
Abstract
Breast cancer is the most commonly diagnosed cancer in the USA. Although advances in treatment over the past several decades have significantly improved the outlook for this disease, most women who are diagnosed with estrogen receptor positive disease remain at risk of metastatic relapse for the remainder of their life. The cellular source of late relapse in these patients is thought to be disseminated tumor cells that reactivate after a long period of dormancy. The biology of these dormant cells and their natural history over a patient’s lifetime is largely unclear. We posit that research on tumor dormancy has been significantly limited by the lack of clinically relevant models. This review will discuss existing dormancy models, gaps in biological understanding, and propose criteria for future models to enhance their clinical relevance.
Similar content being viewed by others

The clinical problem of breast cancer dormancy
Nearly 300,000 new breast cancer cases are diagnosed each year in the USA1. Although breast cancer affects many women, those diagnosed with disease localized to the breast have a very favorable prognosis, with a ten-year survival rate above 95%1. However, the ten-year survival rate does not fully capture the long-term outcomes for these women. In particular, in women whose breast cancers that express the estrogen receptor (ER+), late relapses are common. This contrasts with those with estrogen receptor negative (ER-) cancer where the risk of recurrence peaks in year 2 post diagnosis (Fig. 1a). Late relapse in women with ER+ breast cancer is thought to result from the activation of dormant tumor cells at metastatic sites such as the bone marrow2; and about 20% of women with ER+ disease have a recurrence 15 years or more after initial diagnosis3. Even more striking is that the relative risk of recurrence is constant over at least 20 years, and this risk is proportional to the initial nodal status (Fig. 1b). These data indicate that women with ER+ breast cancer face a lifelong risk of recurrence. The central role of tumor dormancy in mediating these late recurrences highlights the importance of developing effective approaches to target dormant tumor cells. The development of such approaches will depend on a more thorough understanding of the biology of dormancy, including cell-intrinsic and microenvironmental factors that maintain dormancy or that facilitate escape from dormancy. Research focused on tumor dormancy has been limited by the lack of clinically relevant models. This article will review existing in vitro and mouse models of breast cancer dormancy, highlight their advantages and limitations, identify gaps in the current knowledge, and suggest criteria to enhance clinical relevance for future studies.

Disseminated tumor cells: Clinical samples and systems biology
At present, there is no widely used method to monitor the dormant state (also called minimal residual disease) or to predict the probability of late recurrence in women with newly diagnosed breast cancer. Several multigene expression signatures (Oncotype DX, MammaPrint, Genomic Grade Index) are used clinically to stratify patients based on the risk of recurrence and to inform the use of chemotherapy in early-stage breast cancer4,5. Although intrinsic molecular subtypes identified by these assays are predictive of proliferative potential, the risk of early recurrence, and treatment efficacy, they cannot discern the risk of late recurrence. There have been reports that certain molecular signatures of primary tumors can distinguish between early and late recurrence risk, but these have yet to enter clinical practice6,7,8. Furthermore, the minimal overlap between these gene lists, suggests the possibility of overfitting and lack of universal biological relevance9.
A more direct assay predictive of tumor recurrence involves the serial monitoring of circulating tumor cells (CTCs) or cell-free DNA in serum samples. CTCs are shed from the primary tumor and/or occult metastatic sites into the bloodstream and can be identified in a simple blood draw. The persistence or appearance of CTCs following adjuvant therapy is associated with significantly lower disease-free and overall survival. The 4-year disease-free survival rate for CTC-negative patients was 92.9%, whereas those with more than 5 CTCs in 30 mL of blood had a 71.9% 4-year disease-free survival10. CTCs have also been found in patients who were disease free up to 22 years after mastectomy11. This implies that there is a source of tumor cells that are shed into the circulation even when a patient is asymptomatic. Similarly, circulating cell-free DNA with copy number variations associated with the primary tumor have been detected 12 years after diagnosis despite no other evidence of disease12. As with CTCs, detection of cell-free DNA is predictive of overall survival and progression-free survival13.
Another clinically accessible source of tumor cells in patients with minimal residual disease is the bone marrow. Tumor cells that have disseminated to an organ such as bone marrow are known as disseminated tumor cells (DTCs). DTCs can be detected in bone marrow biopsies or bone marrow aspirates by virtue of their expression of epithelial markers such as EpCAM and pan-cytokeratin. Approximately 60% of ER+ patients with metastatic relapse exhibit bone metastasis as the first site of disease recurrence14. The propensity of patients with ER+ disease to suffer bone metastasis, as well as the presence of DTCs in the bone marrow of breast cancer patients, suggests that DTCs in the bone may be the major source of CTCs and cell-free DNA shedding and may cause late relapse. When one considers the sampling error inherent in taking 1–4 mL of an organ of approximately 3 L total volume15 (0.03–0.12%), it is likely that a much higher percentage of patients with ER+ breast cancer harbor DTCs in the bone marrow.
Approximately 30% of patients with ductal carcinoma in situ (DCIS) harbor detectable DTCs in the bone marrow16, and these patients have a much higher risk of mortality than the general population which persists for at least 15 years following DCIS diagnosis17. The number of DTCs and their mutational burden are very similar between patients with ductal carcinoma in situ and with invasive breast cancer18. These observations suggest that bone marrow dissemination is an early event in breast carcinogenesis, a finding with important implications for early detection and screening. Although detection of DTCs is associated with an increased risk of recurrence, these cells may remain dormant for long periods: In one study, 30% of patients with detectable DTCs at diagnosis did not suffer cancer recurrence within 15 years19.
There are two hypothesized types of cancer dormancy. The cellular dormancy model posits that tumor cells exit the cell cycle and remain in a growth-arrested state. An alternative hypothesis, referred to as tumor mass dormancy, holds that dormancy occurs when the proliferation rate of DTCs is countered by an equivalent rate of cell loss that results in a stable microscopic tumor mass. Support for the first model is provided by several studies that have demonstrated the quiescent nature of DTCs20. In contrast, there is little clinical evidence for tumor mass dormancy. Defining the underlying characteristics of tumor dormancy in patients is critical to developing pre-clinical models that accurately reflect this biology.
