
Abstract
Despite the explosion of interest in topological materials over the last decades, their applications remain limited due to challenges in growth and incorporation with today’s microelectronics. As a potential bridge to close this gap, we investigate the group-IV alloy Ge1–xPbx, in the Ge-rich condition using density functional theory and show that relatively low concentrations of Pb (~9.4%) can lead to a topological phase transition. Furthermore, the calculation of the Z2 invariant for both the random alloy and the alloy with short-range order (SRO) indicate that the topological phase of the material can be directly modified by the degree of SRO. These findings are understood in terms of local structural relaxation, which decreases the bandgap in the random alloy. However, in the SRO case, the mutual avoidance of Pb leads to minimal structural relaxation, alleviating strain. Our findings not only highlight the emerging importance of SRO in alloy properties but also indicate the possibility of constructing topological interfaces between materials of identical composition (and nominally identical structure). Moreover, they uncover a viable avenue toward the monolithic integration of quantum materials with today’s semiconductor industry.
Similar content being viewed by others

Introduction
Recent years have witnessed rapid progress in topological quantum physics, as it provides a dimension in solid-state physics with high promises of technological breakthroughs. These range from massless Dirac fermions of high mobility like that of graphene1,2, exotic chiral Weyl fermions3,4,5, to one-way transport without back scattering6,7, to quantum spin Hall conductance8,9,10, quantum anomalous Hall conductance11, which offers a distinct mechanism for dissipationless carrier transport12,13 aside from superconductivity, as well as topological superconductivity14,15, and consequently could serve as the physics foundation for the topological quantum devices in microelectronics16.
As a forward-looking vision, a feasible architecture to implement such microelectronics would naturally involve the integration of topological quantum material-based electronic devices16 for critical functions and Si-CMOS circuits for supporting functions, with the whole chip manufactured through the well-established microelectronic industry infrastructure for large-scale and low cost. A similar philosophy for “CMOS monolithic integration” was the original motivation behind decades-long Si-photonics17 development and has served as the major driving force to complete the whole suite of integrated photonics components, including waveguides18,19, modulators20,21, detectors22, and lasers23 all based on group-IV materials, which has led to the blooming of SiGeSn semiconductor alloy research.
This historical development track suggests the importance of the study of “Group-IV Topological Quantum Alloys” (IVTQAs) SixGeySnzPb1−x-y-z to close the gap between the available topological quantum materials (which have typically been exotic, but lesser-known materials) and silicon-based technology. While α-Sn is known to be a topological semimetal24 and can incorporate several per cent Ge25, significant challenges exist in incorporating it into Si-based devices, e.g. lattice mismatch and its tendency to transition to the β phase. To truly be compatible with Si-based technology, it is advantageous to explore the topological properties of group-IV alloys, which contain high concentrations of Si or Ge. Ironically, most binary group-IV solids form alloys, rather than compounds. The previous studies of topological properties in alloy systems showed that the concentration of the alloy components can induce topological phase transitions26,27,28,29. However, the degree of ordering in alloys and their relationship to the topological degree of freedom, should it exist, are also open questions to address. Recently, we have extensively studied the physics of short-range ordering (SRO) in Si-Ge-Sn alloys30,31,32, showing a great deal of tunability of the bandgap and band structure as functions of the degree and type of order. This SRO effect is expected to be even larger in Pb-containing alloys (due to the increased atomic size mismatch). This and the pronounced spin-orbit coupling of Pb suggest SixGeyPb1−x-y systems could pave the way to viable IVTQAs and establish a connection between the topological physics and SRO.
