
Abstract
Molecular knots and entanglements form randomly and spontaneously in both biological and synthetic polymer chains. It is known that macroscopic materials, such as ropes, are substantially weakened by the presence of knots, but until now it has been unclear whether similar behaviour occurs on a molecular level. Here we show that the presence of a well-defined overhand knot in a polymer chain substantially increases the rate of scission of the polymer under tension (≥2.6× faster) in solution, because deformation of the polymer backbone induced by the tightening knot activates otherwise unreactive covalent bonds. The fragments formed upon severing of the knotted chain differ from those that arise from cleavage of a similar, but unknotted, polymer. Our solution studies provide experimental evidence that knotting can contribute to higher mechanical scission rates of polymers. It also demonstrates that entanglement design can be used to generate mechanophores that are among the most reactive described to date, providing opportunities to increase the reactivity of otherwise inert functional groups.

Similar content being viewed by others
Main
Upon tightening, knots weaken macroscopic strands until they fracture at the entrance to the entanglement1,2. This reduces the tensile strength of knotted materials from ropes used in sailing and mountaineering to fishing lines. Computer simulations3, theory4,5 and intuition5 suggest that similar weakening may occur at the nanoscale, but such processes have not previously been explored experimentally. The effects of mechanical pulling6,7,8,9 on the conformation of molecular knots10,11,12,13 have been probed by force microscopies, but the smallest-scale knot-breaking experiments to date were carried out on actin filaments 0.4–0.8 μm in diameter using optical tweezers14. In polymer mechanochemistry, polymers are used to stretch mechanosensitive molecular structures (mechanophores) at the nanoscale15. This can be achieved in solution using ultrasound-induced cavitation because of the elongational flow generated in the vicinity of collapsing bubbles16. This technique has been used to investigate the effect of interlocking components on the mechanical strength of covalent bonds17 in catenanes18,19 and rotaxanes20,21,22,23 (which respectively reduce or enhance tensional stress). Recent advances in molecular knot synthesis24,25,26,27,28,29,30,31,32,33,34 mean that small-molecule knots are now accessible structural motifs that can be integrated into more complex molecular systems35,36,37.
We decided to incorporate a knotted molecular building block into a polymer to investigate the effect of a structurally well-defined overhand (trefoil, 31) knot, the simplest and most abundant knot formed spontaneously in linear polymer chains13,38, on the rate of scission of a polymer chain under tension (Fig. 1a), and to determine which bonds were broken. The study was carried out using sonication on polymers in solution, but the order of scission rates of functional groups and other structural elements in solution mechanochemistry generally correlates with the order of their breaking rates in the condensed phase39.

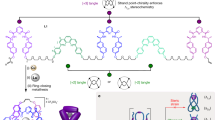
a, Cavitation-induced elongational flow is used to stretch a gated overhand knot (a trefoil knot) derivatized with actuating polymer chains. b, Scission of the Diels–Alder gate in trefoil knot 1 reveals a transient overhand knot that contracts and eventually breaks upon continuous elongation. Red arrows indicate the direction of the force. Plain and dashed reaction arrows indicate covalent and non-covalent processes, respectively. Potential scissile bonds of the knot are shown in red.
The central region of a polymer experiences the largest forces during sonication17, making it the most suitable location to incorporate a knot for these experiments. However, open knots, such as the overhand trefoil knot, are dynamic structures that can expand and contract or translocate along the polymer backbone13,40,41. To avoid unknotting occurring before the polymer chain was under tension, we gated42 the overhand knot with a mechanically labile unit (a mechanophore built around a furan/maleimide Diels–Alder adduct) that would maintain the integrity of the knotted architecture in the form of a closed-loop trefoil knot (Fig. 1)43. Upon stretching, tension builds up along the polymer backbone, ultimately reaching the knot. Because the gate mechanophore is situated along the shortest path connecting the two sides of the polymer, it will be activated before covalent bonds in other regions of the knot19. Dissociation of the gate converts the closed-loop knot into an open overhand knot and enables the polymer to further stretch and tighten the knot until the eventual rupture of a covalent bond occurs, breaking the polymer chain (Fig. 1a). This outcome can only be realized if this sequence takes place during the same elongation event (that is, the overhand knot does not have time to unravel).
