
Abstract
Despite progress, 2-year pancreatic cancer survival remains dismal. We evaluated a biomarker-driven, combination/N-of-one strategy in 18 patients (advanced/metastatic pancreatic cancer) (from Molecular Tumor Board). Targeted agents administered/patient = 2.5 (median) (range, 1–4); first-line therapy (N = 5); second line, (N = 13). Comparing patients (high versus low degrees of matching) (matching score ≥50% versus <50%; reflecting number of alterations matched to targeted agents divided by number of pathogenic alterations), survival was significantly longer (hazard ratio [HR] 0.24 (95% confidence interval [CI], 0.078–0.76, P = 0.016); clinical benefit rates (CBR) (stable disease ≥6 months/partial/complete response) trended higher (45.5 vs 0.0%, P = 0.10); progression-free survival, HR, 95% CI, 0.36 (0.12–1.10) (p = 0.075). First versus ≥2nd-line therapy had higher CBRs (80.0 vs 7.7%, P = 0.008). No grade 3–4 toxicities occurred. The longest responder achieved partial remission (17.5 months) by co-targeting MEK and CDK4/6 alterations (chemotherapy-free). Therefore, genomically matched targeted agent combinations were active in these advanced pancreatic cancers. Larger prospective trials are warranted.
Similar content being viewed by others

Introduction
Despite advances in the management of advanced pancreatic cancer, outcomes remain dismal. With current systemic therapy options, 2-year overall survival (OS) is less than 10% in patients with metastatic disease, with substantial toxicity from systemic chemotherapy1,2. Frontline cytotoxic chemotherapy options rely on FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, oxaliplatin) and gemcitabine/nab-paclitaxel backbones2. There is a clear paucity of targeted therapy options.
The most common genomic alterations noted in metastatic pancreatic cancer specimens are in KRAS, TP53, CDKN2A, and SMAD4 genes. The products of these genes are believed to be difficult to inhibit with targeted therapies3. Several phase II/III trials of targeting agents (bevacizumab4, cetuximab5, trametinib6, selumetinib7, and tipifarnib8) failed to show efficacy. Importantly, however, these trials accepted all-comers and patients were not specifically selected for targeted treatment by their tumor’s genomic anomalies. While the combination of gemcitabine and the targeted epidermal growth factor receptor (EGFR) inhibitor erlotinib showed a statistically significant improvement in medial overall survival (OS) compared to gemcitabine alone9, the improvement in median OS was less than two weeks and patients had considerable skin toxicity. On the other hand, the PARP inhibitor olaparib has shown activity in pancreatic cancers harboring germline BRCA mutations10. Taken together, most previous pancreatic cancer targeted therapy trials occurred in patient populations unselected for their specific genomic alterations which, in turn, may have significantly diluted the targeted agent’s clinical efficacy11.
Several studies suggest that matching genomic alterations to targeted therapy can improve outcomes12,13,14,15. However, it is plausible that, in malignancies such as pancreatic cancer, there may be more than one genomic driver. One strategy to overcome this challenge is to treat with combinations of matched agents, with the intention to target multiple genomic alterations at once16.
Here, we describe a cohort of patients with advanced pancreatic adenocarcinoma who were treated with individualized matched therapy as part of our precision medicine program after discussion at our molecular tumor board. An illustrative case given a chemotherapy-free regimen of targeted agents highly matched to her genomic alterations achieved a partial remission lasting 17.5 months. Our observations suggest that larger cohorts of patients with advanced pancreatic cancer should be studied prospectively with a precision paradigm approach.
