
Abstract
In the present study, the dense Cr and Cr0.92Si0.08 coatings have been deposited on Zircaloy-4 substrates by the magnetron sputtering technique. The high-temperature oxidation resistance of coatings is evaluated in 1200 °C steam for 1–4 h. The Cr0.92Si0.08 coating shows better oxidation resistance than the Cr coating. The in-situ formed Zr2Si diffusion barrier inhibits the mutual diffusion of Cr and Zr. And the formation of a Cr2O3/SiO2 double-layer scale effectively improves the oxidation resistance of a single Cr2O3 layer. The formation of the in-situ diffusion barrier, Cr2O3/SiO2 scales and oxidation protection mechanisms of the Cr0.92Si0.08 coating have been discussed in detail.
Similar content being viewed by others

Introduction
Zirconium alloys (Zircaloy) have been widely used as the cladding materials in light water reactors (LWRs) on account of their low neutron capture cross-section, good mechanical properties and corrosion resistance1,2. However, Zircaloy claddings will react with high-temperature steam rapidly when loss of coolant accidents occurs, such as the Fukushima-Daiichi nuclear accident in 20113. Therefore, accident-tolerant fuel (ATF) has been proposed to increase the safety margin of LWRs under normal operation and accident conditions4. Among the ATF strategies, coating is one of the most attractive ways to enhance the oxidation resistance of Zr alloys because of high efficiency and low cost5. Many coatings have been studied, including metallic coatings (Cr6, FeCrAl7,8, high-entropy alloys9), ceramic coatings (CrN10, MAX phase11) and multilayered coatings12,13. Cr coating has been considered as the most attractive candidate owing to its strong adhesion with Zr substrate, good corrosion resistance, and high-temperature oxidation resistance14,15.
However, it has been found that the Cr2O3 oxide layer will be reduced by Zr after the Cr coating was completely oxidized into Cr2O3 since the formation of ZrO2 is thermodynamically favorable16,17. Furthermore, the interdiffusion between Cr coating and Zr substrate will occur during the oxidation process. On one hand, the Cr coating will be consumed by the formation of a ZrCr2 layer at the coating-substrate interface as well as solid solution and precipitates in the Zr substrate18. On the other hand, the outward diffusion of Zr facilitates the growth of Zr–Cr intermetallics and ZrO2 stringers within the Cr coating, which are the short-circuit diffusion paths for oxygen to penetrate into the Zr substrate16,19. Therefore, the interdiffusion of Cr and Zr plays a negative effect in the Cr-coated Zircaloy system.
It has been reported that suitable diffusion barriers can minimize the interdiffusion between the coating and substrate. For instance, Wang et al. introduced a ZrO2 layer between zirconium alloy and Cr coating by plasma electrolytic oxidation, which enhanced the oxidation resistance of Cr coating because the ZrO2 layer effectively prevent O and Cr diffusion into Zr alloy in 900–1100 °C20,21. Syrtanov et al. indicated that a Mo sublayer between Cr coating and Zr substrate can hinder the interdiffusion of Cr and Zr22. In addition, CrN ceramic layer was also used as the diffusion barrier between Cr coating and Zr substrate12. However, using extra diffusion barriers cause other issues, such as increasing structural complexity, adding process cost, and sometimes cracking or spalling. Another way to hinder the severe interdiffusion is the in-situ formation of a dense diffusion barrier. Huang et al. found that an in-situ formed Ti5Si3 diffusion barrier can prevent the fast depletion of Al in the coating, which improved the oxidation resistance of AlSi coated-TiAl alloy23. Li et al. also indicated that FeCrAlMoSiY-coated Zr alloy has shown good oxidation resistance because an in-situ Zr2Si diffusion barrier can inhibit the mutual diffusion of Fe, Al, and Zr24.
In our previous study, it has been found that Cr–Si coatings show better oxidation resistance than pure Cr coating in 1200 °C steam environment, which may result from an in-situ formation of a Zr–Si interdiffusion layer25. However, the results are lack of adequate experimental evidence and characterizations. In this paper, more characteristic and systematic experiments are performed. The formation of an in-situ formed Zr–Si barrier and oxidation mechanisms of the Cr0.92Si0.08 coatings have been discussed in detail.
Results
Coating microstructures and phases
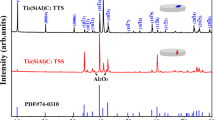
Figure 1a, b shows the surface morphologies of the Cr and Cr–Si coatings. Both coatings are dense, free of holes and micro-cracks. The Cr coating shows needle-like morphologies, whereas the Cr–Si coating presents a flaky structure (Fig. 1a, b). EDS analysis shows that the Cr/Si atomic ratios are 92.3/7.7. Therefore, the Cr–Si coating is denoted as the Cr0.92Si0.08 coating for simplicity. Figure 1c, d shows the cross-sectional morphologies of the Cr and Cr0.92Si0.08 coatings. No cracks and spalls are observed at the interface between the coating and the substrate, and the thickness of Cr and Cr0.92Si0.08 coatings are 16 ± 0.5 μm and 15.4 ± 0.3 μm, respectively. Figure 2 shows the XRD patterns of the as-grown Cr and Cr0.92Si0.08 coatings. Both coatings consist of Cr phase with the bcc crystal structure. Si-containing phases are not found in the Cr0.92Si0.08 coating, which indicates Si forms solid solution within the Cr crystal matrix.