In this work, through first-principles density functional theory (DFT), we examine SRO in Ge1−xPbx alloys and the role it plays in the electronic and topological properties of the material. Monte Carlo (MC) sampling of Ge1−xPbx alloy configurations reveals a strong tendency of Pb to avoid each other in both first and second atomic shells, leading to SRO, which exhibits substantially less Pb clustering than a random solid solution. This SRO has a significant effect on the electronic properties, exhibiting a substantially larger bandgap than the random alloy, which is understood in terms of decreased band broadening due to less structural relaxation in the more homogenous environment of SRO. Moreover, while the bandgap of both the SRO and random alloy reduces with increasing Pb content, the smaller gap of the random alloy leads to band inversion with an accompanying topological phase transition at lower Pb concentrations than with SRO. We show that ~9% Pb content, the random alloy and SRO are associated with distinct topological phases, namely, a strong topological insulator and a normal insulator, respectively. This indicates that controlling SRO may prove to be a powerful platform for harnessing topological properties, enabling the construction of interfaces between ordinary and topological phases of the same material system without introducing either lattice or chemical mismatch at the interface.
Results
SRO in Ge1−xPbx alloy
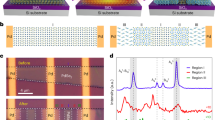
The ensemble-averaged, equilibrium structure of Ge1−xPbx alloy is obtained by combining Metropolis MC/DFT to sample alloy’s configurations according to their Boltzmann factors (see Methods for details). As shown in Fig. 1, the calculated SRO parameter \({\alpha }_{{ij}}^{m}\) for atomic pair i−j in the mth atomic shell (see Methods for details) clearly indicates a strong SRO behavior in an equilibrium Ge1−xPbx alloy at room temperature. Specifically, the SRO in Ge1−xPbx is reflected by the substantially reduced numbers of both first and second Pb-Pb nearest neighbors with respect to those of a truly random distribution. It is noted that this deviation from random distribution is only of short range, as neither Ge nor Pb is found to preferentially occupy a particular lattice site of diamond cubic structure. The depletion of Pb-Pb pairs in the first and second shells is found largely compensated by the enhancement of the third Pb-Pb nearest neighbors. Since this Pb-Pb repulsion resembles the SRO behaviors previously identified in SiSn alloys and SRO was found to substantially modify the electronic band gaps of SiSn and GeSn alloys30,31, we expect SRO to yield a significant change in the underlying electronic band structures of Ge1–xPbx alloys.

Calculated SRO parameter \({\alpha }_{{\rm{PbPb}}}^{m}\) for the 1st (m = 1), 2nd (m = 2), and 3rd (m = 3) Pb-Pb nearest neighbors in Ge1–xPbx alloy as a composition of Pb composition. A random alloy has an SRO parameter of zero (magenta dashed line). A positive/negative \({\alpha }_{{\rm{PbPb}}}^{m}\) indicates a depletion/enhancement of Pb-Pb pairs of the mth nearest neighbors, respectively.
Band structure and topological phase of Ge1–xPbx alloy
In order to examine how the band structure evolves with increased Pb content and the role of SRO on the electronic properties of the alloy, we randomly select configurations from our MC study which have a high statistical weight in order to represent the SRO alloy at a particular Pb concentration. These are then contrasted with special quasi-random structures (SQS)33, which represent the random solid solution. As bulk Ge is a semiconductor and Pb is metallic, the incorporation of Pb into Ge naturally tends to shrink the bandgap with increasing Pb content, evolving from the Ge band structure to Pb band structure, as the Pb concentration increases from 0 to 100%.