Results
Baseline characteristics
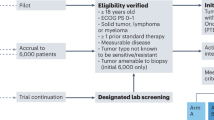
Of 6831 patients in the UCSD institutional PREDICT database, 18 patients with advanced pancreatic cancer who received at least one matched targeted therapy (immunotherapy excluded) were identified (Supplementary Fig. 1). At the time of matched therapy initiation, 88.9% (N = 16/18) had metastatic disease (while the others had advanced unresectable disease) and 72.2% (N = 13/18) of the cohort had received prior lines of therapy, predominately cytotoxic chemotherapy (Table 1). The median number of therapies prior to treatment with matched therapy was one. With regard to the genomic-sequencing data, 83.3% of patients (N = 15/18) underwent tissue NGS and, while several platforms were utilized, the majority of samples were tested with the FoundationOne CDx assay. Blood-based ctDNA NGS testing was performed in 77.8% of patients (N = 14/18), with the majority of samples run by Guardant360 ctDNA assay. Outcomes are shown in Figs. 1–3.

a PFS in 18 patients. b PFS in 5 patients who received targeted therapy as first line versus 13 patients who received it as ≥2nd line. c PFS in 11 patients with matching score ≥50% versus 7 patients with matching score <50%. CI confidence interval, PFS progression-free survival.

a OS in 18 patients. b OS in 5 patients who received targeted therapy as first line versus 13 patients who received it as ≥2nd line. c OS in 11 patients with matching score ≥50% versus 7 patients with matching score <50%. CI confidence interval, OS overall survival.

a Clinical benefit (SD ≥ 6 months/PR) and objective response rate in 18 patients. b Clinical benefit (SD ≥ 6 months/PR) and objective response rate in 5 patients who received targeted therapy as first line versus 13 patients who received it as ≥ 2nd line. c Clinical benefit (SD ≥ 6 months/PR) and objective response rate in 11 patients with matching score ≥50% versus 7 patients with matching score <50%. MS matching score, PR partial response, SD stable disease.
Patients with higher degrees of genomic matching had longer OS
There was no significant difference in age, gender, or number of patients receiving first line versus later lines of therapy in patients with matching scores <50 versus ≥50% (Supplementary Table 1).
Median OS of the cohort was 4.8 months (Fig. 2a). When stratified by line of therapy, median OS was 9.4 months versus 4.3 months (P = 0.13) for patients treated in the first line versus second line or later, respectively (Fig. 2b). (Patients treated with chemotherapy also do better in first versus second line of therapy). Median OS was 6.8 and 3.3 months (P = 0.016) for a matching score of ≥50 versus <50%, respectively (Fig. 2c).
Patients with higher degrees of genomic matching trended towards longer PFS and higher CBR
Among the cohort of 18 patients, median PFS was 1.9 months (Fig. 1a). When stratified by matching score, dichotomized by a score of ≥50 versus <50%, median PFS was 3.9 versus 1.8 months (P = 0.075) (Fig. 1c). When stratified by line of therapy in terms of treatment with matched therapy in the first line versus the second line or later, median PFS was 7.8 months versus 1.8 months (P = 0.011), respectively (Fig. 1b).
The clinical benefit rate ([CBR], SD ≥ 6 months/PR/CR) was 27.8% (5 of 18 patients). All five of these patients received regimens that were chemotherapy-free and all five had a matching score ≥50%. CBR for patients with a matching score of <50% was 0 and 45.5% in patients with a matching score ≥50% (N = 5/11) (P = 0.10) (Fig. 3c). When stratified by line of therapy, the CBR for patients treated in the first line was 80.0% (four of five patients) and 7.7% (1 of 13 patients) for patients treated in the second line or later (P = 0.008) (Fig. 3b).
Toxicity
Among the cohort, no grade 3 or 4 drug-related toxicities were noted with the matched therapy regimens when dosed according to Methods. The most common grade 1 to 2 toxicities were rash (seen with trametinib) and diarrhea (seen with erlotinib and trastuzumab and cetuximab), diarrhea (seen with trametinib) and myelosuppression (seen with palbociclib). At the doses used (see Methods), only the trametinib and everolimus combination required early discontinuation for chronic side effects (rash/mucositis).