From a systems biology perspective, dormancy is a phenotypic choice. In order to become dormant and survive in the foreign microenvironment of the metastatic niche, cells must have plasticity; they cannot be irreversibly wedded to a given physiological state but instead must undergo dynamic transitions. Such plastic cells, including cancer stem cells (CSCs), exist in primary tumors21 and may arise via de-differentiation during the epithelial-mesenchymal transition (EMT)22. Recent work, utilizing computational modeling and direct experimentation, has demonstrated a nuanced connection between EMT status and plasticity. Cells that exhibit hybrid epithelial/mesenchymal (E/M) features23 have a higher probability of attaining stemness24,25,26,27, relative to cells exhibiting pronounced mesenchymal features.
p38 kinase and p21 function have been implicated in the cellular dormancy program, p21 is required to maintain relevant classes of stem cells, and mathematical models of the cell cycle predict that inhibitors such as p21 can block proliferation28,29. There is also significant literature on the analysis of circuits that control stemness versus differentiation30. Various investigators have formulated network models of drug resistance, for example, to PI3K inhibitors in breast cancer31. Finally, there have been major advances in the computational modeling of EMT and how it couples to factors that control stemness24,32. Somewhat surprisingly, there has been little effort to put these pieces together to create a quantitative picture of how these circuits interact and how they collectively respond to the chemical and mechanical microenvironment. Thus, to date, computational studies have not adequately addressed the most critical questions facing the dormancy field, such as the key differences between cell autonomous processes and key microenvironmental signals that distinguish between early metastasis or long-term dormancy with the possibility of recurrence. This is a key area for further research that will complement and inform experimental approaches to understand dormancy.
Dormancy, cancer stem cells, and metabolic plasticity
The functional definition of a CSC is a cell capable of tumor initiation and generation of functional heterogeneous cell populations. There is substantial evidence that breast cancers display a hierarchical cellular organization driven by CSCs33. CSCs also drive tumor metastasis and contribute to treatment resistance33. Just as normal tissue stem cells are tightly regulated by their microenvironment or “niche”, CSCs that are disseminated in distant tissues may be regulated by signals originating from their niche. This suggests that CSCs entering into or exiting from dormancy may be dependent on the crosstalk between tumor-intrinsic and microenvironmental factors including stromal cells, the vascular system (angiogenic dormancy), and immune cells (immunologic dormancy) as described in the review by Clements et al.34.
EMT is a likely contributor to tumor cell dissemination from the primary site, though there has been some debate in the literature as to whether it is absolutely necessary35. There is, however, a well-established connection between EMT and the acquisition of stem-like properties22,24. Two different subsets of breast CSCs (BCSC) have been identified by our group. Mesenchymal BCSCs (M-BCSC, CD44+/CD24-) have a slow proliferating, quiescent phenotype, whereas the epithelial BCSCs (E-BCSC, ALDH+) are more proliferative36. This is consistent with the observation that DTCs in patients have the M-BCSC phenotype indicative of quiescence37. We note in passing that whether EMT states are best described as forming a continuum or just a rich spectrum of hybrid versus fully differentiated phenotypes is still uncertain; this does not affect our assertion that as stem-like cells move towards the epithelial end of the spectrum, they adapt their metabolic strategy in well-defined manners. Interestingly, recent evidence suggests these different subsets have different metabolic profiles and differential sensitivity to glycolysis or redox metabolism inhibition38. Specifically, M-BCSCs have low levels of reactive oxygen species (ROS) and enhanced glycolytic regulatory enzyme expression, whereas E-BCSCs harbor significantly elevated levels of ROS and enhanced mitochondrial oxidative phosphorylation and deploy NRF2-mediated antioxidant defense mechanisms. Moreover, oxidant stressors (i.e., 2-DG, H2O2, hypoxia) or antioxidants (i.e., N-acetyl cysteine) are able to induce a transition between M-BCSCs and E-BCSCs. These redox-regulated CSC state transitions are closely linked to the expression or stabilization of key redox-regulated proteins including AMPK, HIF-1, and NRF2.
Computational studies have contributed to our understanding of basic metabolic processes in cancer cells. The most common approach, known as Flux Balance Analysis39, aims to solve the fluxes traversing comprehensive networks of metabolic reactions under the assumption that the cell optimizes some predetermined quantity such as biomass production rate. One of the goals of these models has been to explain the Warburg effect, which is the observation that cancer cells are characterized by aerobic glycolysis40. These efforts have been only partially satisfactory41 as the models need to be augmented by additional somewhat arbitrary assumptions before agreeing with results of experimental efforts42. Importantly, computational frameworks are insufficient for addressing some of the most relevant questions that arise in the context of dormant cells. That is, cancer metabolism is adaptive in response to external conditions and changes in cell phenotype. Thus, altering the metabolic state is a crucial part of enabling survival in the dormant stem-like state (as has been readily established in the context of drug resistance43,44) and subsequently enabling transitions back to growth and differentiation under specific additional stimuli.
Studying the plasticity of CSCs and DTCs requires the coupling of genetic decision-making circuits to metabolic processes. A mathematical formalism to enable such modeling has recently been proposed with an initial focus on the interplay between the master regulators AMPK and HIF1 in controlling the balance between glycolysis and oxidative phosphorylation (OXPHOS)45 (Fig. 2). One of the outcomes of the study was the recognition that cells need not have a binary choice but can mix and match processes to generate needed energy and biomolecular building blocks as they react to local conditions. This model predicted the existence of a metabolically inactive phenotype that is characterized by low levels of HIF-1 and AMPK and low activity of glycolysis and OXPHOS, termed low-low, that was subsequently verified in the context of drug-tolerant melanoma cells43. The model also predicts the existence of a hybrid metabolic state in which cells exhibit high levels of HIF-1 and AMPK and high activity of glycolysis and OXPHOS. This hybrid metabolic state has been associated with the hybrid E/M phenotype. Whether the low-low phenotype corresponds to M-BCSCs entry into dormancy in vivo remains to be clarified. Interestingly, the model’s prediction that the antioxidant protein NRF2 is highly expressed in the hybrid E/M phenotype46 is consistent with the observation that the levels of NRF2 are high in the proliferative E-BCSCs that effectively uses OXPHOS.

