Abstract
Despite their intriguing photophysical and photochemical activities, naturally occurring photoenzymes have not yet been repurposed for new-to-nature activities. Here we engineered fatty acid photodecarboxylases to catalyse unnatural photoredox radical C–C bond formation by leveraging the strongly oxidizing excited-state flavoquinone cofactor. Through genome mining, rational engineering and directed evolution, we developed a panel of radical photocyclases to facilitate decarboxylative radical cyclization with excellent chemo-, enantio- and diastereoselectivities. Our high-throughput experimental workflow allowed for the directed evolution of fatty acid photodecarboxylases. An orthogonal set of radical photocyclases was engineered to access all four possible stereoisomers of the stereochemical dyad, affording fully diastereo- and enantiodivergent biotransformations in asymmetric radical biocatalysis. Molecular dynamics simulations show that our evolved radical photocyclases allow near-attack conformations to be easily accessed, enabling chemoselective radical cyclization. The development of stereoselective radical photocyclases provides unnatural C–C-bond-forming activities in natural photoenzyme families, which can be used to tame the stereochemistry of free-radical-mediated reactions.

Similar content being viewed by others


Main
Naturally occurring photoenzymes represent an intriguing enzyme family requiring a steady influx of light for catalysis1,2. Although natural evolution has largely opted against photoenzymatic catalysis, the high-energy intermediates confined in photoenzymes provide fertile ground for elucidating unusual photophysics and photochemistry as well as discovering novel photoenzymatic reactions through excited-state biocatalysis. Compared to previously investigated natural photoenzymes including protochlorophyllide oxidoreductases3,4,5,6,7,8 and DNA photolyases9,10,11, the recent groundbreaking discovery and mechanistic elucidation of algae-derived fatty acid photodecarboxylases (FAPs)12,13,14,15 underscored their synthetic potential, as these flavin adenine dinucleotide (FAD)-dependent photoenzymes allow the efficient photodecarboxylation of abundant and/or easily accessible carboxylic acids via a radical mechanism. Important previous work on FAPs has focused on expanding the substrate scope of their native protodecarboxylation activity16,17,18,19,20,21,22,23. However, to date, new-to-nature activities of flavin-dependent FAPs have remained elusive.
The past decade has witnessed the advent and rapid development of stereoselective new-to-nature radical biocatalysis. Due to the challenge of imposing stereocontrol over free-radical intermediates, achieving catalytic asymmetric radical transformations has long remained a daunting task facing synthetic organic chemists. By repurposing and engineering natural enzymes to catalyse unnatural reactions involving an open-shell mechanism, radical biocatalysis provides a promising strategy to tame fleeting radical species for asymmetric catalysis. In this emerging field, our group24,25,26 and the Huang group27 introduced the concept of metalloredox radical biocatalysis to perform atom transfer chemistry in a stereoselective fashion using evolved metalloenzymes. Very recently, we advanced synergistic photoredox–pyridoxal radical biocatalysis to convert pyridoxal-5′-phosphate-dependent enzymes into radical enzymes for the stereoselective synthesis of non-canonical amino acids28. Notably, the pioneering work of Hyster29,30,31,32,33,34,35,36,37,38,39 and Zhao40,41,42 converted ketoreductases and ene reductases into unnatural photoenzymes, enabling reductive radical coupling using reduced flavin or nicotinamide. Nevertheless, in previously described photobiocatalysis based on flavoenzymes, the use of the strongly oxidizing excited-state flavoquinone cofactor for single electron oxidation-triggered radical C–C bond formation has remained unexplored.
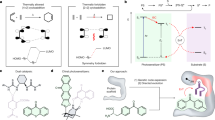
We reasoned that if natural FAPs could be engineered to permit the interception of the nascent carbon-centred radical with a pendant C=C double bond, we would be able to develop new-to-nature radical photocyclases (RAPs) to promote stereocontrolled radical cyclization reactions (Fig. 1). In these processes, easily accessible carboxylic acids are used as the radical precursor, allowing overall redox-neutral radical cyclization reactions to be developed, thereby eliminating the need for stoichiometric reductants. At the commencement of our envisioned catalytic cycle (Fig. 1a), visible light irradiation produces a photoexcited strongly oxidizing flavoquinone FADq*. Single electron transfer from the carboxylate substrate to FADq* followed by rapid decarboxylation would generate an alkyl radical and a flavosemiquinone FAD•− (FADsq). At this stage, this nucleophilic alkyl radical species would undergo addition to a pendent α,β-unsaturated carbonyl to furnish a new carbon-centred radical. Subsequent electron-transfer/proton-transfer (ET/PT) or proton-coupled electron transfer (PCET) involving the flavosemiquinone FAD•− and conserved amino acid residue(s)12,13 would furnish the product in a stereocontrolled manner and regenerate the FAD cofactor, thus completing the catalytic cycle.

a, Proposed catalytic cycle of RAPs. hν denotes light energy. b, RAP-catalysed chemo- and enantioselective decarboxylative radical cyclization. c, RAP-catalysed diastereo- and enantiodivergent decarboxylative radical cyclization giving rise to all four possible stereoisomers of a stereochemical dyad.