Illustrative case among patients achieving partial response to matched therapy
Table 217,18,19 lists patients who achieved clinical benefit (CBR, defined as SD ≥ 6 months/PR/CR).
The longest responder was patient #18, a 65-year-old woman with a history of chronic obstructive pulmonary disease and hypertension who was diagnosed with de novo metastatic pancreatic adenocarcinoma with lung metastases (Fig. 4). Her tissue NGS showed KRAS G12D, KRAS G12R, CDKN2A loss exons 1–2, CDKN2B loss, SMAD4 deletion exon 11, TP53 R267W. Based on the patient’s genomic profiling, the patient was started on matched targeted therapy with the MEK inhibitor trametinib (1 mg orally daily) for SMAD4 and KRAS alterations both of which activate the MEK pathway20,21,22, the CDK4/6 inhibitor palbociclib (75 mg orally 3 weeks on, 1 week off) for CDKN2A exons 1–2 loss and CDKN2B loss which can upregulate CDK4/623, and the VEGF-A antibody bevacizumab (7.5 mg/kg intravenously every 3 weeks) for TP53, which can activate the VEGF/VEGFR pathway24,25. On matched therapy, her CA-19-9 decreased significantly (Fig. 4b) from a peak level of 349 to a nadir level of 35 (normal range, 30–42 U/mL) and scans showed ~37% regression of pancreatic and lung metastases (Fig. 4a). Her partial response lasted 17.5 months without progression of disease. However, the patient passed away due to the complications of underlying chronic lung disease. There were no serious drug-related side effects.

Sixty-four-year-old woman with pancreatic cancer with KRAS G12D, KRAS G12R, CDKN2A loss exons 1–2, CDKN2B loss, SMAD4 deletion exon 11, TP53 R267W on tissue NGS treated with palbociclib (targets CDK4/6 upregulated by CDKN2A/B loss), trametinib (targets MEK, upregulated by KRAS and SMAD4 mutations), and bevacizumab (targets VEGF, upregulated by TP53 mutations). There were no serious drug-related side effects. She achieved partial response with PFS of 17.5 months. Patient died from complications of a chronic obstructive pulmonary disease exacerbation, which was felt to be unrelated to her cancer or her therapy. At the time of death, patient was free from progression. a Serial CT scans of primary pancreatic mass. b CA-19-9 trend on therapy (reference range, 30–42 U/mL).
Discussion
There have been limited successes with the use of targeted therapy in pancreatic cancer, perhaps because only a minority of pancreatic cancer trials use a biomarker for enrolling patients26. It is clear that a biomarker-driven approach has driven advances for other cancers. Given clinical responses seen with N-of-One combination matched therapy in the tumor-agnostic I-PREDICT trial16, there was an interest in this approach in a pancreatic cancer cohort.
The population of patients evaluated in this study was diverse, and the majority of individuals had received prior lines of systemic therapy. With matched targeted therapy, there was significantly longer overall survival among patients with a high matching score, reflecting a high degree of matching, compared to those with a low degree of matching of drugs to molecular alterations. A similar trend was observed with improved PFS and CBR (SD ≥ 6 months/PR/CR). When stratified by line of matched therapy, PFS and CBR were significantly better among patients treated with matched therapy as first-line therapy compared to those treated in the second line and beyond. Similar trends were seen with overall survival, although the overall survival differences in first versus second line or greater did not reach statistical significance. There were no grade 3–4 toxicities at least possibly drug related reported with the matched therapy combinations administered. The rates of OS and PFS reported among this cohort, particularly in the first line and high matching score cohorts, are notable as they are similar to those seen with cytotoxic chemotherapy: FOLFIRINOX with median OS of 11.1 months, and nab-paclitaxel plus gemcitabine with median OS of 8.5 months1,2,27. However, without a randomized trial, these results are not comparable.