The simulator couples key redox-sensing proteins (HIF-1, AMPK, MYC) with main metabolic pathways (glucose, glutamine, and fatty acid). Gray solid arrows represent positive regulations and gray bar-headed arrows represent negative regulations. Pathways labeled in red are up-regulated and those in blue are down-regulated. The transition from M-BCSC to E-BCSC and the reverse can be induced by alteration of cellular ROS levels. The simulator can be adapted to incorporate additional features to study, for example, the effects of inhibiting antioxidant factors. pMET: partial MET (mesenchymal-epithelial transition, the reverse of EMT); pEMT: partial EMT; mtROS: mitochondrial reactive oxygen species; noxROS: NADPH oxidase-derived reactive oxygen species; GSH: glutathione. This figure is adapted from Jia et al. 201945.
Abundant data have established relationships between EMT and metabolism. For example, the EMT transcription factor SNAIL directly represses expression of the gene encoding the fructose bisphosphatase FBP147. FBP1 repression leads to reduced oxygen consumption and ROS production and to increased glycolysis and biomass synthesis. The fact that EMT can lead to an increased reliance on glycolysis is consistent with findings that partial EMT can lead to stem-like behaviors. The genetic circuits encoding dormancy, stemness, and EMT need to be integrated with metabolism models. Unfortunately, unlike the relative maturity of single-cell transcriptomics, single-cell metabolomics is still in its infancy, and data to build these models will have to be inferred from less direct measurements. It is clear that metabolic switches play important roles in EMT and production of CSCs, that therefore constitutes an area of research crucial for both understanding and targeting dormancy.
The role of the microenvironment
The microenvironment at the metastatic site plays a critical role in the establishment and maintenance of dormancy as well as exit from the dormant state. There is evidence that DTCs occupy various niches in the bone marrow including those generally occupied by developing hematopoietic stem cells48, bone endosteal surfaces49,50, and perivascular regions51,52 (Fig. 3). Evidence for immune regulation of tumor dormancy has been recently reported and summarized in a review authored by Ombrato et al.53. It is clear that immune cells provide both stimulatory and inhibitory cues to DTCs, which in turn interact with immune-regulatory cells54. From a conceptual perspective, there are two different but connected issues: First, tumor cells are identified as foreign by the effector arms of the immune response55 and, second, a complex web of tumor cells of various phenotypes and different immune cells establish the tumor-immune interplay56.

Schematic representation of perivascular, hematopoietic, and endosteal bone marrow niches. It is not well established in humans what niche the tumor cells occupy while in the dormant state. In the proliferative state, bone metastasis is associated with the cycle of bone resorption that occurs in the endosteal niche.
Cytotoxic CD8+ T cells can recognize cancer cells via MHC-I-dependent presentation of antigens that can arise via mutational events (cancer neoantigens) or over-expression57. The recognition of over-expressed factors relies on the lack of perfect negative selection in the thymus58. It is likely that both different clones and different phenotypes within a given clone vary in their degree of visibility. For example, it has been established that non-small cell lung cancer cells down-regulate antigen presentation when undergoing EMT59. Dormant cells in ER+ breast cancer may evade immune recognition, which could be crucial for maintaining a dormant cell population. Various algorithms (the best known is NetMHCpan60) based on machine-learning approaches exist that can reasonably predict the binding of peptides to MHC. Also, attempts are underway to devise computational approaches to determine binding specificities between displayed peptides and T cell receptors61. To date, none of these new methods have been applied in the context of the differential recognition of dormant versus proliferative cells.
The dynamics of cancer cell recognition by the immune system can be modeled as a population-level competition between the tumor and the immune system. This process has typically been studied using coupled ordinary differential equations (ODEs) that govern the temporal evolution between the populations62,63,64. These models predict a transition between an equilibrium state, where tumor cell proliferation is balanced by immune cell killing (analogous to bulk dormancy), and immune escape where immunity fails. Effects such as T cell exhaustion and therapeutic attempts to counter it can be studied within this context. But there are many limitations. These models typically cannot consider complete cancer cell extinction, requiring attention to effects absent in the ODE treatment. Extensions of the basic model include mixed populations of growing and non-growing cells65, but these models neglect the possibility that dormant cells can transition back to a proliferative state. These models also do not consider any spatial aspects of the problem and hence are only applicable to cases where T cell infiltration is not limiting66,67. Finally, these models are also highly phenomenological and do not attempt to connect the proposed interaction terms with specific molecular pathways.
When one takes into account the mutual interactions of components of the immune system (the full immune “ecology”), it becomes clear that there can be a large-scale switch in operating mode from “attack” to “recover”, which, in the context of cancer, corresponds to a switch from “anti-tumor” to “pro-tumor” mode. For example, the well-known polarization transition of macrophages from M1 to M2 plays a key role in this functionality switch68. Some modeling efforts have attempted to consider this extra level of complexity56,69, but these have not been coupled to the phenotypic degrees of freedom relevant in the dormancy problem. Given experimental evidence regarding the role of regulatory T cells as well as myeloid-derived suppressor cells (MDSCs) in setting the immune balance in the metastatic bone niche, this is an important direction for future work. These efforts will be complicated by the influence of systemic factors on the immune interaction with cancer cells. For example, modifications to the immune cells of the bone marrow in patients with untreated breast cancer as well as those undergoing chemotherapy or hormone therapy have been described70,71. This suggests that metastases may be influenced by distant primary disease. One such mechanism is the induction of MDSCs72 by primary breast tumors. Systemic effects associated with psychological and physiological stress73 have also been associated with increased risk of breast cancer relapse74. Thus, there is a clear need to extend this modeling framework to address these important aspects.