Results and discussion
The gated overhand knot (the closed-loop trefoil knot) was assembled from a lanthanide-complexed overhand knot26, a versatile motif that has previously been used for the assembly of complex knots31,34 and investigation of their properties44,45. The overhand knot was macrocyclized to form the trefoil knot by connection to the gate through successive Cu-catalysed azide–alkyne cycloaddition and ring-closing olefin metathesis reactions (Supplementary Section 3.4). The chain-centred knot (1, Mn (number average molecular weight) = 62 kDa, Đ (dispersity) = 1.27) was obtained by single electron transfer living radical polymerization46 of methyl acrylate initiated from both sides of the gating unit (Fig. 1b).
Sonication kinetics
To assess the effect of the knot on the mechanical strength of a polymer chain, we compared the rate of dissociation upon mechanical activation of chain-centred knot 1, gate unit 2 (Mn = 71 kDa, Đ = 1.23) and (unknotted) linear ligand 3 (Mn = 67 kDa, Đ = 1.13) (Fig. 2a–c). Mechanical activation was performed in acetonitrile at 5–10 °C, using high-intensity ultrasound (20 kHz, 11.5 W cm−2, 1 s on/2 s off, 180 min). The progress of the reaction was monitored by gel-permeation chromatography (Fig. 2b) and the conversion plotted using the Nalepa method47 (Fig. 2f). The apparent dissociation rate (k*) is obtained from the slope of the traces in Fig. 2f. Comparison of the reaction rates (k* = 8.1 ± 0.7, 7.7 ± 0.5 and 3.0 ± 0.1 min−1 kDa−1 105 for 1, 2 and 3, respectively) show that knot 1 and gate 2 cleave at similar rates, whereas the dissociation of the polymer containing the linear ligand (3) is substantially slower. These results show that the structure of the linear ligand used to assemble the knot is mechanically stronger than gate 2. Indeed, 2 can readily cleave via a retro-cycloaddition pathway to restore the furan and maleimide rings composing the initial Diels–Alder adduct, whereas the ligand segment in 3 does not contain any obviously weak bonds and cleaves in the poly(methyl acrylate) backbone (Supplementary Section 4.3). We found that once embedded in knot 1, the same segment cleaves substantially faster. In other words, the knot architecture accelerates the dissociation of this segment, and the cleavage of the gating mechanophore is the rate-determining step of the dissociation process. This also indicates that in knot 1, both the gate and the resulting overhand knot must cleave in the same elongation event because the intermediate overhand knot (Fig. 1) would unravel under force-free conditions and the resulting linear ligand would then cleave at a slower rate.

a, Mechanical activation of chain-centred knot 1 and isolation of the resulting fragments. Conditions: (i) ultrasound (20 kHz, 11.5 W cm−2, 1 s on/2 s off), CH3CN, 5–10 °C, 180 min; (ii) NaOH. b, The overlay of gel-permeation chromatography traces at various sonication times of chain-centred knot 1 (tetrahydrofuran, 1 ml min−1) is consistent with a rupture in the central region of the polymer. c, Partial 1H NMR (500 MHz, CDCl3) spectra of knot 1 before (i) and after (ii) sonication, along with a reference compound (iii), indicate opening of the gate adduct through mechanical activation. d, Mass spectrometry (electrospray ionisation high-resolution mass spectrometry, negative ion mode) identification of fragments 4, 5 and 6 in the hydrolysed post-sonication mixture. e, Structure of chain-centred gate (2) and linear ligand (3). f, Dissociation kinetics of chain-centred knot (1), gate (2) and linear ligand (3). Mt = Mn at time t, M0 = Mn at 0 min. Solid lines correspond to a linear fit; R2 (goodness of fit) = 0.963, 0.976 and 0.988 for 1, 2 and 3, respectively. Each point corresponds to the average of three sonication experiments. Data are presented as means ± s.e.m. Coloured areas indicate 95% confidence levels.