Altogether, five of 18 patients achieved clinical benefit (SD ≥ 6 months/PR/CR). These patients were treated with chemotherapy-free regimens. The key similarity among these five patients with diverse genomic alterations and therapies was a higher degree of matching of genomic alterations to targeted therapy. From this study and the larger I-PREDICT trial16, the ability to match therapy to a high proportion of detected alterations appears to be a significant factor in the efficacy of matched therapy. Of note, patient #22 (PFS = 13.6 months) was treated with trametinib monotherapy, an agent which failed to show benefit in combination with gemcitabine in a biomarker unselected population6; the molecular alterations in this patient included anomalies in GNAS, KRAS, and NF1, all of which can activate the MEK pathway28,29,30,31. Hence, this patient had multiple activating mutations in the MEK/ERK pathway, perhaps explaining their response to trametinib32. This is supported by reports of benefit with trametinib in gastrointestinal malignancies harboring GNAS alterations19.
More recently, there have been attempts at biomarker-driven clinical trials in advanced pancreatic cancer. For example, the phase III POLO trial tested the PARP inhibitor olaparib versus placebo as maintenance therapy in patients with germline BRCA1/2 alterations who achieved at least stable disease after cytotoxic chemotherapy induction. The trial showed progression-free survival (PFS) benefit with the addition of olaparib maintenance10. While the results of this trial are promising and practice changing, the utility of olaparib is limited to a particular subset of BRCA-mutant patients who have a favorable response to cytotoxic chemotherapy10.
Another example is the phase I pan-cancer trial of KRAS G12C inhibitor sotorasib enrolled ten pancreatic cancer patients whose tumors harbored KRAS G12C alterations. Stable disease was seen in six patients (60%), while four (40%) had progressive disease33. The TAPUR trial treated patients with pancreatic cancer harboring CDKN2A loss or mutation with the CDK4/6 inhibitor palbociclib, but failed to show any clinical response with palbociclib monotherapy34. As mentioned above, PARP inhibitors in germline BRCA1/2-mutated pancreatic cancer has been one targeted therapy success. The rate of germline DNA damage repair alterations in ATM, BRCA1, and BRCA2 has been noted to be as high as 10% in patients with advanced pancreatic cancer35 and these alterations confer sensitivity to PARP inhibitors. Lastly, NRG1 gene fusions in KRAS wild-type pancreatic adenocarcinoma may be a clinically meaningful target36, as neuroregulin family proteins such as NRG1 act on the EGFR receptors. Although NRG1 fusions are rare, there are reports of clinical responses to afatinib HER2/HER3 inhibition in pancreatic cancer36,37.
A previous study implemented a precision oncology approach for patients with pancreatic cancer giving targeted therapies based on patients NGS reports38. This approach reported a significant PFS benefit in patients who received a targeted therapy in addition to chemotherapy; however, there were no combination therapies given. The authors of this study reported a median of four alterations per NGS report. The previously reported study proved that it was feasible to perform NGS on pancreatic tumors and to give patients with pancreatic cancer targeted therapies and improve PFS; our study builds on such experience by also assessing matching scores and combination targeted therapies.
There are several key limitations of our study. This was a pilot study with a small group of patients; therefore, these results require prospective validation with a larger randomized cohort. Moreover, given that therapy selection is based on a patient’s unique genomic profile, the benefit of targeting a specific set of alterations inherently differs from patient to patient. Even so, the strategy of combinatorial matched therapy among a group of patients with differing combinations of alterations has been shown to be a viable strategy across many tumor types, enhanced by molecular tumor board discussions13,39,40,41. While this study excluded patients treated with checkpoint inhibitors, a recent study showed that, among pancreatic cancer patients with alterations in chromatin remodeling genes, treatment with immunotherapy was associated with response42. Given these data, immunotherapy may also have a role in matched therapy in pancreatic cancer. Subsequent lines of therapy may also influence survival, and ultimately a randomized trial is warranted. Finally, more research is needed, as many patients did not respond or responded inadequately, especially in later lines of therapy. Methodologies such as transcriptomics, immunomics, and proteomics should be explored, in order to uncover additional molecular drivers and better matched therapeutic options and to better understand resistance mechanisms in pancreatic cancer, especially in patients whose tumors are refractory to prior treatment regimens.