In vivo models of breast cancer dormancy
When developing models of tumor dormancy in breast cancer, it is important to critically evaluate their clinical relevance. Bone marrow samples obtained from women with ER+ breast cancer provide the most clinically relevant source of DTCs. The use of biopsies that maintain tissue architecture rather than aspirates will facilitate study of both DTCs and their regulatory microenvironment. To maximize their clinical relevance, in vitro and mouse models of dormancy should attempt to recapitulate the biology of the DTC in patients. This includes utilizing ER+ models, simulation of a bone marrow microenvironment, and incorporation of an intact immune system. As described below, there are no current breast cancer dormancy models that fulfill all of these requirements. Despite much interest in studying tumor dormancy, the lack of clinically relevant models has limited progress in the field.
To gain insight into tumor dormancy a variety of in vivo mouse models, including genetically engineered mouse models, orthotopic tumor models, tumor resection models, as well as experimental metastasis mouse models have been used75,76. For instance, experimental metastasis mouse models have revealed the existence of a dormant state in cancer cells delivered to a metastatic organ site in vivo77,78. Upon transplantation into virgin female mice, pregnancy-dependent tumors induced by insertional mutations of mouse mammary tumor virus (MMTV) remain dormant for at least 300 days79. Tumor cell dormancy has also been described in transgenic mouse models for breast cancer, in which polyoma middle-T antigen or ERBB2 signaling was studied in mammary gland tissue that was devoid of β1 integrin80. The MMTV-based, doxycycline-inducible mouse model (MMTV–rTA; TetO–NEU-NT mice) is a valuable model to study dormancy as well. This model permits the loss of NEU (ERBB2) expression in a pre-established NEU-driven mammary tumor, and therefore provides researchers with a unique tool for study of the molecular mechanisms that control tumor dormancy and re-emergence from the dormant state81.
ER+ models of breast cancer dormancy
Until recently, most studies of hormonal regulation of metastasis have utilized human breast cancer cell lines such as MCF7 or T47D that are implanted into immune-deficient mice (Table 1). These systems have generated important information on hormone-dependent processes, but the necessity of utilizing immunodeficient mice means that these models cannot be used to study the immune system’s role in dormancy. Patient-derived xenograft (PDX) models may recapitulate greater intra- and inter-patient heterogeneity than human cell lines82,83, however, these also must be studied in an immunocompromised host34. Humanized mice are an emerging technology that may provide a route to better study human cancer in the context of a human immune system84, but these have yet to be utilized to investigate metastatic dormancy. In order to circumvent this limitation, mouse models involving syngeneic immunocompetent mice have been developed. The vast majority of these syngeneic mouse breast cancer cell lines have been reported to be ER-. However, there is evidence that some of these cell lines have a PAM50 subtype that is Luminal A/B. This is the case for the 4T1 line, which is commonly described as a model of triple-negative breast cancer85. It is important to underscore that human breast cancers may be classified as ER+ when as few as 1% of cells express ER86, though most ER+ tumors display >90% positivity87. Despite the low expression of ER in tumors with 1% positivity, molecular profiling demonstrates that some display a luminal phenotype and are sensitive to hormone therapy86. The same criteria have not been routinely applied to murine breast cancers. To our knowledge only one murine cell line, E0771, has been reported in the literature to have sensitivity to hormone therapy (fulvestrant)88. The first ER+ mouse mammary carcinoma models that spontaneously and consistently metastasize to the bone were reported using the SSM2 and SSM3 cell lines derived from spontaneous tumors formed in STAT1-knockout mice. These models represent a significant advance, since there is a latency period of up to 7 weeks before overt metastases are detected89. Dormancy has not been explicitly studied using this model, however. There are conflicting reports in the literature regarding hormone status of murine breast cancer cell lines, which may be due to the use of different methods (immunostaining, PCR, or RNA-seq), lack of expected cross-reactivity between antibodies for ER that are primarily used on human samples, or simply the absence of a standardized cutoff for positivity. We believe this is a crucial problem in the use of murine breast cancer cell lines and suggest it be solved by the following methods. First, two or more methods should be used to identify hormone status (e.g. immunostaining and PCR for ER/PR). Second, appropriate positive and negative controls should be used and reported in these assays (e.g. MCF-7 and 231 cells for a human/mouse cross-reactive antibody, or healthy murine mammary gland tissue). Finally, any murine cell line that is found to be ER+/PR+ should be tested for responsiveness to estrogen/progesterone in vitro and in vivo via appropriate administration of hormones or hormone therapy.
Sites of breast cancer dormancy
A number of mouse models of dormancy involve the study of DTCs in the lung. Lung metastasis is readily studied due to the ease of delivery of tumor cells to the lung via tail vein injection as compared to the delivery of tumor cells to the bone marrow via intracardiac or intratibial injection. Although the clinical relevance of the lung as a site of tumor dormancy is unclear, dormancy models in the lung have provided important evidence for immune regulation of dormancy. For example, De Lara et al. demonstrated that the CD39+PD-1+ subtype of CD8+ T cells mediated metastatic dormancy in breast cancer using the syngeneic mouse lines 4T07 (dormant) and 4T1 (non-dormant)90. Syngeneic mouse models with dormancy phenotypes and manipulation of the immune background in transgenic mice represent a powerful tool to investigate immune regulation of dormancy.