Nevertheless, achieving this new-to-nature biocatalytic cycle requires several challenges to be overcome. First, the interception of the photoenzymatically formed carbon-centred radical with a pendant C=C double bond moiety needs to outcompete the native PCET process as the native PCET process leads to undesired protodecarboxylation products. Accomplishing this chemoselective radical C–C bond formation is challenging, as FAPs were naturally evolved to facilitate protodecarboxylation. Second, the development of stereoselective RAPs requires both the C–C-bond-forming radical addition step and the proton transfer step to occur with excellent stereocontrol. Such FAP engineering is a non-trivial task, as directed evolution of FAPs has hitherto remained difficult, largely due to the limited stability of natural FAPs posing significant challenges to high-throughput experimentation. Furthermore, despite the elegant prior biochemical and biophysical studies on the native FAP catalysis, the proton donor of the PCET step remains to be further elucidated. While Scrutton and Heyes proposed that residue C432 of Chlorella variabilis FAP (CvFAP) serves as the hydrogen atom donor43, a study from Beisson and coworkers indicated the critical role of R451 and associated water molecules13. Thus, it remains unclear whether it is feasible to engineer FAPs to allow for stereocontrolled proton transfer. In this study, we describe our efforts combining enzyme mining, rational enzyme engineering and directed evolution to develop a collection of stereoselective RAP variants. This study constitutes the successful application of directed evolution to FAP engineering, culminating in a panel of fully enantio- and diastereodivergent radical C–C-bond-forming enzymes.
Results and discussion
Discovery of RAP activity
We focused our initial effort on the enantioselective decarboxylative radical cyclization of α,β-unsaturated ester–carboxylic acid substrate 1a (Fig. 2), as the targeted 5-exo-trig radical cyclization is kinetically facile, in accordance with Baldwin’s rules44,45,46. In addition to CvFAP, extensively studied in the native protodecarboxylation photoenzymatic reaction, we also built an in-house library of FAP homologues for the identification and development of effective stereoselective RAPs. On the basis of our BLASTP search in the non-redundant protein sequence database, using CvFAP as the template, putative FAPs from the glucose–methanol–choline oxidoreductase superfamily possessing functionally essential conserved cysteine and arginine residues corresponding to C432 and R451 of CvFAP12,13 were selected to provide an initial FAP homologue library. Our mined putative FAPs showed an identity ranging from 45% to 73% relative to CvFAP, thus constituting a structurally diverse initial screening library for the development of unnatural decarboxylative photoenzymatic reactions. The phylogenetic tree of a subset of our selected FAP homologues is delineated in Fig. 2a.

a, Phylogenetic analysis of FAP homologues used in this study. Cr, Chlamydomonas reinhardtii; Csc, Chlamydomonas schloesseri, Gp, Gonium pectorale; Vc, Volvox carteri f. nagariensis; Rs, Raphidocelis subcapitata; Cv, Chlorella variabilis; Mc, Micractinium conductrix; Csu, Coccomyxa subellipsoidea strain C-169; Es, Ectocarpus siliculosus; Ccr, Chondrus crispus; Cm, Cyanidioschyzon merolae; Ng, Nannochloropsis gaditana; Ct, Chrysochromulina tobinii. b, Evaluation of FAP homologues and their ‘Y466A’ single mutants for the decarboxylative radical cyclization of 1a using cell-free lysates. DMSO, dimethyl sulfoxide; r.t., room temperature. c, Directed evolution of CvFAP as chemo- and stereoselective RAPs CvRAP1 (CvFAPAAGAA: Y466A/G462A/S574G/S464A/T131A) and CvRAP2 (CvFAPAAAAA: Y466A/G462A/S574A/S464A/T131A). Results with purified enzymes are shown. Active site illustration was made on the basis of the crystal structure of wild-type (WT) CvFAP (Protein Data Bank (PDB) no. 6YRU).
At the outset of this study, we hypothesized that if we could direct substrate 1a towards the near-attack conformation of radical cyclization using the protein scaffold, we would be able to effectively suppress the undesired premature PCET termination of the carbon-centred radical formed from photoenzymatic decarboxylation with photoexcited FADq*. However, wild-type FAPs are naturally evolved to accommodate hydrophobic long-chain fatty acids in an extended linear conformation. In the crystal structure of wild-type CvFAP in complex with the fatty acid substrate (Fig. 2b), the conformationally flexible fatty acid substrate is forced to adopt a slightly kinked but largely extended binding mode in the hydrophobic tunnel of CvFAP. In this binding conformation, the carboxylate radical precursor and the α,β-unsaturated radical acceptor are distal to each other, disfavouring our proposed radical cyclization. To overcome this problem, we posited that if we could further expand the substrate binding pocket by replacing bulky aromatic residues with smaller residues, the carboxylate–olefin substrate would be allowed to freely sample possible conformations, thereby promoting the desired radical cyclization. Upon examining the crystal structure of wild-type CvFAP, the tyrosine residue at 466 (Y466) was found to have a critical role in shaping the substrate tunnel (Fig. 2b). Replacing this tyrosine with an alanine would significantly broaden the active site of CvFAP, thereby facilitating radical cyclization. We thus performed site-directed mutagenesis to generate CvFAP(Y466A) and analogous tyrosine-to-alanine mutants of all our FAP homologues and tested their activity towards photobiocatalytic radical cyclization of 1a (Fig. 2a). Initial photoenzyme activity evaluation was performed using the cell-free lysates of FAP variants.