Matched targeted therapy may offer a more tolerable toxicity profile compared to cytotoxic chemotherapy and may be a better suited option for patients with marginal performance status or organ dysfunction who would otherwise be poor chemotherapy candidates. The results of this analysis suggest that, when genomic-directed matched therapy can achieve a high degree of matching, and especially in first-line settings, clinical outcomes can be improved, even with regimens that exclude chemotherapy. These observations support our prior reports that combinations of targeted agents, such as matched CDK4/6 inhibitors and MEK inhibitors (given when cognate pathway co-alterations such as CDKN2A/B loss and KRAS mutations are present), may have activity, even when single agents are ineffective43. The current results also reflect the need for implementation of multi-omic and functional testing for all patients with advanced pancreatic cancer, perhaps earlier in the course of the disease, to further identify actionable alterations26,44. Prospective trials of this strategy are warranted.
Methods
Patients
This was a single-center analysis of real-world patients with advanced pancreatic cancer treated with matched therapy at the University of California San Diego (UCSD) Moores Cancer Center for Personalized Cancer Therapy. The patients were analyzed according to the guidelines of the PREDICT (Profile Related Evidence Determining Individualized Cancer Therapy) protocol (NCT02478931) and any investigational interventions/therapies for which all patients gave written informed consent. Protocols were approvaed by the UCSD Internal Review Board. Patients underwent genomic profiling of tissue (somatic) and/or blood using next-generation sequencing (NGS) and were treated with targeted therapy based on their individual genomic profiling. The turnaround time for an NGS report was roughly 3–4 weeks. All patients’ genomic profiling were reviewed at a Molecular Tumor Board (MTB) where the targeted therapy regimen was suggested based on the basis of the MTB expert opinion as well as published guidelines such as OncoKB (https://www.oncokb.org/)22,39. The UCSD MTB is a tumor-agnostic (electronic and face-to-face) tumor board comprised of medical oncologists, radiation oncologists, surgeons, radiologists, pathologists, basic scientists, bioinformatics specialists, clinical study coordinators, patient navigators, and drug acquisition specialists39 that focuses on discussing therapies based on patients’ tumor multi-omic results45. However, final treatment choices were the prerogative of the physician who was managing the patient. As previously described in the I-PREDICT trial16, patients were started at ~50% of the usual dose for two-drug combinations and at ~33% of the usual dose for three-drug combinations to avoid overlapping toxicities46. Doses were escalated to tolerance by the individual oncologist. Evaluable patients had at least one follow up visit. Patients treated with immunotherapy were excluded from this analysis (Supplementary Fig. 1). Patients treated had ECOG Performance Status Scale 0–2. Targeted therapies were obtained via the MTB drug acquisition specialists through insurance approval (i.e., prior authorization approval, denial appeal approvals), patient assistance subsidy programs through the manufacturer, or as compassionate use donated from the manufacturers. Patients could also be navigated to secondary clinical trials. Individual patient toxicities were assessed on approximately a weekly basis utilizing the Common Terminology Criteria for Adverse Events (CTCAE) v3.0 toxicity scoring system.
Next-generation sequencing (NGS)
NGS was completed by commercially available clinical laboratory improvement amendment (CLIA) platforms including Foundation One (343–352 genes) (https://www.foundationmedicine.com), Caris (140 genes) (https://www.carislifesciences.com), Tempus (595 genes) (https://www.tempus.com), and a University of California San Diego institutional assay (397 genes). Although specific gene alterations analyzed differ between each assay, there is a strong degree of overlap47,48.
Blood derived circulating-tumor DNA (ctDNA) NGS analyses were done through Guardant Health (73 genes) (https://guardant360.com) and Foundation One Liquid (67–77 genes) (https://www.foundationmedicine.com)32,49,50,51. Only non-synonymous alterations that were not variants of unknown significance were analyzed in this study.