a, c Cr coating; b, d Cr0.92Si0.08 coating.

XRD patterns of the as-deposited Cr and Cr0.92Si0.08 coatings.
Surface morphologies of the oxide layer
Figure 3 shows the surface morphologies of Cr and Cr0.92Si0.08 coatings after oxidized at 1200 °C steam for 1–4 h. For the Cr coatings, after oxidation for 1–3 h, the surface is flat and without typical features (Fig. 3a–c). However, after oxidation for 4 h, the surface morphology presents worm-like structure, pores and white phase (Fig. 3d). EDS point analyses (P1-P5) indicate that the chemical compositions of the oxidation products all agree with the stoichiometric Cr2O3 (Table 1). For the Cr0.92Si0.08 coatings, the surface morphologies show dense and compact oxide scale after oxidation for 1–4 h (Fig. 3e–h), which is determined as Cr2O3 oxide according to the EDS point P6-P10, as listed in Table 1. Figure 4 shows the XRD patterns of Cr and Cr0.92Si0.08 coatings after oxidation. The oxidation products of all Cr and Cr0.92Si0.08 coatings are indexed as Cr2O3. The peaks of Cr can be detected for both coatings because the oxide layer was thin, and the X-ray can penetrate into the Cr coating, which is also reported in the literature17. The XRD results are consistent with the surface EDS point analysis (Table 1).

a–d Cr coatings; e–h Cr0.92Si0.08 coatings.

XRD patterns of the oxidized Cr and Cr0.92Si0.08 coatings after oxidation in 1200 °C steam for 1–4 h.
Cross-sectional morphologies of the oxidized coating
Figure 5 shows the cross-sectional morphologies of uncoated Zry-4 alloy, Cr and Cr0.92Si0.08 coated Zry-4 alloys after 1200 °C steam oxidation for 1–4 h. Figure 5a1–a4 shows that the thickness of ZrO2 layer formed on the uncoated Zry-4 alloys after oxidation for 1 h, 2 h, 3 h, and 4 h is ~147 μm, 226 μm, 281 μm, 327 μm, respectively. For the Cr coatings, the substrates are well protected by the Cr coatings for 1–3 h (Fig. 5b1–b3). While after oxidation for 4 h, the inset image in Fig. 5b4 presents the formation of a zirconia layer (~15 μm) underneath the oxidized Cr coating. For the Cr0.92Si0.08 coatings, the substrates are prevented from oxidizing even for 4 h (Fig. 5c1–c4), indicating better oxidation resistance than the Cr coating in 1200 °C steam environment.