It was found that CvFAP was capable of promoting radical cyclization with low levels of initial activity and modest enantioselectivity (0.5% yield and 42:58 enantiomeric ratio (e.r.) of 2a; Fig. 2b, entry 1). Several other FAPs also catalysed this radical cyclization. Among these wild-type FAPs, Raphidocelis subcapitata FAP (RsFAP) showed an enantio-preference for (S)-2a over (R)-2a (Fig. 2b, entry 5), which is complementary to CvFAP and highlights the value of the genome mining of FAPs. Furthermore, consistent with our hypothesis, the introduction of the Y466A mutation into CvFAP furnished a threefold improvement in total activity (1.5% yield) and chemoselectivity (Fig. 2b, entry 6). Interestingly, CvFAP(Y466A) also displayed a reversed enantio-preference (67:33 e.r. in favour of (S)-2a), allowing the opposite enantiomer to form upon radical cyclization. Importantly, the incorporation of this tyrosine-to-alanine mutation to other FAP homologues was found to be a universal activating mechanism to convert FAPs into RAPs (Fig. 2b, entries 6–10). For example, while wild-type Chlamydomonas reinhardtii FAP (CrFAP)12, Coccomyxa subellipsoidea FAP (CsuFAP)23 and Gonium pectorale FAP (GpFAP)23 were unable to facilitate the radical cyclization to provide 2a, CrFAP(Y427A), CsuFAP(Y431A) and GpFAP(Y428A) furnished 2a in 0.1% yield, 39:61 e.r. (Fig. 2b, entry 7); 0.1% yield, 69:31 e.r. (Fig. 2b, entry 8); and 0.1% yield, 34:66 e.r. (Fig. 2b, entry 9), respectively. Among these single mutants, CrFAP(Y427A) and GpFAP(Y428A) displayed opposite enantio-preferences compared to the others (Fig. 2b, entry 9). The highest enantioselectivity in our initial screening was observed with the previously uncharacterized RsFAP Y431A, which afforded 2a in 0.8% yield and 94:6 e.r., further showcasing the utility of enzyme variants derived from newly mined FAPs. Together, among these FAP analogues, the tyrosine-to-alanine mutation led to at least threefold enhancement in total activity, underscoring its universal activating effect. Finally, control experiments using free FAD (1.0 mol%) in lieu of the FAP provided no cyclization product (Supplementary Table 8). Performing this biocatalytic reaction in the absence of light also afforded no product formation, confirming the photoenzymatic nature of this transformation (Supplementary Table 8). Visible light irradiation at 440 nm provided higher yields of 2a than other wavelengths (Supplementary Table 5).
Chemo- and enantioselective RAPs
With these initial results in hand, we sought to perform directed evolution to improve the activity, chemoselectivity and enantioselectivity of RAPs derived from FAPs (Fig. 2c). We focused our initial effort on CvFAP(Y466A), in part due to the availability of its crystal structure in complex with fatty acid substrates. The directed evolution of FAPs has not been reported, presumably due to their limited stability precluding convenient processing in a high-throughput manner. Indeed, upon the completion of FAP expression, a canonical high-throughput cell lysis protocol using lysozyme or various detergents led to minimal enzyme activity, suggesting FAP denaturation under these cell lysis conditions. The use of intact Escherichia coli cells harbouring FAP also furnished reduced yields and enantioselectivities (Supplementary Table 2). To circumvent these issues, we optimized a mild and high-throughput cell lysis protocol using a 24-tip horn sonicator (Supplementary Figs 2 and 3). Additionally, we assembled an in-house set-up to perform 96 photoenzymatic reactions in parallel at ambient temperature. Together, our newly developed protocol allowed high-throughput experimentation to be carried out in a 96-well plate format, thereby permitting directed evolution of FAPs to be successfully performed (Supplementary Figs 2 and 4).
With these advances, we performed iterative rounds of site-saturation mutagenesis and screening to evolve new-to-nature RAPs. In each round of engineering, four active site residues were randomized to provide a total of four single site-saturation libraries. Molecular docking studies with AutoDock were used to guide the selection of target residues for site-saturation mutagenesis. For each site-saturation mutagenesis library, 88 clones were expressed in 24-well plates. Using CvFAP(Y466A) as the template, directed evolution allowed beneficial mutations including G462A, S574G/S574A, S464A and T131A to be identified, leading highly efficient RAPs CvFAPAAGAA and CvFAPAAAAA to be developed. Herein, CvFAPAAGAA and CvFAPAAAAA are designated as CvRAP1 and CvRAP2, respectively. Evolved CvRAP1 afforded radical cyclization product 2a in 91% yield, 96:4 e.r. and 95:5 chemoselectivity. CvRAP2 provided 2a in 92% yield, 94:6 e.r. and 96:4 chemoselectivity. Among these beneficial mutations, G462A played an essential role in enhancing the cyclization chemoselectivity (Fig. 2c, entry 2; 2a/3a = 98:2). This mutation was previously found to promote the decarboxylation of short-chain carboxylic acids20. Thus, in our evolutionary lineage, the newly engineered CvFAP(Y466A/G462A) was the first variant to effectively suppress the undesired native protodecarboxylation reactivity. Beneficial mutations G462A, S464A and Y466A all reside in the 461–472 α-helix proximal to the substrate. This α-helix was found to play a critical role in modulating enzyme activity (vide infra). T131A is located in the 129–134 α-helix of the active site, while S574G/A resides in a flexible loop close to both the substrate and the FAD cofactor. From a mechanistic perspective, the successful evolution of RAPs demonstrated that the proton transfer step in the catalytic cycle could be rendered highly enantioselective, shedding light on the identity and activity of the proton donor in FAPs. Further studies confirmed that both R451 and C432 remained essential for the non-native FAP function (Supplementary Table 26).