Endpoints, statistical methods, matching score, and case studies
Descriptive statistics were used to summarize the patient characteristics. Key endpoints of the study included OS, PFS, objective response rate (ORR), and CBR (defined as stable disease (SD) ≥ 6 months or partial response (PR) or complete response (CR)). OS was calculated from the time of initiation of therapy to death or last follow up. PFS was calculated from the time of initiation of targeted therapy to progression or death. First therapy after MTB was considered. OS and PFS were stratified by line of matched therapy (1st line vs 2nd line or greater) and matching score (<50 vs ≥50%). Survival analysis was done using Kaplan–Meier analysis and stratified survival curves were compared using the log-rank test. Patients still progression-free or alive at last follow up for PFS and OS, respectively, were censored on that date. ORR and progression of disease were defined by RECIST v1.1 per physician assessment52. The CBR was compared between subgroups using Fishers exact test. As previously described in detail11,16, the matching score roughly describes the proportion of targeted alterations over the total number of deleterious alterations detected; it reflects the degree to which drugs are matched to genomic alterations. Matching score was determined by investigators who were blinded to outcome at the time of calculation. Kaplan–Meier analysis and log-rank test were used to compare subgroups of patients. P-values ≤ 0.05 were considered significant. Statistical analyses were performed with SPSS version 25 software (IBM Corporation, Armonk, NY).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available in Table 2 and Supplementary Table 2. For further information please contact the corresponding authors. Next-generation sequencing was performed by the following CLIA-certified labs: Foundation Medicine, Caris Life Sciences, Tempus, and Guardant 360. Accessing sequencing data beyond what was published would require a data usage agreement with the corresponding commercial company.
Code availability
Statistical analyses were performed with SPSS version 25 software (IBM Corporation, Armonk, NY).
References
Conroy, T. et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 379, 2395–2406 (2018).
Von Hoff, D. D. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013).
Zarkavelis, G. et al. Genetic mapping of pancreatic cancer by targeted next-generation sequencing in a cohort of patients managed with nab-paclitaxel-based chemotherapy or agents targeting the EGFR axis: a retrospective analysis of the Hellenic Cooperative Oncology Group (HeCOG). ESMO Open 4, e000525 (2019).
Kindler, H. L. et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 28, 3617–3622 (2010).
Philip, P. A. et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 28, 3605–3610 (2010).
Infante, J. R. et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 50, 2072–2081 (2014).
Bodoky, G. et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest. N. Drugs 30, 1216–1223 (2012).
Van Cutsem, E. et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 22, 1430–1438 (2004).
Moore, M. J. et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 25, 1960–1966 (2007).
Golan, T. et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 381, 317–327 (2019).
Wheler, J. J. et al. Cancer therapy directed by comprehensive genomic profiling: a single center study. Cancer Res. 76, 3690–3701 (2016).
Tsimberidou, A. M. et al. Initiative for molecular profiling and advanced cancer therapy (IMPACT): an MD Anderson Precision Medicine Study. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00002 (2017).
Tsimberidou, A. M., Fountzilas, E., Nikanjam, M. & Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 86, 102019 (2020).
Tsimberidou, A. M. et al. Long-term overall survival and prognostic score predicting survival: the IMPACT study in precision medicine. J. Hematol. Oncol. 12, 145 (2019).
Massard, C. et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 7, 586–595 (2017).
Sicklick, J. K. et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat. Med. 25, 744–750 (2019).
Ardito, C. M. et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22, 304–317 (2012).
Yap, Y. S. et al. The NF1 gene revisited—from bench to bedside. Oncotarget 5, 5873–5892 (2014).
Parish, A. J. et al. GNAS, GNAQ, and GNA11 alterations in patients with diverse cancers. Cancer 124, 4080–4089 (2018).
Principe, D. R. et al. TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 36, 4336–4348 (2017).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014).