a1–a4 Zry-4 alloys; b1–b4 Cr coatings; c1–c4 Cr0.92Si0.08 coatings.
Higher magnifications of the Cr and Cr0.92Si0.08 coatings after oxidation are shown in Fig. 6, and the corresponding EDS line scans are shown in Fig. 7. For the Cr coating, after oxidation for 1 h, Cr2O3 oxide layer is formed with a thickness of 11.2 μm, and a 2.3-mirco thick ZrCr2 diffusion layer is formed (Figs. 6a and 7a). However, as shown in Figs. 6b and 7b, the thickness of the Cr2O3 layer decreases to 5.2 μm at 2 h, and the thickness of the ZrCr2 layer increases to 3.1 μm. After oxidation for 3 h, a 5.3- μm-thick Cr2O3 layer is observed on the surface. As the oxidation time increased to 4 h, as shown in Figs. 6d and 7d, the thickness of the Cr2O3 layer is about 1.0 μm. A layer of ZrO2 forms underneath the coating (Fig. 5b4), and the ZrCr2 layer cannot be clearly detected. The morphology evolution of Cr coating in our study well agrees with the reported literature16. For the Cr0.92Si0.08 coating, after oxidation for 1 h, the thickness of Cr2O3 layer is about 4.0 μm, which is thinner than that of the Cr coating. In addition, both the ZrCr2 and Zr–Si interdiffusion layers are observed on the interface, and their thicknesses are about 1.3 μm and 2.6 μm, respectively, as shown in Figs. 6e and 7e. After oxidation for 2 h, the structure of the oxidized Cr0.92Si0.08 coating is similar to that of the 1 h except for the thickness, including a top Cr2O3 layer (~3.8 μm), a residual coating (~7.5 μm) and thicker interdiffusion ZrCr2 layer (~1.6 μm) and Zr–Si layer (~3.0 μm). An average 3.3 μm-thick Cr2O3 layer is formed on the coating surface at 1200 °C for 3 h, and the thickness of Zr–Si diffusion layer is about 4.1 μm. After oxidation for 4 h, the thickness of Cr2O3 layer is about 2.0 μm, and the diffusion Zr–Si layer seems to be not uniform (Figs. 6h and 7h), with an average thickness of 6.3 μm. According to the EDS point analyses, as list in Table 2, the interdiffusion layer is composed of Zr and Si with atomic ratio of ~7/3, which is determined as Zr2Si. In addition, with the increase of oxidation time, the other phase is observed in the Zr2Si diffusion layer (Fig. 6h1). The composition is mainly Zr element accompanied with ~25 at.% O (P20, P22 in Table 2). For comparison, the thicknesses of the Cr2O3 scale, residual coatings, and interdiffusion layers of Cr and Cr–Si coatings are plotted in Fig. 8 as a function of oxidation time. It can be seen that the thickness of ZrCr2 layer in Cr0.92Si0.08 coatings are thinner than that of Cr coatings. In addition, according to the thickness of the Cr2O3 layer, Si-doped Cr coating shows a slower oxidation rate than the pure Cr coating, especially after 1200 °C oxidation for 1 h.

a–d Cr coatings; e–h, e1–h1 Cr0.92Si0.08 coatings.

a–d Cr coatings; e–h Cr0.92Si0.08 coatings.