Figure 2 shows the band structures calculated for Pb concentrations in the vicinity of the gap closure, namely, 6.25%, 9.375%, and 12.5%, for the SRO structure (a–c) and for the SQS structure (d–f). At 6.25% Pb concentration, while the bandgap of both SRO (0.33 eV) and SQS (0.18 eV) have been reduced substantially from the bandgap of pure Ge, calculated to be 0.7 eV at the Meta-GGA level34, the SRO gap is significantly larger. Despite this, the band structures are qualitatively similar and exhibit the breaking of the three-fold degeneracy of the 3p derived Ge valence band maximum (VBM) and the conduction band minimum (CBM) is associated with Ge 4 s antibonding orbitals (s*). As the Pb concentration is increased to 9.375%, however, the orbital character of the band structures for SRO (Fig. 2b) and SQS (Fig. 2e) become qualitatively different. While the SRO alloy band structure maintains the same character as bulk Ge, there is inversion of the bands of the random alloy (in the vicinity of Γ), where the CBM takes on p character and the state below the VBM takes on s* character. When Pb content is increased further to 12.5%, as shown in Fig. 1c, f, the s*-orbitals of both SRO and SQS continue to move downward relative to the p-orbitals, leading to band inversion, even in the case of SRO. For the case of random alloy, these larger Pb concentrations lead to multiple conduction band states crossing with the VBM, resulting in a metal. These results indicate that increasing the Pb concentration continuously lowers the conduction bands relative to the valence bands, first causing a shrinking of the bandgap, then an inverted bandgap, and finally a metallic state. As the SRO alloy has a larger gap relative to the random alloy, band inversion occurs at lower Pb concentrations in the random alloy than in the SRO alloy. Considering the variation of alloy structure, the band structure calculations are also performed in other randomly picked SRO and Random configurations at 6.25%, 9.375%, and 12.5%, Pb concentrations. All these results are in agreement with the results shown in Fig. 2 [see Supplementary Fig. 1]. The average value of the topological gap in SRO alloy with 12.5% Pb and random alloy with 9.375% Pb are 42 meV and 46 meV, respectively. Here, we note that while the strong spin-orbit coupling (SOC) of Pb also contributes to band inversion by pushing two of the p-states upward, the magnitude of this effect is quite similar for both the SRO and SQS configurations, as detailed in the Supplementary Discussion and Supplementary Figs. 2–4.

Band structure of \({\rm{G}}{{\rm{e}}}_{{\it{1}}-{{x}}}{\rm{P}}{{\rm{b}}}_{{{x}}}\) alloys with degree of projection onto Ge s*-orbitals and Ge p-orbitals indicated by size of the red circles and green triangles, respectively. Pb content is increasing from left to right, with x = 0.0625, 0.09375, and 0.125, respectively. a–c correspond to the SRO alloy, and the bottom d–f correspond to the random alloy (SQS).
To investigate if the band inversion is associated with a change in topological properties, topological invariants were calculated based on the hybrid Wannier charge center (HWCC) method, which can be used in both semiconductors and metallic systems35. The alloys still have the time-reversal symmetry, so the net Chern number is always 0 and the Z2 invariant36,37 is used to determine the topological feature. The classification of the 3D topological phases relays on the Z2 invariant for 3D systems \({v}_{0};\left({v}_{1},{v}_{2},{v}_{3}\right)\), which are defined by the 2D Z2 invariant in time-reversal invariant planes in first Brillouin zone (BZ)37. For trivial insulators, v0 and vi are all zero. If v0 = 0, but any of vi equals to 1, the system is called weak topological insulator. If v0 = 1, the system is called a strong topological insulator. The calculated topological properties for the configurations of the Ge1−xPbx alloy are listed in Table 1 (flows of the HWCCs are shown in Supplementary Fig. 5). These results are consistent with the band inversion leading to a strong topological insulator state. Due to the delay of band crossing, the Ge0.90625Pb0.09375 with SRO structure is topologically trivial, while the random alloy with the same Pb concentration is topologically non-trivial. After increasing the Pb concentration to 12.5%, the SRO structure becomes a topological insulator, and the random alloy becomes metal due to further band crossing. These results point toward the possibility of controlling the topological properties of materials and designing interfaces based on the ability to manipulate the SRO of the alloy. For example, for a Pb concentration of 9.375%, these results predict the emergence of topologically protected interfacial states at the boundary between regions of SRO and random alloy.