Additionally, we evolved photocyclases from RsFAP using RsFAP(Y431A) as the parent (Supplementary Table 12). To identify key active-site residues of RsFAP, we analysed multiple sequence alignment results of RsFAP, CvFAP and other FAP homologues (Supplementary Fig. 1). We also used the AlphaFold2 model of RsFAP to assist in this enzyme engineering. The final variant RsRAP carried three beneficial mutations relative to RsFAP, including Y431A, Y434F and Q429D, all of which reside in the α-helix that aligns with the critical 461–472 α-helix of CvFAP (vide supra). RsRAP (RsFAP(Y431A/Y434F/Q429D)) catalysed radical cyclization of 1a in 48% yield and 96:4 e.r. with lower chemoselectivity (2a/3a = 51:49) relative to that of CvRAP.
With a set of evolved RAPs in hand, we next surveyed the substrate scope of this photoenzymatic radical cyclization (Fig. 3). In addition to ethyl ester 2a, methyl ester (2b), n-propyl ester (2c) and 2-bromoethyl ester (2d) represented excellent substrates for the photobiocatalytic radical cyclization, leading to cyclized products with good to excellent levels of enantioselectivity. Lactone 2e could also be converted with good yield and enantiocontrol. Importantly, α-halogen-substituted α,β-unsaturated esters were also excellent radical acceptors, giving rise to α-fluoroester (2f), α-chloroester (2g) and α-bromoester (2h) in a highly enantiomerically enriched form. Thus, these evolved RAPs afford a new method to prepare enantioenriched α-haloesters in a catalytic asymmetric manner. Increasing the size of the α-substituent to an ethyl (2i) did not lower the enzymatic enantiocontrol over the radical cyclization process. In addition to α,β-unsaturated esters, α,β-unsaturated amide 2j could also be transformed into the cyclized product, albeit with lower conversion and enantioselectivity. The α,β-unsaturated ketones (2k) underwent a smooth photoenzymatic transformation, but the cyclization product formed in a racemic fashion. Furthermore, in addition to five-membered cyclization products (2a–2k), six-membered products could be readily prepared without additional photoenzyme engineering (2l–2o). Diverse substituents of the cyclohexyl moiety, including difluoromethyl (2m), gem-dimethyl (2n) and an oxygen (2o), were compatible with this 6-exo-trig photoenzymatic cyclization. Taken together, these results demonstrated the utility of the photoenzyme in controlling the α-stereochemistry of cyclized products through proton transfer (Fig. 1a).

LED, light-emitting diode; a2 mol% CvRAP1 was used. bCvRAP2 was used in lieu of CvRAP1. cRsRAP was used in lieu of CvRAP1.
Diastereo- and enantiodivergent RAPs
We further challenged our engineered RAPs to set two contiguous stereogenic centres at the α and β positions in a stereodivergent fashion (Fig. 4). The highly stereoselective formation of stereochemical dyads would require the photocyclase to exert exquisite control over both the C–C-bond-forming radical cyclization step and the subsequent proton transfer step. Towards this end, 1p was selected as the model substrate to provide product 2p with adjacent α- and β-stereocentres. Starting from CvRAP2 (CvFAPAAAAA), three additional rounds of site-saturation mutagenesis and screening led to CvRAP2(G431W/I130F/A131V) (CvRAP3) with further improved diastereo- and enantioselectivity towards the formation of (2S,3R)-2p (83% yield, 90:10 d.r., >99:1 e.r.). Additionally, evaluation of the CvRAP2 evolution lineage on the photobiocatalytic cyclization of 1p revealed that the intermediate variant CvFAPAAAA (CvFAP(Y466A/G462A/S574A/S464A)) and the final variant CvFAPAAAAA displayed similar diastereo- and enantioselectivities, surpassing those with CvRAP1 (CvFAPAAGAA). Further directed evolution was thus also carried out using CvFAPAAAA as the starting variant. It was found that by inverting the β-stereochemistry, CvFAPAAAA(T465S) resulted in the reversal of diastereoselectivity along with a significant improvement in enantioselectivity for (2S,3S)-2p (72% yield, 33:67 d.r., 98:2 e.r.). Further protein engineering led to CvFAPAAAA(T465S/T131S) (CvRAP4), giving rise to (2S,3S)-2p in 86% yield, 20:80 d.r. and >99:1 e.r.

Directed evolution of orthogonal photoenzyme variants CvRAP3–CvRAP6 for stereodivergent asymmetric radical cyclization of 1p with CvRAP2–CvRAP6.