Fragments analysis
Because the knotted architecture of 1 clearly enhances the mechanochemical reactivity of its constituent covalent bonds (the unknotted polymer, 3, is much more slowly cleaved), we then sought to determine the scission point(s) to establish the origin of the enhanced reactivity. The scission of the Diels–Alder gate was confirmed by 1H NMR spectroscopy (Fig. 2c), but the structural complexity of the various components (knot, polymer, mechanophore, fragments and so on) prevented further insights from NMR. Hence, we attempted to isolate the fragments of the broken knot, because hydrolysis of the post-sonication mixture should lead to removal of the polymer arms and gate elements. The post-sonication mixture was hydrolysed with NaOH and then adjusted to pH 3 and the resulting solution extracted with CH2Cl2. Three major species (4, 5 and 6) (Fig. 2a) were identified by high-resolution mass spectrometry from the CH2Cl2 fraction (Fig. 2d). Their structures are indicative of mechanical scission of the C–O bond adjacent to one of the naphthyl groups (Fig. 1b), resulting in naphthol (5) and ethylene glycol (4, 6) fragments (Fig. 2a). We were able to isolate fragments 4 and 6 and confirm their identity by 1H NMR spectroscopy and high-resolution mass spectrometry (Supplementary Section 4.9). The fact that only the naphthyl ethers connected to the ethylene glycol linkers are observed to cleave suggests that the knot contracts mainly around the central pyridyl unit (Fig. 1b). We did not isolate or detect any scission products that would occur from migration of the knot along the strand while it is under tension during the elongation event, although we cannot rule out that occurring to a minor extent.
Calculations
Constrained geometries simulate external force (CoGEF)48 calculations gave further insight into the scission process. The extension of the knot was first simulated by molecular mechanics (Merck molecular force field (MMFF)) on a model knot lacking the gate unit (Supplementary Section 9). The actual scission of the knot was simulated on a shorter model extracted from a contracted intermediate of the MMFF profile. More specifically, the intermediate stretched by 40 Å was isolated and excised of the unknotted sections of the ligand (caused by contraction of the knot; Supplementary Section 9). The elongation of this shorter model was then simulated by density functional theory (UB3LYP/6-31G*, gas phase) until bond scission occurred (Fig. 3a). Although the CoGEF method does not allow for a detailed mechanistic interpretation of the dissociation process (because it does not account for dynamic or thermal effects), it has proved successful in mapping19,21,22 conformation and bond deformation, as well as in identifying the scissile bond(s) in mechanophores49. The CoGEF profile displays a steady increase in energy from the starting structure (Fig. 3a(i)) to the state of maximal deformation (Fig. 3a(ii)), which is followed by a heterolytic bond scission (Fig. 3d(iii)). As the central cavity of the knot contracts, the mobility of the ethylene glycol linkers becomes increasingly restricted, and deformation of the backbone increases. Notably, the C–O–C angle (denoted as α in Fig. 3a,c) containing the scissile bond is distorted to ~140° (from ~109° in the tension-free knot) and the naphthyl groups (denoted as β in Fig. 3a,c) are bent to ~145° (from ~180° in the tension-free knot) in the state of maximal deformation (Fig. 3a(ii),d(ii)). Ultimately, scission occurs at the C–O bond (denoted as a in Fig. 3a,b) of the external naphthyl group.

a, CoGEF simulation (UB3LYP/6-31G*, gas phase) on a model knot; anchor atoms are shown in red. b, Tensile deformation of scissile (labelled a) and non-scissile (labelled b and c) C–Onapht bonds. c, Angular deformation around the scissile bond (α) and a naphthyl group (β). d, CoGEF structures at onset (i), maximal deformation (ii) and after scission (iii) correspond to the states highlighted in a.