Thickness of the Cr2O3 layer, residual coating, ZrCr2 and Zr2Si diffusion layers for Cr and Cr0.92Si0.08 coatings after oxidation in 1200 °C steam as a function of oxidation time.
To further study the morphologies and structures evolution of the oxidized Cr0.92Si0.08 coating, TEM/STEM specimens are prepared and analyzed. Figure 9a is the TEM image showing the overview cross-sectional morphology of the Cr0.92Si0.08 coating after 1200 °C oxidation for 2 h. As shown in Fig. 9a, four distinct layers are observed, including top Cr2O3 layer, middle residual coating, ZrCr2 interdiffusion layer, and bottom Zr2Si interdiffusion layer, which agrees with the SEM observations (Fig. 6e–h). The structure analysis of the Cr2O3 layer is shown in Fig. 9b, b1, b2. Figure 9b is the enlarged TEM image of the oxide layer. Figure 9b1 is a HAADF-STEM image from the oxide scale as marked in Fig. 9b, where the lattice spacing agrees with the Cr2O3. The SAED diffraction further confirms the Cr2O3 structure, which can be indexed well along the [001] zone axis, as shown in Fig. 9b2. The structure analysis of the ZrCr2 interdiffusion layer is shown in Figs. 9c, c1, c2. Figure 9c is an enlarged TEM image of the coating/ZrCr2 interface, where the upper part is the residual Cr–Si coating and the lower part is the ZrCr2 layer. As shown in Fig. 9c1, c2, both the lattice spacings and SAED diffraction can be indexed as ZrCr2 along the [011] zone axis. Figure 9d is an enlarged TEM image of the Zr2Si interdiffusion layer. Figure 9d1, d2 is the HAADF-STEM image and SAED diffraction of the Zr2Si layer, respectively, marked with red square in Fig. 9d. Both the lattice spacings and diffractogram can be indexed well with Zr2Si in the [-113] zone axis. Figure 9d3 shows the EDS spectrum of Zr2Si layer, which mainly consists of 66.14 at% Zr and 25.44 at% Si elements. Figure 10 shows the TEM analysis on the Cr0.92Si0.08 coating after 1200 °C oxidation for 4 h. Four layers can also be seen on the overview cross-section image after oxidation, and SiO2 particles are observed under the surface Cr2O3 scale (Fig. 10a). Similarly, the HAADF-STEM and SAED diffraction images are shown to determine the structure and phase of the ZrCr2 and Zr2Si layer (Figs. 10b, b1, c, c1). In addition, the EDS spectrum also confirms the composition of the Zr2Si layer (Fig. 10c2). The TEM results are consistent with the SEM/EDS results (Figs. 6 and 7 and Table 2).

a TEM image of the overview cross-sectional of Cr0.92Si0.08 coating after oxidation at 1200 °C for 2 h; b TEM image of the Cr2O3 layer, b1 HAADF-STEM image of the Cr2O3 layer and b2 corresponding SAED image; c TEM image of the coating/ZrCr2 layer interface, c1 HAADF-STEM image of the ZrCr2 layer and c2 corresponding SAED image; d TEM image of the Zr2Si layer, d1 HAADF-STEM image of the Zr2Si layer and d2, d3 corresponding SAED image and EDS spectrum, respectively.

a HAADF-STEM image of the overview cross-sectional of Cr0.92Si0.08 coating after oxidation at 1200 °C for 4 h; b HAADF-STEM image of the ZrCr2 layer and b1 corresponding SAED image; c HAADF-STEM image of the Zr2Si layer and c2, c3 corresponding SAED image and EDS spectrum, respectively.
Discussion
The above results show that the Cr0.92Si0.08 coating presents better oxidation resistance than the Cr coating in 1200 °C steam environment. Especially after oxidation for 4 h, the underneath substrate is oxidized in the Cr-coated sample, while the Cr0.92Si0.08 coated sample is not (Fig. 5). It is obvious that the Si element plays an important role on the oxidation resistance of the Cr-–Si coating. The oxidation behavior of the Cr coating has been extensively studied by researches16,19,26,27. Therefore, in this study, the formation of the oxide scales and the Zr2Si layer in the Cr0.92Si0.08 coatings will be discussed in detail.
Normally, mutual interdiffusion between the coating and substrate will occur at elevated temperatures because of the gradient of chemical composition. In the present study, for the Cr0.92Si0.08 coatings, Cr and Si elements in the coating diffuse into the substrate while Zr element in the substrate diffuses conversely, forming the interdiffusion layer in the coating-substrate interface. In order to understand the evolution process of the ZrCr2 and Zr2Si interdiffusion layer, a group of shorter-term oxidation experiments have been carried out, as shown in Fig. 11. The EDS point analysis is listed in Fig. 11g. After oxidation for 5 min, the diffusion layer is Zr–Cr–Si (Fig. 11a). With the prolonged time, the ZrCr2 and Zr2Si interdiffusion layers are formed (Fig. 11b–f). After oxidation for 1–4 h, similar structures of ZrCr2 and Zr2Si interdiffusion layers are formed in the interface (Figs. 6, 9, and 10). On one hand, in view of diffusivity, an estimate of the diffusion coefficient of Cr in Zr at 928–1206 °C is 5.8 × 10–12 m2 s−1 (see ref. 28), and the diffusivity of Si in Zr in the Ti3SiC2–Zr system at 1100–1300 °C is 7.0 × 10–15 m2 s−1 (see ref. 29), which indicates the diffusion rate of Si in Zr is slower than that of Cr. On the other hand, from the view of thermodynamics, the following two reactions will occur30:

a 5 min; b 10 min; c 20 min; d 30 min; e 40 min; f 50 min; g EDS point analysis in the diffusion layer.
Because the Gibbs free energy of reaction (2) is more negative, Zr2Si is thermodynamically preferred. Therefore, the formation of the diffusion layer is a dynamic balance between thermodynamically and kinetically controlled diffusion processes.
Therefore, from the cross-sectional images of Cr0.92Si0.08 coatings after oxidation, the ZrCr2 layer followed by Zr2Si layer can be observed in the interface. Once the continuous Zr2Si layer forms, it can further hinder the outward diffusion of Zr. As shown in Fig. 12, it is clear that the Zr veins through the Cr coating, especially after oxidation for 3 h and 4 h (Fig. 12a1–a4). While for the Cr0.92Si0.08 coating, the outdiffusion of Zr is limited (Fig. 12b1–b4). Thus, compared to the pure Cr coating, the reduction reaction between Cr2O3 and Zr can be limited in the Cr0.92Si0.08 coating, and the diffusion path formed by the ZrO2 phase within the coating can be also reduced16,17,19,31. In addition, The thickness of ZrCr2 layer in the Cr0.92Si0.08 coating is thinner than that of the pure Cr coating (Fig. 8). Therefore, the oxidation resistance of Cr0.92Si0.08 coating is improved by the in-situ formation of Zr2Si layer. For the Zr2Si interdiffusion layer in the Cr0.92Si0.08 coating, another phenomenon is that with the oxidation time increases to 4 h, some Zr-rich phases encapsulated by the Zr2Si matrix are observed (Fig. 6h1 and Table 2). The main reason for this phenomenon is the depletion of Si in the Cr–Si coating, and the residual Si cannot form a thicker and continuous Zr2Si layer.

a1–a4 Cr coatings; b1–b4 Cr0.92Si0.08 coatings; c EDS maps of (a2); d EDS maps of (b1).
For the Cr0.92Si0.08 coating, in the initial oxidation stage, the O in the environment reaches the surface and reacts with Cr, then a continuous Cr2O3 layer forms on the surface. Once the dense oxide layer forms, it will serve as a diffusion barrier to prevent the inward diffusion of O. However, for example, after oxidation for 1 h, the thickness of the Cr2O3 scale (4 μm) for Cr0.92Si0.08 coating is thinner than that of the pure Cr coating (11.2 μm). Moreover, the Cr2O3 scale of the Cr0.92Si0.08 coating seems denser than the pure Cr coating. In order to analyze the reasons, high-magnification SEM images and the corresponding EDS mapping at the interface between Cr2O3 scale and coating are observed at 1200 °C for 20 min and 1 h, as shown in Fig. 13. It can be seen that the SiO2 sublayer is formed after oxidation for 20 min and 1 h. In view of thermodynamics, SiO2 (−644 kJ mol−1) is preferred to form in 1200 °C because it presents more negative Gibbs free energy than that of 2/3Cr2O3 (−500 kJ mol−1)30. However, the formation of Cr2O3 is kinetically favored since the content of Cr (92.3 at.%) in the coating is much higher than that of Si (7.7 at.%). Therefore, firstly, the Cr2O3 layer forms on the surface. As the oxidation time prolonged, the Si element in the coating will diffuse and react with O, and the SiO2 sublayer forms, which can further hinder the diffusion rate of Cr and O. Therefore, the Cr2O3 layer in the Cr0.92Si0.08 coating is thinner and dense than the pure Cr coating. However, with the increase of oxidation time, due to the lack of Si content in the coating, the continuous growth of SiO2 scale cannot be maintained. Therefore, the SiO2 scale becomes discontinuous after oxidation for 2–4 h (Fig. 12b2–b4).