Moreover, empowered by directed evolution, both the β- and α-stereochemistry could be reversed, and all the four possible stereoisomers of 2p could be accessed in an enzyme-controlled fashion. Specifically, starting from CvRAP4, through iterative site-saturation mutagenesis and screening, CvRAP4(A466F/S465M/V463A) (CvRAP5) was advanced, providing (2R,3S)-2p with inverted α-stereochemistry in 55% yield, 59:41 d.r. and 90:10 e.r. (Fig. 4). Furthermore, the fourth stereoisomer (2R,3R)-2p could also be synthesized with another evolved RAP, CvFAPAAAA(I130F/T484S/V463N) (CvRAP6) in 89% yield, 78:22 d.r. and 96:4 e.r. (Fig. 4). The biocatalyst-controlled, fully diastereo- and enantiodivergent access of all four possible stereoisomers reported herein represents an example in the field of asymmetric radical biocatalysis with all enzyme classes, highlighting the exceptional tunability of natural FAP photoenzymes. Additionally, CvRAP6 allowed the conversion of 1a into 2a in 86% yield and 25:75 e.r. with inverted enantioselectivity (Fig. 5a). Together, CvRAP1 and CvRAP6 constitute an orthogonal set of biocatalysts for the enantiodivergent synthesis of cyclization products with a single stereocentre. Furthermore, using CvRAP5, tetrasubstituted olefin 1q could be transformed into highly enantioenriched product 2q with a quaternary carbon (Fig. 5b).

a, Enantiodivergent biotransformation of 1a with CvRAP1 and CvRAP6. b, Enantioselective biotransformation of tetrasubstituted olefin 1q to produce 2q with a quaternary carbon.
Stereoconvergent photobiocatalytic radical cyclization
During the course of this study, it was found that (E)- and (Z)-olefins were transformed with almost identical yields and enantioselectivities favouring the same major enantiomeric products, permitting α,β-unsaturated substrates as an E/Z mixture to be transformed in a stereoconvergent fashion (Extended Data Fig. 1a). To further investigate the nature of this stereoconvergent photobiotransformation, we first evaluated the photoactivity of (E)- and (Z)-1a in the presence of free FAD under blue light (440 nm) irradiation (Extended Data Fig. 1b). We found that (E)-1a maintained its geometric integrity in the presence of free FAD and did not undergo radical cyclization. By contrast, (Z)-1a underwent Z/E scrambling under these conditions, and the photochemical equilibration largely favoured (E)-1a. Starting from (Z)-1a, no radical cyclization product 2a was observed when FAD was used as the photocatalyst. Next, using our evolved CvRAP1, we measured the E/Z ratio of recovered (E)-1a and (Z)-1a at varying conversions (Extended Data Fig. 1c). Similar to our findings with free FAD, when (E)-1a was applied, no erosion of olefin configuration was observed. With (Z)-1a, stereochemical scrambling of 1a was observed, and a notable amount of (E)-1a was present in recovered 1a. Together, these results demonstrate that under photochemical conditions, both the engineered photoenzyme CvRAP1 and the free FAD cofactor can catalyse the olefin isomerization of (Z)-1a, but not (E)-1a (refs. 47,48). Furthermore, using CvRAP1, we compared the initial rate of this photobiocatalytic decarboxylative radical cyclization with (E)-1a and (Z)-1a. Interestingly, although (Z)-1a was found to isomerize to (E)-1a under visible light irradiation as evidenced by a lower Z/E ratio of recovered 1a (Z/E = 72:28), the conversion of (Z)-1a into the cyclized product 2a was approximately twice as fast as that with (E)-1a, as revealed by the higher initial rate of the 1a → 2a conversion with (Z)-1a ((Z)-1a, 6.4 μmol l–1 min–1; (E)-1a, 3.4 μmol l–1 min–1). These data reveal that although (Z)-1a undergoes photoisomerization to (E)-1a, using CvRAP1, it can also be directedly converted into 2a at a faster rate. Finally, a deuterium incorporation experiment using 1a in D2O showed 95% incorporation of deuterium into (S)-2a-d1 (Supplementary Section X for details).
Computational insights from molecular dynamics simulations
To gain further insights into the substantially enhanced chemoselectivity of CvRAP1 favoring C–C-bond-forming radical cyclization over premature PCET, we carried out classical molecular dynamics (MD) simulations to explore the conformational space of alkyl radical 4a formed from the photoenzyme-catalysed decarboxylation of 1a (Fig. 6). Our MD simulations reveal that in the active site of wild-type CvFAP, the alkyl radical intermediate 4a prefers an anti–anti conformation with a long distance between the radical centre (C7) and the β-carbon (C3) (distance d = 4.8–5.3 Å; Fig. 6b,d and Supplementary Fig. 5). By contrast, because of its significantly expanded active site compared to wild-type CvFAP (Supplementary Fig. 6), in the evolved RAP CvRAP1, alkyl radical 4a is much more conformationally flexible. With CvRAP1, the most populated gauche−–anti conformation has a shorter C3–C7 distance of ~4.1–4.7 Å (Fig. 6b,e and Supplementary Fig. 5). The difference between the conformational profiles of 4a in the active site of wild-type CvFAP and evolved CvRAP1 is corroborated by Gaussian accelerated MD enhanced sampling simulations49 (Fig. 6c), indicating a preference for the anti–anti conformation in wild-type CvFAP and gauche−–anti conformation in CvRAP1. Both conventional MD and Gaussian accelerated MD simulations indicate that the near-attack conformations50 (d(C3–C7) ≤ 3.1 Å) are rarely populated in wild-type CvFAP (<0.3% population and >5.5 kcal mol–1 less stable than the most stable conformer), but are present in a relatively large population in evolved CvRAP1 (2.1% and 1.5 kcal mol–1). Representative snapshots from conventional MD simulations (Fig. 6d,e) indicate that in wild-type CvFAP, residues Y466 and Q486 block both π-faces of the C=C double bond of 4a, disfavouring radical cyclization to the C=C double bond. Therefore, the key Y466A mutation introduced to CvRAP1 increases the size of the binding pocket, allowing the radical intermediate to adopt the bent near-attack conformation and removing the steric shield preventing radical cyclization in wild-type CvFAP.

