically coherent interlayers on SnO2 electrodes. Nature 598, 444–450 (2021).
Chen, S. et al. Stabilizing perovskite-substrate interfaces for high-performance perovskite modules. Science 373, 902–907 (2021).
Degani, M. et al. 23.7% efficient inverted perovskite solar cells by dual interfacial modification. Sci. Adv. 7, eabj7930 (2021).
Bube, R. H., Buch, F., Fahrenbruch, A. L., Ma, Y. Y. & Mitchell, K. W. Photovoltaic energy conversion with n-CdS—p-CdTe heterojunctions and other II–VI junctions. IEEE Trans. Electron Devices 24, 487–492 (1977).
Sutton, R. J. et al. Cubic or orthorhombic? Revealing the crystal structure of metastable black-phase CsPbI3 by theory and experiment. ACS Energy Lett. 3, 1787–1794 (2018).
Marronnier, A. et al. Anharmonicity and disorder in the black phases of cesium lead iodide used for stable inorganic perovskite solar cells. ACS Nano 12, 3477–3486 (2018).
Eperon, G. E. et al. Inorganic caesium lead iodide perovskite solar cells. J. Mater. Chem. A 3, 19688–19695 (2015).
Wang, Y. et al. Thermodynamically stabilized beta-CsPbI3-based perovskite solar cells with efficiencies >18%. Science 365, 591–595 (2019).
Stoumpos, C. C. & Kanatzidis, M. G. The renaissance of halide perovskites and their evolution as emerging semiconductors. Acc. Chem. Res. 48, 2791–2802 (2015).
Wang, Y., Zhang, T., Kan, M. & Zhao, Y. Bifunctional stabilization of all-inorganic α-CsPbI3 perovskite for 17% efficiency photovoltaics. J. Am. Chem. Soc. 140, 12345–12348 (2018).
Ye, Q. et al. Stabilizing γ-CsPbI3 perovskite via phenylethylammonium for efficient solar cells with open-circuit voltage over 1.3 V. Small 16, 2005246 (2020).
Wang, Y. et al. The role of dimethylammonium iodide in CsPbI3 perovskite fabrication: additive or dopant? Angew. Chem. Int. Ed. 58, 16691–16696 (2019).
Chang, X. et al. Printable CsPbI3 perovskite solar cells with PCE of 19% via an additive strategy. Adv. Mater. 32, 2001243 (2020).
Du, Y. et al. Ionic liquid treatment for highest-efficiency ambient printed stable all-inorganic CsPbI3 perovskite solar cells. Adv. Mater. 34, 2106750 (2022).
Yoon, S. M. et al. Surface engineering of ambient-air-processed cesium lead triiodide layers for efficient solar cells. Joule 5, 183–196 (2021).
Fu, S. et al. Humidity-assisted chlorination with solid protection strategy for efficient air-fabricated inverted CsPbI3 perovskite solar cells. ACS Energy Lett. 6, 3661–3668 (2021).
Wang, Y., Chen, Y., Zhang, T., Wang, X. & Zhao, Y. Chemically stable black phase CsPbI3 inorganic perovskites for high-efficiency photovoltaics. Adv. Mater. 32, 2001025 (2020).
Zhang, Z. et al. Efficient thermally evaporated γ-CsPbI3 perovskite solar cells. Adv. Energy Mater. 11, 2100299 (2021).
Wang, K. et al. In-situ hot oxygen cleansing and passivation for all-inorganic perovskite solar cells deposited in ambient to breakthrough 19% efficiency. Adv. Funct. Mater. 31, 2101568 (2021).
Zhao, B. et al. Thermodynamically stable orthorhombic γ-CsPbI3 thin films for high-performance photovoltaics. J. Am. Chem. Soc. 140, 11716–11725 (2018).
Tessler, N. Adding 0.2 V to the open circuit voltage of organic solar cells by enhancing the built-in potential. J. Appl. Phys. 118, 215501 (2015).
Choi, H. et al. Cesium-doped methylammonium lead iodide perovskite light absorber for hybrid solar cells. Nano Energy 7, 80–85 (2014).
Wu, T. et al. Efficient and stable CsPbI3 solar cells via regulating lattice distortion with surface organic terminal groups. Adv. Mater. 31, 1900605 (2019).
Wang, Q. et al. Stabilizing the α-phase of CsPbI3 perovskite by sulfobetaine zwitterions in one-step spin-coating films. Joule 1, 371–382 (2017).