a 20 min; b 1 h.
The Cr0.92Si0.08 coating shows better oxidation resistance than the Cr coating. The schematic diagram of microstructure evolution is shown in Fig. 14. In the initial oxidation stage, Cr2O3 scale forms on the surface. Cr and Si elements diffuse inward simultaneously and react with Zr, forming the Zr–Cr–Si layer first in the interface (Fig. 14b). With the prolonged oxidation, the Si element in the coating reacts with the inward diffusion of O, and a continuous SiO2 sublayer is formed. The Cr2O3/SiO2 bilayer shows good oxidation resistance. Si in the Zr–Cr–Si layer diffuse into the Zr substrate, forming the ZrCr2 layer and Zr2Si diffusion layer in the interface (Fig. 14c). The outward diffusion of Zr is hindered by the in-situ formation of the Zr2Si diffusion barrier. And the redox reaction between Cr2O3 and Zr is mitigated. Similar interdiffusion layers have also been observed in other coating-substrate or diffusion couple systems, such as Ti5Si3 layer23, Zr2Si layer24 and Nb5Si3 layer32. When the oxidation time is further increased, the SiO2 scale becomes discontinuous, and the Zr2Si layer is embedded with the Zr-rich phases. In addition, because the redox reaction between Cr2O3 and Zr has occurred in this stage, the thickness of Cr2O3 decreases (Fig. 14d).