Kato, S. et al. Prognostic implications of RAS alterations in diverse malignancies and impact of targeted therapies. Int. J. Cancer 146, 3450–3460 (2020).
Witkiewicz, A. K., Knudsen, K. E., Dicker, A. P. & Knudsen, E. S. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 10, 2497–2503 (2011).
Wheler, J. J. et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: implications for targeted therapeutics. Mol. Cancer Ther. 15, 2475–2485 (2016).
Li, A. M., Boichard, A. & Kurzrock, R. Mutated TP53 is a marker of increased VEGF expression: analysis of 7525 pan-cancer tissues. Cancer Biol. Ther. 21, 95–100 (2020).
Adashek, J. J., Goloubev, A., Kato, S. & Kurzrock, R. Missing the target in cancer therapy. Nat. Cancer 2, 369–371 (2021).
Conroy, T. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825 (2011).
Ang, C. et al. Clinical benefit from trametinib in a patient with appendiceal adenocarcinoma with a GNAS R201H mutation. Case Rep. Oncol. 10, 548–552 (2017).
Wilson, C. H., McIntyre, R. E., Arends, M. J. & Adams, D. J. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene 29, 4567–4575 (2010).
Ideno, N. et al. GNAS(R201C) induces pancreatic cystic neoplasms in mice that express activated KRAS by inhibiting YAP1 signaling. Gastroenterology 155, 1593–1607.e1512 (2018).
Hattori, S. et al. Antibody against neurofibromatosis type 1 gene product reacts with a triton-insoluble GTPase activating protein toward ras p21. Biochem. Biophys. Res. Commun. 177, 83–89 (1991).
Patel, H. et al. Clinical correlates of blood-derived circulating tumor DNA in pancreatic cancer. J. Hematol. Oncol. 12, 130 (2019).
Hong, D. S. et al. CodeBreak 100: Phase I study of AMG 510, a novel KRASG12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). J. Clin. Oncol. 38, 3511–3511 (2020).
Al Baghdadi, T. et al. Palbociclib in patients with pancreatic and biliary cancer with CDKN2A alterations: results from the targeted agent and profiling utilization registry study. JCO Precis. Oncol. 3, 1–8 (2019).
Dudley, B. et al. Germline mutation prevalence in individuals with pancreatic cancer and a history of previous malignancy. Cancer 124, 1691–1700, https://doi.org/10.1002/cncr.31242 (2018).
Jones, M. R. et al. NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin. Cancer Res. 25, 4674–4681 (2019).
Laskin, J. J. et al. Afatinib as a novel potential treatment option for NRG1 fusion-positive tumors. J. Glob. Oncol. 5, 110–110 (2019).
Pishvaian, M. J. et al. Molecular profiling of patients with pancreatic cancer: initial results from the know your tumor initiative. Clin. Cancer Res. 24, 5018–5027 (2018).
Kato, S. et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 11, 4965 (2020).
Heestand, G. M. & Kurzrock, R. Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget 6, 4553–4561 (2015).
Schwaederle, M. et al. Molecular tumor board: the University of California-San Diego Moores Cancer Center experience. Oncologist 19, 631–636 (2014).
Botta, G. P. et al. SWI/SNF complex alterations as a biomarker of immunotherapy efficacy in pancreatic cancer. JCI Insight https://doi.org/10.1172/jci.insight.150453 (2021).
Kato, S. et al. Concomitant MEK and cyclin gene alterations: implications for response to targeted therapeutics. Clin. Cancer Res. 27, 2792–2797 (2021).
Kato, S. et al. Functional measurement of mitogen-activated protein kinase pathway activation predicts responsiveness of RAS-mutant cancers to MEK inhibitors. Eur. J. Cancer 149, 184–192 (2021).
Persha, H. E. et al. Osteosarcoma with cell-cycle and fibroblast growth factor genomic alterations: case report of Molecular Tumor Board combination strategy resulting in long-term exceptional response. J. Hematol. Oncol. 15, 119 (2022).