Mechanisms of breast cancer dormancy in established immunocompetent murine models
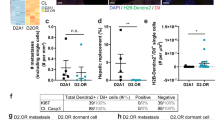
Table 1 summarizes the characteristics of the existing possible in vivo models of dormancy and their clinically relevant characteristics. All cell lines included are murine, to facilitate the study of dormancy in a fully immunocompetent organism, with the exception of MCF7 and T47D which are shown to illustrate ER + cell lines that demonstrate bone metastasis and dormancy. Of these murine breast cancer cell lines, 8 of 22 demonstrate metastasis to bone while 19 of 22 demonstrate metastasis to the lung. Of those 19 that metastasize to lung, 4 have demonstrated dormancy in the lung. Interestingly each cell line (67NR, 4T07, EMT6, and D2.OR) has demonstrated some degree of immune dependence for the mechanism of lung dormancy. Specifically, 67NR dormancy can be abrogated by LPS administration91, 4T07 dormancy is controlled by CD39 + PD-1 + CD8 + T cells90, EMT6 lung dormancy is controlled by the interplay between granulocytic-MDSC and CD8 + T cells92, and D2.OR cells can be awakened from dormancy via LPS administration93 or induction of fibrosis94. Only one study has demonstrated dormancy in the bone marrow in an immunocompetent host using chemotherapy treatment of 4T07 cells and identified the role of the perivascular niche in DTC chemotherapy resistance52. These findings highlight the importance of the immune system and use of immunocompetent models in the study of breast cancer dormancy.
Challenges for in vivo models of tumor dormancy moving forward
Although mouse models have provided valuable information on the regulation of tumor dormancy, all the existing models have limitations. There are currently no in vivo models that faithfully replicate the clinical situation of dormant breast cancer cells residing in the bone marrow of a fully immunocompetent host. The development of models that are ER+, immunocompetent, and dormant in bone marrow are of the utmost importance for the field moving forward.
In vitro models of breast cancer dormancy
Understanding how DTCs remain dormant and how environmental cues awaken them is essential for developing novel treatment approaches. Study of DTCs has been hampered, in part, by the lack of in vitro experimental models that recapitulate the bidirectional interactions of DTCs and their complex, dynamic microenvironment. Some of the in vitro experimental models widely used to study tumor dormancy and strategies to increase the complexities of these in vitro models to more faithfully mimic clinical dormancy are discussed in this section (Fig. 4).

Engineered 3D models generally exhibit the greatest geometric and cellular complexity combining both multicellular models and engineered 3D geometry.
2D and 3D monoculture models
A simple model of tumor cell dormancy in vitro can be generated using mitogen (i.e., serum) deprivation of 2D cultures for an extended period (up to 14 days), which selects for a quiescent population. With this model, Barney et al. recently demonstrated a role of TGFβ-mediated fibronectin deposition in the promotion of the FAK-ERK survival signaling axis and maintenance of a dormant state95. In another study, Tivari et al. developed a 2D in vitro dormancy model of ER+ MCF7 cells by plating at clonogenic density on fibronectin-coated surfaces to select for a quiescent population in the presence of FGF-296. In addition to fibronectin and FGF-2, chronic hypoxia or cobalt chloride treatment of metastatic breast cancer cells MDA-MB-231 or ER+ MCF7 cells has been reported to induce reversible quiescence or dormancy97,98. Although these models are useful for investigating the molecular mechanisms that result in the establishment and survival of dormant cells, 2D culture models often do not recapitulate in vivo findings due to the lack of relevant cell–cell and cell–matrix interactions that only occur in 3D. 3D Cultrex® basement membrane extract has been employed to model in vivo growth characteristics of cells that exhibit either dormant or proliferative metastatic behavior in vivo in more biologically relevant 3D culture94,99. Models using transglutaminase crosslinked collagen gels100, fibrin hydrogels101, tunable PEG hydrogels102, and engineered poly(ε-caprolactone) scaffolds with aligned or random fibers103 have also been developed to model breast cancer dormancy in 3D culture. The selection of specific subtypes of tumor cells is another method for studying dormancy in vitro. For example, M-BCSCs have been shown to represent a more quiescent subpopulation of tumor cells than E-BCSCs or bulk tumor cells36.
Multicellular co-culture models
To model tumor dormancy in the metastatic bone niche, Marlow and colleagues reported a multicellular co-culture model. Their models are based on co-cultures of breast cancer cell lines in 3D collagen biomatrix with HS-5 human bone marrow stromal cells, which generate a supportive niche, or multicellular stromal cells consisting of HUVEC, fetal osteoblasts, and HS-5, which generate an inhibitory niche104. Similarly, by creating organotypic models of lung or bone marrow microvascular niches consisting of lung fibroblasts or mesenchymal stem cells along with HUVEC, Ghajar et al. showed that stable microvasculature constitutes a dormant niche, whereas sprouting neovasculature sparks micro-metastatic outgrowth of breast cancer cells51. Although these 3D co-culture systems support quiescent breast cancer cells, the assay format used in these models precludes large-scale screening of compounds that selectively inhibit growth of dormant tumor cells. To overcome this issue, Cavnar et al. reported the development of a 384-well 3D spheroid model in which cancer cells are reversibly arrested in the G1/G0 phase of the cell cycle due to co-culture with bone marrow stromal cells; this system was used to model selective elimination of dormant tumor cells from bone marrow105.
Tissue-engineered models
The rapid development of sophisticated tissue-engineered ex vivo biomimetic platforms together with co-culture of multiple types of tissue stromal cells and cancer cells have led to a better understanding of how the bone, liver, and lung alveolar microenvironments regulate breast cancer dormancy and reactivation. This emerging area of research is the focus of a thorough literature review from Montagner et al.106. For instance, using metastasis-indolent MDA-MB-231BRMS1 cells co-cultured in osteoblast bioreactors that generate a multilayered bone-like structure, Sosnoski et al. showed that cancer cells attached to the bone matrix produced by MC3T3-E1 osteoblasts are dormant until addition of bone-remodeling mediators including TNFα, IL1-β, IL6, and PGE2, which stimulated cell proliferation50. Using lung alveolar type 1 and type 2 cells and fibroblasts co-cultured with the metastatic indolent D2.OR mouse mammary tumor cells on a gas-permeable substrate in mitogen-low glucose-low medium, Montagner et al. demonstrated that the indolent behavior of D2.OR cells in the lung is determined by their interaction with alveolar epithelial cells, in particular the type 1 cells. The indolent behavior is mediated by frizzled-related protein SFRP2, which is secreted by alveolar epithelial cells. SFRP2 promotes the formation of fibronectin fibrils by indolent cells to drive integrin-dependent pro-survival signals107. Similarly, a 3D hepatic micro-physiological system that reproduces several features of liver physiology results in spontaneous dormancy in a subpopulation of breast cancer cells108,109. These ex vivo models of breast cancer dormancy allow investigators to mimic the pathophysiology of metastatic breast cancer cells more reliably than standard 2D cell culture systems. Notably, due to physiologically relevant cell–cell and cell–extracellular matrix interactions that occur in the ex vivo systems, cells in these models exhibit several different phenotypes of tumors generally not observed in cells cultured in 2D or 3D.