The models suggest a heterolytic scission in which the developing positive and negative charges, on the ethylene glycol carbon and naphthol oxygen, respectively, are stabilized by the pyridine lone pair and the amide NH (Fig. 3d(ii,iii)). The scission occurs where the strand exits the loop formed by the knot (Fig. 3d(ii)). This position is similar to that predicted computationally in previous studies3,4. The knot topology dramatically reduces the force required to break the otherwise unreactive covalent bond. The calculated force at maximum deformation (Fmax) is reduced from ≥5.6 nN for models of the linear ligand (Supplementary Sections 9.3 and 9.4) to 2.9 nN for the same ligand in a knotted topology, where the cleaved bond differs from the linear analogue (suggesting an even larger difference in reactivity between the scissile bond observed in the knot and the same bond in the linear ligand). In fact, the looped arrangement of the knot causes the activation of an otherwise unreactive covalent bond because of the force-induced bending and stretching deformations (see above)17. Remarkably, as a mechanophore the knot (Fmax = 2.9 nN) is even more reactive than the Diels–Alder mechanophore used in the gate (Fmax = 3.7 nN) and ranks among the most reactive scissile mechanophores described to date49.
Taken together, these results provide a picture of the overall process. As the chain-centred knot enters the flow field surrounding a collapsing cavitation bubble, the chain is stretched until the gating unit opens. This results in the initial closed-loop trefoil knot opening into an overhand knot. Unlike the closed-loop trefoil knot, the overhand knot is potentially able to expand and diffuse along the polymer chain50. However, the kinetic data indicate that opening of the gate and the knot scission occur in the same elongation event. In other words, the overhand knot formed after the gate opening does not have time to relax and rearrange in the high-strain environment of the cavitation field. Indeed, a cavitation bubble collapses much faster (microsecond timescale51) than the relaxation time of a knot (which, for example, occurs on the seconds timescale for DNA41). Moreover, knot diffusion along a strand is suppressed at relatively low tension52. Other dynamic ‘memory effects’ have previously been observed with cyclic polymers53. The implication for the current system is that the overhand knot is likely to contract around the central region, although not necessarily symmetrically, of the knotted strand (Fig. 1), a picture supported by the CoGEF simulations (Fig. 3). As the knot enters the final stage of its contraction, a large amount of backbone deformation (in the form of bond bending and stretching) is observed, conspicuously, via the substantial bending of the flanking naphthyl groups as well as at the naphthyl/ethylene glycol junction where the strand exits the knot cavity. Ultimately, this leads to the heterolytic scission of a C–O bond in the latter section, where the developing charges are stabilized by hydrogen bonding of the amide and the pyridine lone pair. This nature of the scissile bond was confirmed experimentally by isolation of the knot fragments post-sonication.
Conclusions
Matter often behaves very differently at different length scales. For example, the friction and inertia that both maintain and localize knots in macroscopic strands do not do so at the nanoscale. However, we find that the stress distribution and scission point in a molecular overhand knot under stretching are very similar to that observed for knotted fishing lines and cooked spaghetti2; in other words, overhand knots induce closely related modes of strand weakening across molecular (nm), microscopic (μm)14 and macroscopic (mm)1,2 scales. The effect that knotting has on the mechanical strength of covalent bonds in a polymer chain is dramatic, reducing the scission force from ≥5.6 nN to 2.9 nN, which results in a scission rate at least 2.6× higher than an unknotted counterpart, producing one of the most reactive scissile mechanophores known. Knot activation is so effective that scission of the knotted polymer chain involves a different set of chemical bonds from those that break in an unknotted polymer under mechanical stress.
Knots are found extensively in biomacromolecules10 and form spontaneously13,38,54,55,56,57,58 in many synthetic polymers. However, the probability of finding a randomly formed knot is only significant at molecular masses >1 MDa (refs. 13,38). Many of the polymers that have previously been investigated by sonication are <200 kDa and so have a low frequency of being knotted. Our results provide experimental evidence that knotting may be intrinsically detrimental to the mechanical strength of high molecular mass and other knotted polymers. This study was carried out on polymers in solution, but we note that the order of scission rates of structural elements in solution mechanochemistry generally correlates with the order of their breaking rates in the condensed phase39.
Methods
Detailed methods and protocols are given in the Supplementary Information.
Mechanical activation
The polymer (15 mg) was dissolved in CH3CN (15 ml) and added to a modified Suslick cell. The solution was sonicated using a Sonics VCX 500 ultrasonic processor equipped with a 13-mm-diameter solid probe or replaceable-tip probe (20 KHz, 11.5 W cm−2, 1 s on/2 s off, 5–10 °C). Nitrogen was gently bubbled through the solution as it was sonicated. After 180 min of sonication time, the mixture was concentrated and dried under high vacuum for an extended period (~24 h); the polymer was washed with methanol (5 ml) before drying again.