Role of SRO on the band structure
In Ge1−xPbx alloys, the Pb-Pb bonds are longer than GePb bonds, which are, in turn, longer than Ge-Ge bonds. Due to these bond length differences, Pb atoms tend to push surrounding atoms outward, leading to distortions. While there are both local atomic distortions and overall supercell distortions, we note the supercell distortion is very minor with changes in the lattice vectors (~0.2%), and they have negligible effect on the electronic structure. The local distortions are quantified by comparing the relaxed structures (for a given Pb concentration in either the SQS or SRO configuration) to the same Pb distribution in which each atom is at its ideal position in the diamond lattice. The magnitude of the displacement, shown in Fig. 3a, indicates two clear peaks for both SQS and SRO. The peak at larger displacements is associated with Ge, which is the nearest neighbor to Pb, while the peak with the smaller distortion is primarily associated with Ge, which is further away. Here, it is also seen that the SQS structure has, on average, larger distortions than the SRO structure.

The distribution of a atomic displacements, b bond angles, and c q6 parameters associated with atomic distortion in SQS (in red) and SRO (in green) configurations for a Pb concentration of 9.375%.
Due to the stiffness of the covalent bonding, the larger distortion in SQS primarily manifests as altered bond angles with neighboring atoms. The bond angle distortions with respect to the ideal diamond bond angle (109.5°) of SQS and SRO are shown in Fig. 3b. The most striking differences between SRO and SQS are that the distribution of bond angle distortions is much broader in SQS, reflecting a larger inhomogeneity in its local environment. This inhomogeneity is also quite clear from the sixth-order Steinhardt-type parameter, q6, which measures the degree to which the first coordination shell around an atom is ordered38,39, shown in Fig. 3c. The advantage of using this spherical harmonics-based approach is that it provides a single quantity to represent the degree of distortion for each atom, equaling 1 for an ideal tetrahedron geometry and decreasing as any of the bond angles to neighboring atoms departs from their ideal values of 109.5°. The relative homogeneity of SRO can be traced back to the tendency of Pb atoms to repel each other to relieve stain, leading to a more uniform spacing of Pb atoms. This can be contrasted to SQS, where the random Pb distribution results in higher concentrations of Pb-Pb NNs and Pb-Pb second NNs. As the heterogeneous spatial distribution of Pb does not minimize the strain in SQS, greater local distortions are thus induced.
The band structure of the relaxed and unrelaxed lattice of SQS and SRO configurations is shown in Fig. 4a, b. Despite the difference in the spatial distribution of Pb, if the atomic coordinates are unrelaxed (at the ideal diamond sites), the band structures of SQS and SRO are almost identical. However, after atomic relaxation, there is pronounced splitting of the SQS valence band, leading to a significantly smaller bandgap than the SRO configuration. This can be simply understood in that large bond angle distortions associated with SQS weaken the coupling between atomic states, leading to a higher energy bonding and lower energy antibonding state. This understanding is further bolstered by examining the charge density associated with the VBM, shown in Fig. 4c, d. While the charge density of the VBM for the SRO is quite delocalized, even after atomic relaxation, for SQS it resembles a gap state with much greater spatial localization. As shown in Fig. 4d, the charge density of the VBM is primarily localized in the region of atoms which have a greater degree of local distortion, as indicated by their q6 parameter.

The band structure for relaxed (black) and unrelaxed (red) lattices in a SRO and b SQS configurations without SOC with 9.375% Pb. (c) and (d) show an isosurface of the charge distribution of the VBM for SRO and SQS, respectively. The atoms are colored based on their q6 parameter with darker shade indicating more bond angle distortion.
Discussion
In this study, we confirm the vision and concept of Group-IV Topological Quantum Alloys and show that a relatively small concentration of Pb in Ge can lead to the topological insulating phase. Through systematic calculations of the topological invariant for both random Ge1−xPbx alloys and those exhibiting SRO over a range of Pb concentrations, we have shown the topological properties of Ge1−xPbx alloy can be significantly modified by varying the local arrangement of alloying atoms. The underlying mechanism for such dependence is attributed to local lattice distortions controlled by local ordering of atoms: A random distribution of alloying atoms leads to rather inhomogeneous and large local lattice distortions, whereas SRO yields a more optimized arrangement of alloying atoms that greatly minimizes and homogenizes local distortions. The significant variation in VBM with local lattice distortion thus enables a drastic change in electronic band structures and even band topology due to the variation in atomic order.