Nikanjam, M., Liu, S., Yang, J. & Kurzrock, R. Dosing three-drug combinations that include targeted anti-cancer agents: analysis of 37,763 patients. Oncologist 22, 576–584 (2017).
Weiss, G. J. et al. Evaluation and comparison of two commercially available targeted next-generation sequencing platforms to assist oncology decision making. Onco Targets Ther. 8, 959–967 (2015).
Beaubier, N. et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 10, 2384–2396 (2019).
Frampton, G. M. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031 (2013).
Odegaard, J. I. et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin. Cancer Res. 24, 3539–3549 (2018).
Papanicolau-Sengos, A. et al. Identification of targets for prostate cancer immunotherapy. Prostate 79, 498–505, https://doi.org/10.1002/pros.23756 (2019).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Acknowledgements
This study was funded in part by National Cancer Institute grant P30 CA023100 (S.K., H.P., P.T.F., and J.K.S.).
Author information
Authors and Affiliations
Contributions
J.S., S.K., and R.K. drafted the manuscript; J.S., S.K., and R.K. designed the study; J.S., S.K., G.P.B., and R.K. analyzed the data; J.S., S.K., H.P., P.T.F., G.P.B., J.K.S., and R.K. collected the data. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.K. serves as a consultant for Foundation Medicine. He receives speaker’s fee from Roche and advisory board for Pfizer. He has research funding from ACT Genomics, Sysmex, Konica Minolta and OmniSeq. J.J.A. serves on the advisory board of CureMatch, Inc. G.P.B. serves on the Advisory Board of Natera and as a consultant to TumorGen Inc. and CEND Therapeutics. H.P. worked as consultant/scientific advisory board member for Epinoma. S.K. serves as a consultant for Foundation Medicine. He receives speaker’s fee from Roche and advisory board for Pfizer. He has research funding from ACT Genomics, Sysmex, Konica Minolta and OmniSeq. J.K.S. receives research funding from Amgen Pharmaceuticals and Foundation Medicine, consultant fees from Deciphera, speaker’s fees from Deciphera, Foundation Medicine, La-Hoffman Roche, Merck, MJH Life Sciences, and QED Therapeutics. R.K. received research funding from Biological Dynamics, Boehringer Ingelheim, Debiopharm, Foundation Medicine, Genentech, Grifols, Guardant, Incyte, Konica Minolta, Medimmune, Merck Serono, Omniseq, Pfizer, Sequenom, Takeda, and TopAlliance; as well as consultant and/or speaker fees and/or advisory board for Actuate Therapeutics, AstraZeneca, Bicara Therapeutics, Biological Dynamics, Daiichi Sankyo, Inc., EISAI, EOM Pharmaceuticals, Iylon, Merck, NeoGenomics, Neomed, Pfizer, Prosperdtx, Roche, TD2/Volastra, Turning Point Therapeutics, X-Biotech; has an equity interest in CureMatch Inc., CureMetrix, and IDbyDNA; serves on the Board of CureMatch and CureMetrix, and is a co-founder of CureMatch. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shaya, J., Kato, S., Adashek, J.J. et al. Personalized matched targeted therapy in advanced pancreatic cancer: a pilot cohort analysis. npj Genom. Med. 8, 1 (2023). https://doi.org/10.1038/s41525-022-00346-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-022-00346-5
This article is cited by
-
Genetic ancestry and diagnostic yield of exome sequencing in a diverse population
npj Genomic Medicine (2024)
-
Complete response to alectinib in ALK-fusion metastatic salivary ductal carcinoma
npj Precision Oncology (2023)
-
Exploring the Potential of Compounds Isolated from Laranthus micranthus for the Treatment of Benign Prostatic Hyperplasia: Comprehensive Studies on Spectroscopic, Reactivity, and Biological Activity
Chemistry Africa (2023)