CoGEF calculation
CoGEF calculations were performed on Spartan 14/20 following Beyer’s method48. The knot was constructed in Spartan 14 and minimized using molecular mechanics (MMFF). The distance between the anchor groups was constrained and increased in increments of 0.5 Å. At each step, the energy was minimized by molecular mechanics (MMFF) with Spartan 14 and then density functional theory (UB3LYP/6-31G*, gas phase) with Spartan 20. The relative energy of each intermediate was determined by setting the energy of the unknotted state to 0 kJ mol−1. The Fmax value was determined from the slope of the final 40% of the energy/elongation curve (that is, from 0.6Emax to Emax, where Emax is the maximum relative energy immediately before bond rupture).
Data availability
The data that support the finding of this study are available within the paper and its Supplementary Information or are available from the figshare data repository: https://doi.org/10.6084/m9.figshare.21548319 (ref. 59). Source data are provided with this paper.
References
Ashley, C. W. The Ashley Book of Knots (Doubleday, 1994).
Pieranski, P., Kasas, S., Dietler, G., Dubochet, J. & Stasiak, A. Localization of breakage points in knotted strings. New J. Phys. 3, 10 (2001).
Saitta, A. M., Soper, P. D., Wasserman, E. & Klein, M. L. Influence of a knot on the strength of a polymer strand. Nature 399, 46–48 (1999).
Stauch, T. & Dreuw, A. Knots ‘choke off’ polymers upon stretching. Angew. Chem. Int. Ed. 55, 811–814 (2016).
Beyer, M. K. & Clausen-Schaumann, H. Mechanochemistry: the mechanical activation of covalent bonds. Chem. Rev. 105, 2921–2948 (2005).
Bornschlogl, T. et al. Tightening the knot in phytochrome by single-molecule atomic force microscopy. Biophys. J. 96, 1508–1514 (2009).
Ziegler, F. et al. Knotting and unknotting of a protein in single molecule experiments. Proc. Natl Acad. Sci. USA 113, 7533–7538 (2016).
Wang, H. & Li, H. Mechanically tightening, untying and retying a protein trefoil knot by single-molecule force spectroscopy. Chem. Sci. 11, 12512–12521 (2020).
Calvaresi, M. et al. Mechanical tightening of a synthetic molecular knot by atomic force microscopy. Chem 9, 65–75 (2023).
Lim, N. C. H. & Jackson, S. E. Molecular knots in biology and chemistry. J. Phys. Condens. Matter 27, 354101 (2015).
Fielden, S. D. P., Leigh, D. A. & Woltering, S. L. Molecular knots. Angew. Chem. Int. Ed. 56, 11166–11194 (2017).
Ashbridge, Z. et al. Knotting matters: orderly molecular entanglements. Chem. Soc. Rev. 51, 7779–7809 (2022).
Tubiana, L., Rosa, A., Fragiacomo, F. & Micheletti, C. Spontaneous knotting and unknotting of flexible linear polymers: equilibrium and kinetic aspects. Macromolecules 46, 3669–3678 (2013).
Arai, Y. et al. Tying a molecular knot with optical tweezers. Nature 399, 446–448 (1999).
O’Neill, R. T. & Boulatov, R. The many flavours of mechanochemistry and its plausible conceptual underpinnings. Nat. Rev. Chem. 5, 148–167 (2021).
May, P. A. & Moore, J. S. Polymer mechanochemistry: techniques to generate molecular force via elongational flows. Chem. Soc. Rev. 42, 7497–7506 (2013).
De Bo, G. Mechanochemistry of the mechanical bond. Chem. Sci. 9, 15–21 (2018).
Lee, B., Niu, Z. & Craig, S. L. The mechanical strength of a mechanical bond: sonochemical polymer mechanochemistry of poly(catenane) copolymers. Angew. Chem. Int. Ed. 55, 13086–13089 (2016).
Zhang, M. & De Bo, G. A catenane as a mechanical protecting group. J. Am. Chem. Soc. 142, 5029–5033 (2020).