In a broader sense, this finding reinforces the emerging recognition of the significance of local atomic order in alloys: Beneath the nominal randomness of lattice site occupation in an alloy, there can exist layers of subtlety in the distribution that may profoundly change the properties of alloys. As a result, composition and crystalline structure alone are often insufficient to unequivocally determine alloy’s properties. The distribution of alloying atoms within a short distance needs to be considered to account for the variations in the properties. In this regard, SRO has been recently shown to fundamentally impact mechanical properties40,41, thermoelectric figure of merit42,43, and ion transport44 in multi-principal alloys. The identified variations in band topology induced by SRO contribute to another dimension of property modulation enabled by the ordering in alloys.
Our findings also open an opportunity for harnessing topological properties based on the group-IV alloy platform. Both the ordinary and topological phases can be present in one alloy with the same alloy composition and crystal structure, with the only deciding factor being how alloying atoms distribute themselves within the short distance in the lattice. While the topological gap (~45 meV) is relatively small, it may still allow for room-temperature operation. Furthermore, as the interface between differently ordered phases is both lattice and chemically matched, the resulting Dirac interface state is expected to be clean of defect states. This provides true surface-like topological states within the chemically protected bulk region, which have the potential to be patterned for desired applications. More broadly, such material systems could advance the development of the integration of quantum devices, such as topological lasers, thermoelectric devices, and topological spintronics devices, with conventional electronics, in contrast to other materials platforms, which are still very challenging.
Methods
Monte Carlo sampling
To obtain the ensemble average of alloy structures, we employ Metropolis MC method to sample alloy’s configurational space. From a given alloy configuration i, a pair of Ge and Pb atoms are randomly selected and swapped to create a new configuration j, which then undergoes a full relaxation to obtain its energy Ej. This new configuration is then accepted by the probability \(\min (1,\exp (-({E}_{j}-{E}_{i})/{k}_{B}T))\), where Ei is the energy of configuration i, kB is the Boltzmann constant, and T is the temperature (300 K in this study). For each alloy composition, MC sampling starts with a random alloy configuration and proceeds with ~2000 MC moves. The first 300 configurations are excluded from the ensemble average to account for equilibration.
The total energy calculation is carried out using the Vienna Ab initio Simulation Package45 based on the projector augmented wave method. Local density functional (LDA)46 is employed for the exchange-correlation functional, which has been shown to yield the best agreement with an experiment on pure Ge for geometry optimization (e.g., ~0.2% offset when compared with the experiment). A supercell containing 128 atoms is generated by replicating the primitive cell of the diamond cubic structure four times along each dimension. An energy cutoff of 300 eV and a 2 × 2 × 2 Monkhorst-Pack k-points grid are chosen for structural relaxation, combined with the convergence criteria of 10−4 and 10−3 eV for electronic and ionic relaxations, respectively. The choice of all these parameters has been carefully examined to ensure the total energy difference between two configurations i and j (\(\varDelta E={E}_{j}-{E}_{i}\)), which plays a crucial role in MC sampling, is well converged.
SRO parameters
The SRO parameter is defined as \({\alpha }_{{ij}}^{m}=1-\frac{{p}_{{ij}}^{m}}{{c}_{j}\,}\), where cj is the concentration of element j and \({p}_{{ij}}^{m}\) is the probability of finding element j in the mth shell surrounding element i. According to this definition, an \({\alpha }_{{ij}}^{m}=0\) corresponds to random alloy; an \({\alpha }_{{ij}}^{m}\in [\mathrm{0,1}]\) means a depletion of i, j pairs, with \({\alpha }_{{ij}}^{m}=1\) meaning a complete depletion; and an \({\alpha }_{{ij}}^{m} \,<\, 0\) indicates a preference of i, j pairs. Note that \({\alpha }_{{ij}}^{m}\) is different but related to the commonly adopted Warren-Cowley SRO parameter47.