a As-deposited coating; b the initial oxidation stage; c the in-situ formation of the Zr2Si diffusion barrier stage; and d the final oxidation stage.
In this study, the Cr and Cr0.92Si0.08 coatings have been deposited on Zry-4 substrate by the magnetron sputtering technique. High-temperature oxidation tests have been performed in 1200 °C steam for 1–4 h. Based on the results and discussion, the following conclusions can be drawn:
-
(1)
The Cr0.92Si0.08 coating shows better oxidation resistance than the Cr coating in 1200 °C steam environment.
-
(2)
The formation of SiO2 sublayer in the Cr0.92Si0.08 coating further reduces the inward diffusion of O and outward of Cr, which can improve the oxidation resistance of Si-doped Cr coating.
-
(3)
In the Cr0.92Si0.08 coating, an in-situ formation of Zr2Si layer acted as a diffusion barrier can prevent the interdiffusion of Cr and Zr, enhancing the oxidation resistance of the coating.
Methods
Sample preparation
All coatings were deposited on Zirconium-4 (Zry-4) alloy coupons in size of 10 × 10 × 1.2 mm. Before coating deposition, all substrates were grounded on 2000 grit SiC paper, then cleaned ultrasonically in alcohol and acetone for 15 min, respectively. Coatings were fabricated through direct current magnetron sputtering. The diameter of the targets was 60 mm. As the pressure in the chamber was less than 8.31 × 10–4 Pa, substrate samples were etched by Ar ion under a negative bias voltage of 500 V for about 15 min at Ar pressure of 3–5 Pa in order to remove the surface contaminations of the substrates. The deposition was conducted in the gas of Ar at 0.5 Pa, under a bias voltage of 70 V. A Cr–Si alloy (Si 8.9 at.%, purity 99.95 at.%) target and a Cr (purity 99.95 at.%) target were used to prepare the Cr–Si and Cr coatings, respectively. The power used on the Cr–Si and Cr targets was both 200 W, and the deposition time is 4 h. In order to avoid serious oxidation of the uncoated side of the Zircaloy substrate, the coupons were two-sided deposited where one side first in 400 °C and then the other side in room temperature under the same deposition conditions.
Oxidation experiments
High-temperature steam oxidation tests of the coated coupons were conducted in an alumina tube furnace with a continuous flow of steam (0.5 ml min−1) produced by a steam generator. The steam pressure is 1 atm. When the temperature at the middle of the tube reached the experimental temperature (1200 °C), the steam was introduced into the tube and kept more than 30 min to ensure the tube was full of steam. And then the samples were transferred into the furnace and hold in the furnace for various times. After the oxidation tests had finished, the samples were pulled out from the furnace immediately and then cooled down to room temperature in the air.
Characterization of microstructures and compositions
The phase composition analysis of coatings was conducted by a Bruker diffractometer with a Cu-Kα source (λ = 1.540598 Å). Surface and cross-sectional morphologies were characterized by a ZEISS high-resolution field emission scanning electron microscope (SEM) equipped with energy-dispersive X-ray spectrometry (EDS). To further characterize the oxidation layers and remaining coatings, focused ion beam (FIB) milling was applied to prepare TEM samples. The microstructures at atomic levels were characterized by a Cs-corrected transmission electron microscope (TEM).
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Azevedo, C. R. F. Selection of fuel cladding material for nuclear fission reactors. Eng. Fail. Anal. 18, 1943–1962 (2011).
Hallstadius, L., Johnson, S. & Lahoda, E. Cladding for high performance fuel. Prog. Nucl. Energ. 57, 71–76 (2012).
Ma, X., Toffolon-Masclet, C., Guilbert, T., Hamon, D. & Brachet, J. C. Oxidation kinetics and oxygen diffusion in low-tin Zircaloy-4 up to 1523K. J. Nucl. Mater. 377, 359–369 (2008).
Terrani, K. A. Accident tolerant fuel cladding development: Promise, status, and challenges. J. Nucl. Mater. 501, 13–30 (2018).
Tang, C., Stueber, M., Seifert, H. J. & Steinbrueck, M. Protective coatings on zirconium-based alloys as accident-tolerant fuel (ATF) claddings. Corros. Rev. 35, 141–165 (2017).
Park, J.-H. et al. High temperature steam-oxidation behavior of arc ion plated Cr coatings for accident tolerant fuel claddings. Surf. Coat. Technol. 280, 256–259 (2015).
Yeom, H. et al. Development of cold spray process for oxidation-resistant FeCrAl and Mo diffusion barrier coatings on optimized ZIRLO™. J. Nucl. Mater. 507, 306–315 (2018).
Han, X., Wang, Y., Peng, S. & Zhang, H. Oxidation behavior of Fe Cr Al coated Zry-4 under high temperature steam environment. Corros. Sci. 149, 45–53 (2019).
Zhang, W., Tang, R. & Yang, Z. B. Preparation, structure, and properties of an AlCrMoNbZr high-entropy alloy coating for accident-tolerant fuel cladding. Surf. Coat. Technol. 347, 13–19 (2018).