Challenges for in vitro models of tumor dormancy
Dormant breast cancer cells usually reside in specific microenvironments such as the bone endosteal surfaces49,50 and perivascular regions in the bone marrow, lung, and brain that are infiltrated by immune cells51,52. However, despite the progress made in the development of complex, multicellular, or tissue-engineered systems to model tumor dormancy, none of these models have incorporated immune cells. Given the importance of immune surveillance in regulating tumor behaviors, future studies will need to incorporate immune cells into organotypic or organoid cultures110. Such co-culture systems have been recently developed111, although not yet utilized to study tumor dormancy. The integration of immune cells into organoid models will be of great interest and utility for the study of dormancy in both the primary tumor microenvironment and various metastatic microenvironments that may be able to be recapitulated with organoid systems in vitro.
Breast cancer dormancy models that bridge the gap between in vitro and in vivo systems
Successful integration of in vitro and in vivo systems remains a challenge for tumor dormancy research. Three models have been described that incorporate features of both in vitro and in vivo dormancy models. The first was developed by the Dontu lab and combines human osteoblasts, endothelial cells, and tumor cells in a 3D collagen scaffold as mentioned above104,112. Crucially, the 3D scaffold facilitates the implantation of these dormant microenvironments into the subcutaneous space of mice, resulting in development of an inhibitory (i.e., dormant) or a supportive niche in vivo. The second of these models was developed by the Shea lab and utilized a microporous polymer scaffold and implantation into tumor-bearing mice to generate a niche in vivo to which metastatic tumor cells home113,114,115. This approach facilitates the development and study of the metastatic niche in vivo and can be expanded via explantation of the engineered niche and subsequent culture in vitro116,117. The third model developed by the Lee lab utilizes a combination of the approaches118. In this model, pre-seeded human bone marrow stromal cells form a vascularized niche after implantation to which both human peripheral blood mononuclear cells and prostate cancer cells home in vivo. These engineered niches can be explanted and monitored for tumor cell growth or dormancy ex vivo. Taken together, these models bridge the gap between in vivo and in vitro microenvironments and provide unique opportunities to study how systemic cues resulting from primary tumor development, aging, or immune dysfunction promote or inhibit dormancy on a whole organism scale.
Future directions
Tumor dormancy is a major clinical problem particularly relevant to ER + breast cancer. Most current therapies do not kill non-dividing dormant cancer cells. One therapeutic alternative is to force dormant cancer cells to exit dormancy and then target them as they proliferate. However, this approach depends on the ability to kill all cells as they escape dormancy. Failure to accomplish this may increase the likelihood of metastasis. Theoretically, treatments that maintain the dormant state could be developed. This was the rationale for extending the period of adjuvant hormonal therapy from 5 to 10 years119. Although this extended therapy may delay recurrence in some patients, it is often associated with side effects. The development of effective strategies to target dormant cancer cells raise additional issues: First, it is not clear how to identify patients who require such treatment after completion of adjuvant therapy as well as how to monitor therapeutic efficacy. Future studies need to evaluate the utility of non-invasive assays such as those that detect CTCs or cell-free DNA for identification of women with dormant cancer cells. Second, DTC assessment requires invasive and painful biopsies but may provide sufficiently worthwhile information to justify use. It remains to be determined whether women with pure ductal carcinoma in situ are more likely to harbor dormant cells than women without cancer cells in the ducts. Finally, methods to assess elimination of DTCs must be developed. The complexity of tumor dormancy and the difficulty of conducting clinical trials over two or more decades highlights the need for continued pre-clinical model development (Fig. 5). As highlighted in this review, current models, although valuable, all have limitations. Future development of more clinically relevant models will help elucidate the biology underlying breast cancer dormancy and develop strategies to overcome the clinical challenge of breast cancer relapse.

The successful study of breast cancer dormancy relies on the interplay between in vivo models of dormancy, in vitro manipulation, and patient samples in order to identify mechanisms of dormancy, generate new hypotheses, and develop therapies with the ultimate goal of new clinical trials to study breast cancer dormancy and to assess treatments.
Data availability
No data or code are associated with this manuscript.
References
Howlader, N. et al. SEER Cancer Statistics Review, 1975–2016. National Cancer Institute (2019).
Copson, E. et al. Prospective observational study of breast cancer treatment outcomes for UK women aged 18–40 years at diagnosis: the POSH study. J. Natl Cancer Inst. 105, 978–988 (2013).
Pan, H. et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N. Engl. J. Med. 377, 1836–1846 (2017).
Villarreal-Garza, C. et al. Clinical utility of genomic signatures in young breast cancer patients: a systematic review. NPJ breast cancer 6, 1–9 (2020).
Sparano, J. A. et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. 379, 111–121 (2018).
Cheng, Q. et al. A signature of epithelial-mesenchymal plasticity and stromal activation in primary tumor modulates late recurrence in breast cancer independent of disease subtype. Breast Cancer Res. 16, 407 (2014).
Mittempergher, L. et al. A gene signature for late distant metastasis in breast cancer identifies a potential mechanism of late recurrences. Mol. Oncol. 7, 987–999 (2013).
Kim, R. S. et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 7, e35569 (2012).
Domany, E. Using high-throughput transcriptomic data for prognosis: a critical overview and perspectives. Cancer Res. 74, 4612–4621 (2014).
Rack, B. et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl Cancer Inst. 106, dju066 (2014).
Meng, S. et al. Circulating tumor cells in patients with breast cancer dormancy. Clin. Cancer Res. 10, 8152–8162 (2004).