a, Representative conformations of alkyl radical intermediate 4a. b, Distribution of C3–C7 distance of 4a in the enzyme active sites from classical MD simulations. c, Potential of mean force (PMF) of 4a in the enzyme active sites from Gaussian accelerated MD simulations. d, Overlay of representative conformations of 4a in wild-type CvFAP. e, Overlay of representative conformations of 4a in CvRAP1; a near-attack conformation of 4a is shown in pink. Only the enzyme geometry in the most populated structure is shown for clarity. Carbon dioxide molecules are omitted.
Conclusion
In conclusion, we repurposed and evolved natural FAPs as unnatural RAPs to catalyse stereocontrolled cyclization via an open-shell mechanism. The advancement of a high-throughput experimentation platform allowed directed evolution of FAPs to be carried out, allowing a collection of highly chemo- and stereoselective RAPs to be developed. Notably, a set of diastereo- and enantiodivergent RAP variants were successfully evolved, permitting all the four possible stereoisomers of a stereochemical dyad to be prepared. These results represent an effort to repurpose natural photoenzymes to enable new-to-nature chemistry, highlighting the underappreciated utility of natural photoenzymes in solving outstanding synthetic problems. In the field of new-to-nature photoenzymatic catalysis, in contrast to previously studied flavophotoenzymology with ‘ene’ reductases that operates through a strongly reducing excited-state flavohydroquinone, the RAPs repurposed from natural FAPs capitalize on the strongly oxidizing power of the excited-state flavoquinone to initiate radical biotransformations. Collectively, the promiscuous nature and the exceptional evolvability of FAPs highlights their enormous potential to address longstanding challenges in free-radical chemistry and asymmetric catalysis.
Methods
Expression of FAP variants
E. coli BL21(DE3) cells harbouring recombinant plasmid encoding the appropriate FAP variant were grown overnight at 37 °C and 230 rpm in 25 ml Luria-Bertani (LB) media supplemented with kanamycin (LBkan). Preculture (10 ml, 1% v/v) was used to inoculate 1 l Terrific Broth (TB) media supplemented with kanamycin (TBkan) in a 4 l Erlenmeyer flask. The culture was shaken at 37 °C and 230 rpm for 3 h to reach an optical density measured at 600 nm (OD600) of approximately 1.0. The culture was then cooled on ice for 20 min and induced with 0.1 mM IPTG reagent. Protein expression was performed at 18 °C and 200 rpm for 18 h. The cells were harvested for further use by centrifugation (5,000g, 15 min, 4 °C) using a superspeed centrifuge.
Purification of FAP variants
An E. coli cell pellet was resuspended in NaPi buffer (50 mM NaPi buffer, pH 7.5, 500 mM NaCl, 20 mM imidazole) and lysed by sonication. After centrifugation (12,000g, 20 min, 4 °C), the supernatant was filtered through a 0.45 μm membrane filter, loaded onto a HisTrap HP column (5 ml) and purified with the aid of an ÄKTA Start protein purifier. Proteins were eluted with an increasing gradient of imidazole from 20 to 500 mM in NaPi buffer at a flow rate of 5.0 ml min–1. Fractions containing the desired protein were pooled and subjected to three exchanges of NaPi buffer (50 mM, pH 8.0) to remove the excess salt and imidazole. Concentrated proteins were aliquoted, flash-frozen in liquid nitrogen and stored at –80 °C with 20% glycerol until further use. Protein concentration was determined using a FAD-specific UV–visible assay12.
Stereoselective photobiocatalytic radical cyclization
To prepare the substrate stock solution (100 mM in DMSO), the substrate was added to a 4 ml vial. The vial was then transferred into a Coy anaerobic chamber where degassed DMSO was added. Purified enzyme used for the reaction was kept on ice in the dark before it was transferred into the Coy anaerobic chamber. Vials (4 ml) charged with a stir bar (7 × 2 mm) were transferred into the Coy anaerobic chamber. In the anaerobic chamber, degassed Tris-HCl buffer (50 mM, pH 8.5), a stock solution of the substrate (100 mM in DMSO, 50 μl) and the FAP variant were added into the vial in succession. Final reaction volume was 500 μl; final concentrations were 10 mM substrate, 10% (v/v) DMSO and 0.1–2.0 mol% FAP. The vials were tightly sealed with polypropylene screw caps (with all-purpose silicone/PTFE septa) and parafilm, taken out of the anaerobic chamber and illuminated with a Kessil LED lamp for 20 h. The reaction mixture was then analysed by chiral gas chromatography and/or HPLC.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text and the Supplementary Information.
References
Begley, T. P. Photoenzymes: a novel class of biological catalysts. Acc. Chem. Res. 27, 394–401 (1994).
Taylor, A., Heyes, D. J. & Scrutton, N. S. Catalysis by nature’s photoenzymes. Curr. Opin. Struct. Biol. 77, 102491 (2022).
Schoefs, B. & Franck, F. Protochlorophyllide reduction: mechanisms and evolution. Photochem. Photobiol. 78, 543–557 (2003).
Zhang, S. et al. Structural basis for enzymatic photocatalysis in chlorophyll biosynthesis. Nature 574, 722–725 (2019).