Fu, S. et al. Tailoring in situ healing and stabilizing post-treatment agent for high-performance inverted CsPbI3 perovskite solar cells with efficiency of 16.67%. ACS Energy Lett. 5, 3314–3321 (2020).
Wang, J. et al. Highly efficient all-inorganic perovskite solar cells with suppressed non-radiative recombination by a Lewis base. Nat. Commun. 11, 177 (2020).
Zhang, T. et al. Bication lead iodide 2D perovskite component to stabilize inorganic α-CsPbI3 perovskite phase for high-efficiency solar cells. Sci. Adv. 3, e1700841 (2017).
Tan, S. et al. Temperature-reliable low-dimensional perovskites passivated black-phase CsPbI3 toward stable and efficient photovoltaics. Angew. Chem. 134, e202201300 (2022).
Le Corre, V. M., Sherkar, T. S., Koopmans, M. & Koster, L. J. A. Identification of the dominant recombination process for perovskite solar cells based on machine learning. Cell Rep. Phys. Sci. 2, 100346 (2021).
Zhao, J. et al. Strained hybrid perovskite thin films and their impact on the intrinsic stability of perovskite solar cells. Sci. Adv. 3, eaao5616 (2017).
Yang, S. et al. Stabilizing halide perovskite surfaces for solar cell operation with wide-bandgap lead oxysalts. Science 365, 473–478 (2019).
Cho, Y. et al. Immediate and temporal enhancement of power conversion efficiency in surface-passivated perovskite solar cells. ACS Appl. Mater. Interfaces 13, 39178–39185 (2021).
Moghadamzadeh, S. et al. Spontaneous enhancement of the stable power conversion efficiency in perovskite solar cells. J. Mater. Chem. A 8, 670–682 (2020).
Fassl, P. et al. Fractional deviations in precursor stoichiometry dictate the properties, performance and stability of perovskite photovoltaic devices. Energy Environ. Sci. 11, 3380–3391 (2018).
Becker-Koch, D. et al. Probing charge transfer states at organic and hybrid internal interfaces by photothermal deflection spectroscopy. J. Phys. Condens. Matter 31, 124001 (2019).
Sun, Q. et al. Role of microstructure in oxygen induced photodegradation of methylammonium lead triiodide perovskite films. Adv. Energy Mater. 7, 1700977 (2017).
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (ERC grant agreement number 714067, ENERGYMAPS, Y.V.) and the Deutsche Forschungsgemeinschaft (DFG) in the framework of the Special Priority Program (SPP 2196) project PERFECT PVs (number 424216076, Y.V.). R.J. and Z.Z. are grateful for the financial support by the China Scholarship Council (scholarship numbers 201806070145 and 201806750012, respectively). R.J. thanks Y. Liu, Y. Li, Y.Y. Zheng, X. Jia and S. Xing for useful discussions and M. Deconinck for help with the UPS depth profiling measurements. Y. V. thanks N. Tessler and A.A. Bakulin for fruitful discussions. We also thank S. Reineke for financial support to C.H.
Author information
Authors and Affiliations
Contributions
R.J. conceived the idea, designed experiments and analysed data. R.J. and Z.Z. fabricated perovskite thin films and photovoltaic devices and characterized the SEM for the perovskite thin films. R.J. synthesized the PbI2·xDMAI precursor, carried out the XRD, electroluminescence, conductivity measurements, optical modelling and photovoltaic device testing. C.H. measured n, k values for optical modelling. R.B. performed the PDS measurements and analysis. Y.J.H. performed the UPS depth profiling measurements and analysis. R.J. and F.P. performed the PL measurements and analysis. Y.V. supervised and guided the work. R.J. and Y.V. wrote the manuscript, which has been commented on and edited by all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Energy thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–16 and Tables 1 and 2.
Supplementary Data
Source data for Supplementary Fig. 8.
Supplementary Data
Source data for Supplementary Fig. 10.
Supplementary Data
Source data for Supplementary Fig. 12.
Supplementary Data
Source data for Supplementary Fig. 14.
Source data
Source Data Fig. 3
Source data for Fig. 3.
Source Data Fig. 4
Source data for Fig. 4.
Source Data Fig. 5
Source data for Fig. 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ji, R., Zhang, Z., Hofstetter, Y.J. et al. Perovskite phase heterojunction solar cells. Nat Energy 7, 1170–1179 (2022). https://doi.org/10.1038/s41560-022-01154-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41560-022-01154-y