Band structure and topological invariant calculations
Based on the structures determined from LDA, the band structures are calculated using the VASP code45. Meta-GGA is functional with modified Becke-Johnson (MBJ) potential34,48 is used to correctly describe the bandgap. The energy cutoff of 230 eV and a 2 × 2 × 2 Monkhorst-Pack k-points grid are applied. The validity of the approach is confirmed by its agreement with experiment and higher-level theory. For bulk Ge, for example, the predicted Meta-GGA gap is 0.72 eV versus the experimental gap of 0.74 eV30,34. For GePb alloys, there is no experimental data, but we have tested our method by GW many-body perturbation theory49. For the SRO structure in the Ge 0.9375Pb 0.0625 alloy, for example, the gap at Γ is 0.34 eV in META-GGA and 0.36 eV in GW. The Z2-invariant calculation is performed using Z2pack35,50, based on tracking HWCC for all occupied states. The HWCC is calculated from the overlap matrices computed by the Wannier90 library51, which uses the Bloch wavefunctions from VASP as input. The Z2 invariant is determined from the flow of the HWCC calculated using the Meta-GGA DFT calculation as input and using the maximum movement of an HWCC of 1% of the lattice constant to guarantee the continuity of HWCC flowing. Starting with 7 k-points along the path for overlap matrices calculation, the number of k-points is increased with a step of 2 until the change in Wannier charge centre positions converges to 1% of the lattice constant.
Data availability
The datasets generated and/or analyzed during the current study are available in the NoMaD repository, https://nomad-lab.eu/prod/v1/gui/search/entries/entry/id/w-juiuTpbAsTpzVo9oJVw6SDDjAb.
Code availability
The code used to perform DFT calculations is proprietary and can be licensed at VASP https://www.vasp.at/. Codes used to determine Wannier charge centers and calculate topological properties are open source and freely available at Wannier90 http://www.wannier.org/ and Z2pack https://z2pack.greschd.ch/en/latest/.
References
Novoselov, K. S. et al. Two-dimensional gas of massless Dirac fermions in graphene. nature 438, 197–200 (2005).
Castro Neto, A. H., Guinea, F., Peres, N. M. R., Novoselov, K. S. & Geim, A. K. The electronic properties of graphene. Rev. Mod. Phys. 81, 109 (2009).
Huang, S.-M. et al. A Weyl Fermion semimetal with surface Fermi arcs in the transition metal monopnictide TaAs class. Nat. Commun. 6, 7373 (2015).
Xu, N. et al. Observation of Weyl nodes and Fermi arcs in tantalum phosphide. Nat. Commun. 7, 11006 (2016).
Lv, B. Q. et al. Experimental discovery of Weyl semimetal TaAs. Phys. Rev. X 5, 031013 (2015).
Wang, Z., Chong, Y., Joannopoulos, J. D. & Soljačić, M. Observation of unidirectional backscattering-immune topological electromagnetic states. Nature 461, 772–775 (2009).
Roushan, P. et al. Topological surface states protected from backscattering by chiral spin texture. Nature 460, 1106–1109 (2009).
Sinova, J. et al. Universal intrinsic spin Hall effect. Phys. Rev. Lett. 92, 126603 (2004).
Bernevig, B. A. & Zhang, S.-C. Quantum spin Hall effect. Phys. Rev. Lett. 96, 106802 (2006).
Qian, X., Liu, J., Fu, L. & Li, J. Quantum spin Hall effect in two-dimensional transition metal dichalcogenides. Science 346, 1344–1347 (2014).