Heyes, D. J. et al. Photocatalysis as the ‘master switch’ of photomorphogenesis in early plant development. Nat. Plants 7, 268–276 (2021).
Archipowa, N., Kutta, R. J., Heyes, D. J. & Scrutton, N. S. Stepwise hydride transfer in a biological system: insights into the reaction mechanism of the light‐dependent protochlorophyllide oxidoreductase. Angew. Chem. Int. Ed. 57, 2682–2686 (2018).
Heyes, D. J., Sakuma, M. & Scrutton, N. S. Solvent‐slaved protein motions accompany proton but not hydride tunneling in light‐activated protochlorophyllide oxidoreductase. Angew. Chem. Int. Ed. 48, 3850–3853 (2009).
Dong, C. et al. Crystal structures of cyanobacterial light-dependent protochlorophyllide oxidoreductase. Proc. Natl Acad. Sci. USA 117, 8455–8461 (2020).
Zhang, M., Wang, L., Shu, S., Sancar, A. & Zhong, D. Bifurcating electron-transfer pathways in DNA photolyases determine the repair quantum yield. Science 354, 209–213 (2016).
Brettel, K. & Byrdin, M. Reaction mechanisms of DNA photolyase. Curr. Opin. Struct. Biol. 20, 693–701 (2010).
Li, J. et al. Dynamics and mechanism of repair of ultraviolet-induced (6–4) photoproduct by photolyase. Nature 466, 887–890 (2010).
Sorigué, D. et al. An algal photoenzyme converts fatty acids to hydrocarbons. Science 357, 903–907 (2017).
Sorigué, D. et al. Mechanism and dynamics of fatty acid photodecarboxylase. Science 372, eabd5687 (2021).
Moulin, S. L. Y. et al. Fatty acid photodecarboxylase is an ancient photoenzyme that forms hydrocarbons in the thylakoids of algae. Plant Physiol. 186, 1455–1472 (2021).
Aselmeyer, C. et al. Fatty acid photodecarboxylase is an interfacial enzyme that binds to lipid–water interfaces to access its insoluble substrate. Biochemistry 60, 3200–3212 (2021).
Huijbers, M. M. E., Zhang, W., Tonin, F. & Hollmann, F. Light‐driven enzymatic decarboxylation of fatty acids. Angew. Chem. Int. Ed. 57, 13648–13651 (2018).
Zhang, W. et al. Hydrocarbon synthesis via photoenzymatic decarboxylation of carboxylic acids. J. Am. Chem. Soc. 141, 3116–3120 (2019).
Xu, J. et al. Light‐driven kinetic resolution of α‐functionalized carboxylic acids enabled by an engineered fatty acid photodecarboxylase. Angew. Chem. Int. Ed. 58, 8474–8478 (2019).
Li, D. et al. Engineering fatty acid photodecarboxylase to enable highly selective decarboxylation of trans fatty acids. Angew. Chem. Int. Ed. 60, 20695–20699 (2021).
Xu, J. et al. Light-driven decarboxylative deuteration enabled by a divergently engineered photodecarboxylase. Nat. Commun. 12, 3983 (2021).
Santner, P. et al. Optimization and engineering of fatty acid photodecarboxylase for substrate specificity. ChemCatChem 13, 4038–4046 (2021).
Xu, W. et al. Rational design of fatty acid photodecarboxylase enables the efficient decarboxylation of medium- and short-chain fatty acids for the production of gasoline bio-alkanes. Mol. Catal. 524, 112261 (2022).
Amer, M. et al. Low carbon strategies for sustainable bio-alkane gas production and renewable energy. Energy Environ. Sci. 13, 1818–1831 (2020).
Zhou, Q., Chin, M., Fu, Y., Liu, P. & Yang, Y. Stereodivergent atom-transfer radical cyclization by engineered cytochromes P450. Science 374, 1612–1616 (2021).
Fu, Y. et al. Engineered P450 atom-transfer radical cyclases are bifunctional biocatalysts: reaction mechanism and origin of enantioselectivity. J. Am. Chem. Soc. 144, 13344–13355 (2022).
Fu, W. et al. Enzyme-controlled stereoselective radical cyclization to arenes enabled by metalloredox biocatalysis. Nat. Catal. 6, 628–636 (2023).
Rui, J. et al. Directed evolution of nonheme iron enzymes to access abiological radical-relay C(sp3)−H azidation. Science 376, 869–874 (2022).
Cheng, L. et al. Stereoselective amino acid synthesis by synergistic photoredox-pyridoxal radical biocatalysis. Science 381, 444–451 (2023).
Emmanuel, M. A. et al. Photobiocatalytic strategies for organic synthesis. Chem. Rev. 123, 5459–5520 (2023).
Emmanuel, M. A., Greenberg, N. R., Oblinsky, D. G. & Hyster, T. K. Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 540, 414–417 (2016).
Biegasiewicz, K. F. et al. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–1169 (2019).
Black, M. J. et al. Asymmetric redox-neutral radical cyclization catalysed by flavin-dependent ‘ene’-reductases. Nat. Chem. 12, 71–75 (2020).
Clayman, P. D. & Hyster, T. K. Photoenzymatic generation of unstabilized alkyl radicals: an asymmetric reductive cyclization. J. Am. Chem. Soc. 142, 15673–15677 (2020).