Stoll, R. S., Friedman, D. C. & Stoddart, J. F. Mechanically interlocked mechanophores by living-radical polymerization from rotaxane initiators. Org. Lett. 13, 2706–2709 (2011).
Zhang, M. & De Bo, G. Impact of a mechanical bond on the activation of a mechanophore. J. Am. Chem. Soc. 140, 12724–12727 (2018).
Zhang, M. & De Bo, G. Mechanical susceptibility of a rotaxane. J. Am. Chem. Soc. 141, 15879–15883 (2019).
Muramatsu, T. et al. Rotaxane-based dual function mechanophores exhibiting reversible and irreversible responses. J. Am. Chem. Soc. 143, 9884–9892 (2021).
Prakasam, T. et al. Simultaneous self-assembly of a [2]catenane, a trefoil knot, and a Solomon link from a simple pair of ligands. Angew. Chem. Int. Ed. 52, 9956–9960 (2013).
Ayme, J.-F. et al. Lanthanide template synthesis of a molecular trefoil knot. J. Am. Chem. Soc. 136, 13142–13145 (2014).
Gil-Ramírez, G. et al. Tying a molecular overhand knot of single handedness and asymmetric catalysis with the corresponding pseudo-D3-symmetric trefoil knot. J. Am. Chem. Soc. 138, 13159–13162 (2016).
Cougnon, F. B. L., Caprice, K., Pupier, M., Bauzá, A. & Frontera, A. A strategy to synthesize molecular knots and links using the hydrophobic effect. J. Am. Chem. Soc. 140, 12442–12450 (2018).
Segawa, Y. et al. Topological molecular nanocarbons: all-benzene catenane and trefoil knot. Science 365, 272–276 (2019).
Zhang, H.-N., Gao, W.-X., Lin, Y.-J. & Jin, G.-X. Reversible structural transformation between a molecular Solomon link and an unusual unsymmetrical trefoil knot. J. Am. Chem. Soc. 141, 16057–16063 (2019).
Inomata, Y., Sawada, T. & Fujita, M. Metal–peptide torus knots from flexible short peptides. Chem 6, 294–303 (2020).
Leigh, D. A. et al. Tying different knots in a molecular strand. Nature 584, 562–568 (2020).
Carpenter, J. P. et al. Controlling the shape and chirality of an eight-crossing molecular knot. Chem 7, 1534–1543 (2021).
Leigh, D. A. et al. A molecular endless (74) knot. Nat. Chem. 13, 117–122 (2021).
Ashbridge, Z. et al. Vernier template synthesis of molecular knots. Science 375, 1035–1041 (2022).
Marcos, V. et al. Allosteric initiation and regulation of catalysis with a molecular knot. Science 352, 1555–1559 (2016).
Leigh, D. A., Pirvu, L., Schaufelberger, F., Tetlow, D. J. & Zhang, L. Securing a supramolecular architecture by tying a stopper knot. Angew. Chem. Int. Ed. 57, 10484–10488 (2018).
August, D. P. et al. Self-assembly of a layered two-dimensional molecularly woven fabric. Nature 588, 429–435 (2020).
Virnau, P., Kantor, Y. & Kardar, M. Knots in globule and coil phases of a model polyethylene. J. Am. Chem. Soc. 127, 15102–15106 (2005).
Chen, Y., Mellot, G., van Luijk, D., Creton, C. & Sijbesma, R. P. Mechanochemical tools for polymer materials. Chem. Soc. Rev. 50, 4100–4140 (2021).
Plesa, C. et al. Direct observation of DNA knots using a solid-state nanopore. Nat. Nanotechnol. 11, 1093–1097 (2016).
Klotz, A. R., Soh, B. W. & Doyle, P. S. Motion of knots in DNA stretched by elongational fields. Phys. Rev. Lett. 120, 188003 (2018).
Wang, J., Kouznetsova, T. B., Boulatov, R. & Craig, S. L. Mechanical gating of a mechanochemical reaction cascade. Nat. Commun. 7, 13433 (2016).
Stevenson, R. & De Bo, G. Controlling reactivity by geometry in retro-Diels–Alder reactions under tension. J. Am. Chem. Soc. 139, 16768–16771 (2017).