Liu, J. et al. Oxidation behavior, thermal stability, and the coating/substrate interface evolution of CrN-coated Zircaloy under high-temperature steam. Corros. Sci. 185, 109416 (2021).
Tang, C. et al. Deposition, characterization and high-temperature steam oxidation behavior of single-phase Ti2AlC-coated Zircaloy-4. Corros. Sci. 135, 87–98 (2018).
Sidelev, D. V. et al. Protective Cr coatings with CrN/Cr multilayers for zirconium fuel claddings. Surf. Coat. Technol. 433, 128131 (2022).
Wu, K. et al. Oxidation behavior and chemical evolution of architecturally arranged Zr/Si multilayer at high temperature. Surf. Coat. Technol. 399, 126205 (2020).
Park, D. J. et al. Behavior of an improved Zr fuel cladding with oxidation resistant coating under loss-of-coolant accident conditions. J. Nucl. Mater. 482, 75–82 (2016).
Brachet, J.-C., Idarraga-Trujillo, I. & Flem, M. L. Early studies on Cr-Coated Zircaloy-4 as enhanced accident tolerant nuclear fuel claddings for light water reactors. J. Nucl. Mater. 517, 268–285 (2019).
Han, X. et al. A systematic study of the oxidation behavior of Cr coatings on Zry4 substrates in high temperature steam environment. Corros. Sci. 174, 108826 (2020).
Han, X., Xue, J., Peng, S. & Zhang, H. An interesting oxidation phenomenon of Cr coatings on Zry-4 substrates in high temperature steam environment. Corros. Sci. 156, 117–124 (2019).
Yang, J. et al. High temperature Cr-Zr interaction of two types of Cr-coated Zr alloys in inert gas environment. J. Nucl. Mater. 547, 152806 (2021).
Brachet, J.-C. et al. High temperature steam oxidation of chromium-coated zirconium-based alloys: kinetics and process. Corros. Sci. 167, 108537 (2020).
Wang, X. et al. Enhancement of high temperature steam oxidation resistance of Zr–1Nb alloy with ZrO2/Cr bilayer coating. Corros. Sci. 187, 109494 (2021).
Wang, X. et al. Steam oxidation behavior of ZrO2/Cr-coated pure zirconium prepared by plasma electrolytic oxidation followed by filtered cathodic vacuum arc deposition. J. Alloy. Compd. 883, 160798 (2021).
Syrtanov, M. S., Kashkarov, E. B., Abdulmenova, A. V. & Sidelev, D. V. High-temperature oxidation of Zr1Nb zirconium alloy with protective Cr/Mo coating. Surf. Coat. Technol. 439, 128459 (2022).
Huang, J., Zhao, F., Cui, X., Wang, J. & Xiong, T. Long-term oxidation behavior of silicon-aluminizing coating with an in-situ formed Ti5Si3 diffusion barrier on γ-TiAl alloy. Appl. Surf. Sci. 582, 152444 (2022).
Li, Y., Meng, F., Ge, F. & Huang, F. Improved oxidation resistance through an in-situ formed diffusion barrier: oxidation behavior of amorphous multi-component FeCrAlMoSiY-coated Zr in high-temperature steam. Corros. Sci. 189, 109566 (2021).
Zeng, S. et al. Oxidation behavior of CrSi coatings on Zry-4 substrates in 1200°C steam environment. J. Nucl. Mater. 558, 153310 (2022).
Yeom, H. et al. High temperature oxidation and microstructural evolution of cold spray chromium coatings on Zircaloy-4 in steam environments. J. Nucl. Mater. 526, 151737 (2019).
Weia, T., Zhang, R. & Yang, H. Microstructure, corrosion resistance and oxidation behavior of Cr-coatings on Zircaloy-4 prepared by vacuum arc plasma deposition. Corros. Sci. 158, 108077 (2019).
Neumann, G. & Tuijn, C. Self-diffusion and Impurity Diffusion in Pure Metals: Handbook of Experimental Data, 1st edn. (Pergamon Materials, 2009).
Tallman, D. J., Yang, J., Pan, L., Anasori, B. & Barsoum, M. W. Reactivity of Zircaloy-4 with Ti3SiC2 and Ti2AlC in the 1100–1300 °C temperature range. J. Nucl. Mater. 460, 122–129 (2015).
Barin, I. Thermochemical Data of Pure Substances, 3rd edn. (Wiley-VCH, 1995).
Steinbrück, M. et al. High-temperature oxidation and quenching of chromium-coated zirconium alloy ATF cladding tubes with and w/o pre-damage. J. Nucl. Mater. 559, 153470 (2022).
Prasad, S. & Paul, A. Growth mechanism of phases by interdiffusion and diffusion of species in the niobium–silicon system. Acta Mater. 59, 1577–1585 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants Nos. U20B2027, 91326102, 51532009, 11905194, and 52032011), Haibin Zhang is grateful to the foundation by the Recruitment Program of Global Youth Experts and the Youth Hundred Talents Project of Sichuan Province. The assistance of Haifeng Wang (SEM/EDS), Pengcheng Li, and Huahai Shen (FIB/TEM) are also gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
S.Z.: investigation, writing—original draft. J.-F.L.: formal analysis and validation. C.C., Y.M., and C.-W.Z.: validation and visualization. X.-C.H.: conceptualization, methodology, and writing—review and editing. Y.-W.B.: supervision, conceptualization, and funding acquisition. H.-B.Z.: project administration, supervision, and funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, S., Li, JF., Chen, C. et al. Improved oxidation resistance of Cr-Si coated Zircaloy with an in-situ formed Zr2Si diffusion barrier. npj Mater Degrad 7, 56 (2023). https://doi.org/10.1038/s41529-023-00373-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-023-00373-2