Page, C. G. et al. Quaternary charge-transfer complex enables photoenzymatic intermolecular hydroalkylation of olefins. J. Am. Chem. Soc. 143, 97–102 (2021).
Gao, X., Turek-Herman, J. R., Choi, Y. J., Cohen, R. D. & Hyster, T. K. Photoenzymatic synthesis of α-tertiary amines by engineered flavin-dependent “ene”-reductases. J. Am. Chem. Soc. 143, 19643–19647 (2021).
Nicholls, B. T. et al. Engineering a non‐natural photoenzyme for improved photon efficiency. Angew. Chem. Int. Ed. 61, e202113842 (2022).
Fu, H. et al. An asymmetric sp3–sp3 cross-electrophile coupling using ‘ene’-reductases. Nature 610, 302–307 (2022).
Fu, H., Qiao, T., Carceller, J. M., MacMillan, S. N. & Hyster, T. K. Asymmetric C-alkylation of nitroalkanes via enzymatic photoredox catalysis. J. Am. Chem. Soc. 145, 787–793 (2023).
Page, C. G. et al. Regioselective radical alkylation of arenes using evolved photoenzymes. J. Am. Chem. Soc. 145, 11866–11874 (2023).
Harrison, W., Huang, X. & Zhao, H. Photobiocatalysis for abiological transformations. Acc. Chem. Res. 55, 1087–1096 (2022).
Huang, X. et al. Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 584, 69–74 (2020).
Huang, X. et al. Photoinduced chemomimetic biocatalysis for enantioselective intermolecular radical conjugate addition. Nat. Catal. 5, 586–593 (2022).
Heyes, D. J. et al. Photochemical mechanism of light-driven fatty acid photodecarboxylase. ACS Catal. 10, 6691–6696 (2020).
Jasperse, C. P., Curran, D. P. & Fevig, T. L. Radical reactions in natural product synthesis. Chem. Rev. 91, 1237–1286 (1991).
Gilmore, K. & Alabugin, I. V. Cyclizations of alkynes: revisiting Baldwin’s rules for ring closure. Chem. Rev. 111, 6513–6556 (2011).
Gilmore, K., Mohamed, R. K. & Alabugin, I. The Baldwin rules: revised and extended. WIREs Comput. Mol. Sci. 6, 487–514 (2016).
Neveselý, T., Wienhold, M., Molloy, J. J. & Gilmour, R. Advances in the E → Z isomerization of alkenes using small molecule photocatalysts. Chem. Rev. 122, 2650–2694 (2022).
Litman, Z. C., Wang, Y., Zhao, H. & Hartwig, J. F. Cooperative asymmetric reactions combining photocatalysis and enzymatic catalysis. Nature 560, 355–359 (2018).
Miao, Y., Feher, V. A. & McCammon, J. A. Gaussian accelerated molecular dynamics: unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 11, 3584–3595 (2015).
Soler, J., Gergel, S., Klaus, C., Hammer, S. C. & Garcia-Borràs, M. Enzymatic control over reactive intermediates enables direct oxidation of alkenes to carbonyls by a P450 iron-oxo species. J. Am. Chem. Soc. 144, 15954–15968 (2022).
Acknowledgements
This research is supported by the National Institutes of Health (NIH; R35GM128779 to P.L., R35GM147387 to Y.Y.), American Chemical Society Petroleum Research Fund (ACS PRF; 65807-DNI1) and the National Science Foundation (NSF; OCE-1756947 to D.L.V.). We acknowledge the NSF BioPACIFIC Materials Innovation Platform (MIP; DMR-1933487) and NSF Materials Research Science and Engineering Centers (MRSEC) at University of California, Santa Barbara (UCSB; DMR-2308708) for access to instrumentation. Computational studies were carried out at the University of Pittsburgh Center for Research Computing and the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) programme, supported by NSF award numbers OAC-2117681 and OAC-2138259. We thank Y. Wang for critical reading of this manuscript.
Author information
Authors and Affiliations
Contributions
Y.Y. conceived and directed the project. S.J. performed all the enzyme engineering and substrate scope studies. S.J. and A.V.-E. performed enzyme mining and molecular cloning with Y.Y. and D.L.V., and J.W. provided guidance. D.L., S.J. and X.L. synthesized all the substrates and racemic products for analysis. B.K.M. carried out the computational studies with P.L. providing guidance. Y.Y., P.L., S.J. and B.K.M. wrote the manuscript with the input of all other authors.
Corresponding authors
Ethics declarations
Competing interests
Y.Y. and S.J. are inventors on a patent application submitted by the University of California Santa Barbara (UC case no. 2024-843) that covers compositions, methods and applications of evolved RAPs derived from natural FAPs. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks Dunming Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Origin of stereoconvergent photobiocatalytic radical cyclisation with (E)- and (Z)-1a.
a, Stereoconvergent photobiotransformation of (E)-1a and (Z)-1a. b, Photoisomerisation of (Z)-1a with FAD (free cofactor). c, Initial rate and photoisomerisation with CvRAP1.
Supplementary information
Supplementary Information
Supplementary Figs. 1–6, Discussion and Tables 1–26.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ju, S., Li, D., Mai, B.K. et al. Stereodivergent photobiocatalytic radical cyclization through the repurposing and directed evolution of fatty acid photodecarboxylases. Nat. Chem. (2024). https://doi.org/10.1038/s41557-024-01494-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41557-024-01494-0