Chang, C.-Z. et al. Experimental observation of the quantum anomalous Hall effect in a magnetic topological insulator. Science 340, 167–170 (2013).
Chang, C.-Z. et al. Zero-field dissipationless chiral edge transport and the nature of dissipation in the quantum anomalous Hall state. Phys. Rev. Lett. 115, 057206 (2015).
Ying, Z. et al. Experimental evidence for dissipationless transport of the chiral edge state of the high-field Chern insulator in MnBi2Te4 nanodevices. Phys. Rev. B 105, 085412 (2022).
Alicea, J. New directions in the pursuit of Majorana fermions in solid-state systems. Rep. Prog. Phys. 75, 076501 (2012).
Sato, M. & Ando, Y. Topological superconductors: a review. Rep. Prog. Phys. 80, 076501 (2017).
Jin, K.-H., Jiang, W., Sethi, G. & Liu, F. Topological quantum devices: a review. Nanoscale 15, 12787–12817 (2023).
Soref, R. The past, present, and future of silicon photonics. IEEE J. Sel. Top. Quantum Electron. 12, 1678–1687 (2006).
Heck, M. J. R., Bauters, J. F., Davenport, M. L., Spencer, D. T. & Bowers, J. E. Ultra-low loss waveguide platform and its integration with silicon photonics. Laser Photon. Rev. 8, 667–686 (2014).
Su, Y., Zhang, Y., Qiu, C., Guo, X. & Sun, L. Silicon photonic platform for passive waveguide devices: materials, fabrication, and applications. Adv. Mater. Technol. 5, 1901153 (2020).
Reed, G. T., Mashanovich, G., Gardes, F. Y. & Thomson, D. J. Silicon optical modulators. Nat. Photonics 4, 518–526 (2010).
Rahim, A. et al. Taking silicon photonics modulators to a higher performance level: state-of-the-art and a review of new technologies. Adv. Photonics 3, 024003 (2021).
Michel, J., Liu, J. & Kimerling, L. C. High-performance Ge-on-Si photodetectors. Nat. Photonics 4, 527–534 (2010).
Liang, D. & Bowers, J. E. Recent progress in lasers on silicon. Nat. Photonics 4, 511–517 (2010).
Barfuss, A. et al. Elemental topological insulator with tunable Fermi level: strained α-Sn on InSb (001). Phys. Rev. Lett. 111, 157205 (2013).
Liu, S. et al. Growth of α-Sn on silicon by a reversed β-Sn to α-Sn phase transformation for quantum material integration. Commun. Mater. 3, 17 (2022).
Fu, L. & Kane, C. L. Topological insulators with inversion symmetry. Phys. Rev. B 76, 045302 (2007).
Xu, S.-Y. et al. Observation of a topological crystalline insulator phase and topological phase transition in Pb1-xSnxTe. Nat. Commun. 3, 1192 (2012).
Huang, H., Jin, K.-H. & Liu, F. Alloy engineering of topological semimetal phase transition in MgTa2-xNbxN3. Phys. Rev. Lett. 120, 136403 (2018).
Kim, D. & Liu, F. Topological alloy engineering and locally linearized gap dependence on concentration. Phys. Rev. B 106, 085105 (2022).
Cao, B., Chen, S., Jin, X., Liu, J. & Li, T. Short-range order in GeSn alloy. ACS Appl. Mater. Interfaces 12, 57245–57253 (2020).
Jin, X., Chen, S. & Li, T. Short-range order in SiSn alloy enriched by second-nearest-neighbor repulsion. Phys. Rev. Mater. 5, 104606 (2021).
Jin, X., Chen, S. & Li, T. Coexistence of two types of short-range order in Si–Ge–Sn medium-entropy alloys. Commun. Mater. 3, 66 (2022).
Wei, S.-H., Ferreira, L. G., Bernard, J. E. & Zunger, A. Electronic properties of random alloys: Special quasirandom structures. Phys. Rev. B 42, 9622 (1990).