Katsonis, N. et al. Knotting a molecular strand can invert macroscopic effects of chirality. Nat. Chem. 12, 939–944 (2020).
August, D. P. et al. Transmembrane ion channels formed by a Star of David [2]catenane and a molecular pentafoil knot. J. Am. Chem. Soc. 142, 18859–18865 (2020).
Anastasaki, A. et al. Cu(0)-mediated living radical polymerization: a versatile tool for materials synthesis. Chem. Rev. 116, 835–877 (2016).
Sato, T. & Nalepa, D. E. Shear degradation of cellulose derivatives. J. Appl. Polym. Sci. 22, 865–867 (1978).
Beyer, M. The mechanical strength of a covalent bond calculated by density functional theory. J. Chem. Phys. 112, 7307–7312 (2000).
Klein, I. M., Husic, C. C., Kovács, D. P., Choquette, N. J. & Robb, M. J. Validation of the CoGEF method as a predictive tool for polymer mechanochemistry. J. Am. Chem. Soc. 142, 16364–16381 (2020).
Bao, X. R., Lee, H. J. & Quake, S. R. Behavior of complex knots in single DNA molecules. Phys. Rev. Lett. 91, 265506 (2003).
Lauterborn, W. & Kurz, T. Physics of bubble oscillations. Rep. Prog. Phys. 73, 106501 (2010).
Narsimhan, V., Renner, C. B. & Doyle, P. S. Jamming of knots along a tensioned chain. ACS Macro Lett. 5, 123–127 (2016).
Lin, Y., Zhang, Y., Wang, Z. & Craig, S. L. Dynamic memory effects in the mechanochemistry of cyclic polymers. J. Am. Chem. Soc. 141, 10943–10947 (2019).
Bayer, R. K. Structure transfer from a polymeric melt to the solid state. Part III: influence of knots on structure and mechanical properties of semicrystalline polymers. Colloid Polym. Sci. 272, 910–932 (1994).
Foteinopoulou, K., Karayiannis, N. C., Laso, M., Kroeger, M. & Mansfield, M. L. Universal scaling, entanglements, and knots of model chain molecules. Phys. Rev. Lett. 101, 265702 (2008).
Laso, M., Karayiannis, N. C., Foteinopoulou, K., Mansfield, M. L. & Kroeger, M. Random packing of model polymers: local structure, topological hindrance and universal scaling. Soft Matter 5, 1762–1770 (2009).
Meyer, H., Horwath, E. & Virnau, P. Mapping onto ideal chains overestimates self-entanglements in polymer melts. ACS Macro Lett. 7, 757–761 (2018).
Zhang, J., Meyer, H., Virnau, P. & Daoulas, K. C. Can soft models describe polymer knots? Macromolecules 53, 10475–10486 (2020).
Zhang, M., Nixon, R., Schaufelberger, F., Pirvu, L., De Bo, G. & Leigh, D. A. Mechanical scission of a knotted polymer. figshare https://doi.org/10.6084/m9.figshare.21548319 (2024).
Acknowledgements
We thank the University of Manchester Mass Spectrometry Service Centre for high-resolution mass spectrometry, East China Normal University, the Engineering and Physical Sciences Research Council (EP/P027067/1) and the European Union (European Research Council, Advanced Grant no. 786630) for funding and networking contributions from the COST Action CA17139, European Topology Interdisciplinary Action. G.D.B. is a Royal Society University Research Fellow. D.A.L. is a Royal Society Research Professor. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
M.Z., R.N., F.S. and L.P. planned and carried out the synthetic work. G.D.B. performed the computational investigation. D.A.L. and G.D.B. directed the research. All authors contributed to the analysis of the results and the writing of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information
Supplementary Figs. 1–9, experimental procedures and characterization data, Schemes 1–6, Tables 1 and 2, and Spectra 1–75.
Source data
Source Data Fig. 2
Dissociation kinetics over time.
Source Data Fig. 3
CoGEF simulation over time.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, M., Nixon, R., Schaufelberger, F. et al. Mechanical scission of a knotted polymer. Nat. Chem. (2024). https://doi.org/10.1038/s41557-024-01510-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41557-024-01510-3