Shaw, J. A. et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 22, 220–231 (2012).
Schwarzenbach, H. & Pantel, K. Circulating DNA as biomarker in breast cancer. Breast Cancer Res. 17, 136 (2015).
Smid, M. et al. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 68, 3108–3114 (2008).
Nombela-Arrieta, C. & Manz, M. G. Quantification and three-dimensional microanatomical organization of the bone marrow. Blood Adv. 1, 407–416 (2017).
Sänger, N. et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int. J. Cancer 129, 2522–2526 (2011).
Giannakeas, V., Sopik, V. & Narod, S. A. Association of a diagnosis of ductal carcinoma in situ with death from breast cancer. JAMA Netw. Open 3, e2017124–e2017124 (2020).
Hüsemann, Y. et al. Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68 (2008).
Tjensvoll, K. et al. Detection of disseminated tumor cells in bone marrow predict late recurrences in operable breast cancer patients. BMC Cancer 19, 1131 (2019).
Borgen, E. et al. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 20, 120 (2018).
Bocci, F. et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl Acad. Sci. USA 116, 148–157 (2019).
Mani, S. A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Jolly, M. K. et al. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 5, 155 (2015).
Jolly, M. K. et al. Towards elucidating the connection between epithelial–mesenchymal transitions and stemness. J. R. Soc. Interface 11, 20140962 (2014).
Grosse-Wilde, A. et al. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS ONE 10, e0126522 (2015).
Pastushenko, I. et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018).
Pattabiraman, D. R. & Weinberg, R. A. in Cold Spring Harbor symposia on quantitative biology. 11–19 (Cold Spring Harbor Laboratory Press).
Heldt, F. S., Barr, A. R., Cooper, S., Bakal, C. & Novák, B. A comprehensive model for the proliferation–quiescence decision in response to endogenous DNA damage in human cells. Proc. Natl Acad. Sci. 115, 2532–2537 (2018).
Cheng, T. et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808 (2000).
Huang, B. et al. Decoding the mechanisms underlying cell-fate decision-making during stem cell differentiation by Random Circuit Perturbation. arXiv preprint arXiv:2004.00156 (2020).
Zañudo, J. G. T., Scaltriti, M. & Albert, R. A network modeling approach to elucidate drug resistance mechanisms and predict combinatorial drug treatments in breast cancer. Cancer convergence 1, 5 (2017).
Jolly, M. K., Tripathi, S. C., Somarelli, J. A., Hanash, S. M. & Levine, H. Epithelial/mesenchymal plasticity: how have quantitative mathematical models helped improve our understanding? Mol. Oncol. 11, 739–754 (2017).
Brooks, M. D., Burness, M. L. & Wicha, M. S. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 17, 260–271 (2015).
Clements, M. E. & Johnson, R. W. Breast cancer dormancy in bone. Curr. Osteoporos. Rep. 17, 353–361 (2019).
Williams, E. D., Gao, D., Redfern, A. & Thompson, E. W. Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 19, 716–732 (2019).
Liu, S. et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2, 78–91 (2014).
Balic, M. et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 12, 5615–5621 (2006).
Luo, M. et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. 28, 69–86. e66 (2018).
Orth, J. D., Thiele, I. & Palsson, B. Ø. What is flux balance analysis? Nat. Biotechnol. 28, 245–248 (2010).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Yizhak, K., Chaneton, B., Gottlieb, E. & Ruppin, E. Modeling cancer metabolism on a genome scale. Mol. Syst. Biol. 11, 817 (2015).
Folger, O. et al. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 7, 501 (2011).
Jia, D. et al. Drug-tolerant idling melanoma cells exhibit theory-predicted metabolic low-low phenotype. bioRxiv, 809889 (2020).
Sansone, P. et al. Self-renewal of CD133 hi cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 7, 1–10 (2016).
Jia, D. et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl Acad. Sci. USA 116, 3909–3918 (2019).
Bocci, F. et al. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. 11, 251–263 (2019).
Dong, C. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331 (2013).
Risson, E., Nobre, A. R., Maguer-Satta, V. & Aguirre-Ghiso, J. A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer 1, 672–680 (2020).
McGrath, J., Panzica, L., Ransom, R., Withers, H. G. & Gelman, I. H. Identification of genes regulating breast cancer dormancy in 3D bone endosteal niche cultures. Mol. Cancer Res 17, 860–869, https://doi.org/10.1158/1541-7786.MCR-18-0956 (2019).
Sosnoski, D. M., Norgard, R. J., Grove, C. D., Foster, S. J. & Mastro, A. M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 32, 335–344, https://doi.org/10.1007/s10585-015-9710-9 (2015).
Ghajar, C. M. et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15, 807–817, https://doi.org/10.1038/ncb2767 (2013).
Carlson, P. et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 21, 238–250, https://doi.org/10.1038/s41556-018-0267-0 (2019).
Ombrato, L. & Montagner, M. Technical advancements for studying immune regulation of disseminated dormant cancer cells. Front. Oncol. 10, 2445 (2020).
Goddard, E. T., Bozic, I., Riddell, S. R. & Ghajar, C. M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 20, 1240–1249 (2018).
George, J. T., Kessler, D. A. & Levine, H. Effects of thymic selection on T cell recognition of foreign and tumor antigenic peptides. Proc. Natl Acad. Sci. USA 114, E7875–E7881 (2017).
Arabameri, A., Asemani, D. & Hadjati, J. A structural methodology for modeling immune-tumor interactions including pro-and anti-tumor factors for clinical applications. Math. Biosci. 304, 48–61 (2018).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Davis, M. M. Not-so-negative selection. Immunity 43, 833–835 (2015).
Tripathi, S. C. et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl Acad. Sci. USA 113, E1555–E1564 (2016).
Hoof, I. et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61, 1 (2009).
Lin, X. et al. Rapid Assessment of T-Cell Receptor Specificity of the Immune Repertoire. bioRxiv (2020).
Wilkie, K. P. in Systems biology of tumor dormancy 201-234 (Springer, 2013).