Tran, F. & Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401 (2009).
Soluyanov, A. A. & Vanderbilt, D. Computing topological invariants without inversion symmetry. Phys. Rev. B 83, 235401 (2011).
Kane, C. L. & Mele, E. J. Z2 topological order and the quantum spin Hall effect. Phys. Rev. Lett. 95, 146802 (2005).
Fu, L., Kane, C. L. & Mele, E. J. Topological insulators in three dimensions. Phys. Rev. Lett. 98, 106803 (2007).
Li, T., Donadio, D. & Galli, G. Nucleation of tetrahedral solids: a molecular dynamics study of supercooled liquid silicon. J. Chem. Phys. 131, 224519 (2009).
Li, T., Donadio, D., Russo, G. & Galli, G. Homogeneous ice nucleation from supercooled water. Phys. Chem. Chem. Phys. 13, 19807–19813 (2011).
Zhang, R. et al. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature 581, 283–287 (2020).
Wu, Y. et al. Short-range ordering and its effects on mechanical properties of high-entropy alloys. J. Mater. Sci. Technol. 62, 214–220 (2021).
Jiang, B. et al. High-entropy-stabilized chalcogenides with high thermoelectric performance. Science 371, 830–834 (2021).
Roychowdhury, S. et al. Enhanced atomic ordering leads to high thermoelectric performance in AgSbTe2. Science 371, 722–727 (2021).
Ji, H. et al. Hidden structural and chemical order controls lithium transport in cation-disordered oxides for rechargeable batteries. Nat. Commun. 10, 592 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Ceperley, D. M. & Alder, B. J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 45, 566 (1980).
Cowley, J. M. An approximate theory of order in alloys. Phys. Rev. 77, 669–675 (1950).
Becke, A. D. & Johnson, E. R. A simple effective potential for exchange. J. Chem. Phys. 124, 22 (2006).
Shishkin, M. & Kresse, G. Implementation and performance of the frequency-dependent GW method within the PAW framework. Phys. Rev. B 74, 035101 (2006).
Gresch, D. et al. Z2Pack: numerical implementation of hybrid Wannier centers for identifying topological materials. Phys. Rev. B 95, 075146 (2017).
Mostofi, A. A. et al. wannier90: a tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 178, 685–699 (2008).
Acknowledgements
T.S.Li and S.-Q.Yu would like to thank professor Gregory Salamo and professor Jin Hu at the University of Arkansas for their fruitful early phase discussions to shape the vision of “Group-IV Topological Quantum Alloys”. They would also like to thank the encouragement from Prof. Jifeng Liu at Dartmouth College and Drs. Ezra Bussmann, Tzu-Ming Lu, and Shashank Misra at Sandia to identify the significant scientific merits of this vision. This work was supported by µ-ATOMS, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award DE-SC0023412. We also acknowledge supercomputer time provided by NERSC under DOE Contract No. DEAC02-05CH11231, time provided on TAMU FASTER and Purdue Anvil made available by ACCESS through allocation TG-DMR180114, and the CCI at RPI.
Author information
Authors and Affiliations
Contributions
T.L. and S.Y. conceived the initial idea of Group-IV Topological Quantum Alloys. Under the guidance of T.L., S.C. generated the SQS and SRO structures and performed the initial band structure calculation to demonstrate band inversion, and J.X. wrote the MC sampling code, calculated the SRO parameter, and performed q6 structural analysis. D.W. and S.B.Z. designed the study of the topological properties, analyzed the data, and guided Y.L. Y.L. to perform first-principles calculations of topological properties and developed an understanding of how SRO affects the topological properties. The manuscript was drafted by Y.L. and D.W. with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, Y., Chen, S., Jin, X. et al. Group IV topological quantum alloy and the role of short-range order: the case of Ge-rich Ge1–xPbx. npj Comput Mater 10, 82 (2024). https://doi.org/10.1038/s41524-024-01271-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-024-01271-0