Kuznetsov, V. A., Makalkin, I. A., Taylor, M. A. & Perelson, A. S. Nonlinear dynamics of immunogenic tumors: parameter estimation and global bifurcation analysis. Bull. Math. Biol. 56, 295–321 (1994).
Alvarez, R. F., Barbuto, J. A. & Venegeroles, R. A nonlinear mathematical model of cell-mediated immune response for tumor phenotypic heterogeneity. J. Theor. Biol. 471, 42–50 (2019).
Page, K. & Uhr, J. Mathematical models of cancer dormancy. Leuk. lymphoma 46, 313–327 (2005).
Li, X. et al. Infiltration of CD8+ T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl Acad. Sci. USA 116, 3678–3687 (2019).
Keren, L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373–1387. e1319 (2018).
Shapouri‐Moghaddam, A. et al. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233, 6425–6440 (2018).
Li, X., Jolly, M. K., George, J. T., Pienta, K. J. & Levine, H. Computational modeling of the crosstalk between macrophage polarization and tumor cell plasticity in the tumor microenvironment. Front. Oncol. 9, 10 (2019).
Solomayer, E.-F. et al. Influence of adjuvant hormone therapy and chemotherapy on the immune system analysed in the bone marrow of patients with breast cancer. Clin. Cancer Res. 9, 174–180 (2003).
Feuerer, M. et al. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int. J. cancer 92, 96–105 (2001).
Diaz-Montero, C. M. et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 58, 49–59 (2009).
Mundy-Bosse, B. L., Thornton, L. M., Yang, H.-C., Andersen, B. L. & Carson, W. E. Psychological stress is associated with altered levels of myeloid-derived suppressor cells in breast cancer patients. Cell. Immunol. 270, 80–87 (2011).
Antoni, M. H. et al. Stress management, leukocyte transcriptional changes and breast cancer recurrence in a randomized trial: an exploratory analysis. Psychoneuroendocrinology 74, 269–277 (2016).
Hurst, R. E., Bastian, A., Bailey-Downs, L. & Ihnat, M. A. Targeting dormant micrometastases: rationale, evidence to date and clinical implications. Therapeutic Adv. Med. Oncol. 8, 126–137 (2016).
Marshall, J.-C. A. et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J. Natl Cancer Inst. 104, 1306–1319 (2012).
Heyn, C. et al. In vivo MRI of cancer cell fate at the single‐cell level in a mouse model of breast cancer metastasis to the brain. Magn. Reson. Med.: Off. J. Int. Soc. Magn. Reson. Med. 56, 1001–1010 (2006).
Naumov, G. N. et al. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 62, 2162–2168 (2002).
Gattelli, A. et al. Progression of pregnancy-dependent mouse mammary tumors after long dormancy periods. Involvement of Wnt pathway activation. Cancer Res. 64, 5193–5199 (2004).
White, D. E. et al. Targeted disruption of β1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer cell 6, 159–170 (2004).
Moody, S. E. et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer cell 2, 451–461 (2002).
Sprouffske, K. et al. Genetic heterogeneity and clonal evolution during metastasis in breast cancer patient-derived tumor xenograft models. Computational Struct. Biotechnol. J. 18, 323–331 (2020).
Dobrolecki, L. E. et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 35, 547–573 (2016).
Yao, L.-C. et al. in Target Identification and Validation in Drug Discovery 241–252 (Springer, 2019).
Yang, Y. et al. Immunocompetent mouse allograft models for development of therapies to target breast cancer metastasis. Oncotarget 8, 30621 (2017).
Fujii, T. et al. Revisiting the definition of estrogen receptor positivity in HER2-negative primary breast cancer. Ann. Oncol. 28, 2420–2428 (2017).
Ma, H. et al. Quantitative measures of estrogen receptor expression in relation to breast cancer-specific mortality risk among white women and black women. Breast Cancer Res. 15, 1–12 (2013).
Le Naour, A. et al. EO771, the first luminal B mammary cancer cell line from C57BL/6 mice. Cancer Cell Int. 20, 1–13 (2020).
Capietto, A.-H. et al. Novel ERα positive breast cancer model with estrogen independent growth in the bone microenvironment. Oncotarget 7, 49751 (2016).
de Lara, P. T. et al. CD39+ PD-1+ CD8+ T cells mediate metastatic dormancy in breast cancer. Nature communications 12, 1–14 (2021).
De Cock, J. M. et al. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 76, 6778–6784 (2016).
Piranlioglu, R. et al. Primary tumor-induced immunity eradicates disseminated tumor cells in syngeneic mouse model. Nat. Commun. 10, 1–13 (2019).
Albrengues, J. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361 (2018).
Barkan, D. et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 70, 5706–5716, https://doi.org/10.1158/0008-5472.CAN-09-2356 (2010).
Barney, L. E. et al. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 6, eaaz4157, https://doi.org/10.1126/sciadv.aaz4157 (2020).
Tivari, S., Lu, H., Dasgupta, T., De Lorenzo, M. S. & Wieder, R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun. Signal 16, 48, https://doi.org/10.1186/s12964-018-0259-5 (2018).
Carcereri de Prati, A. et al. Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. J. Cell Biochem. 118, 3237–3248, https://doi.org/10.1002/jcb.25972 (2017).
Lee, H. R., Leslie, F. & Azarin, S. M. A facile in vitro platform to study cancer cell dormancy under hypoxic microenvironments using CoCl2. J. Biol. Eng. 12, 12, https://doi.org/10.1186/s13036-018-0106-7 (2018).
El Touny, L. H. et al. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Invest. 124, 156–168, https://doi.org/10.1172/JCI70259 (2014).
Fang, J. Y. et al. From competency to dormancy: a 3D model to study cancer cells and drug responsiveness. J. Transl. Med. 14, 38, https://doi.org/10.1186/s12967-016-0798-8 (2016).
Liu, Y. et al. Fibrin stiffness mediates dormancy of tumor-repopulating cells via a Cdc42-driven Tet2 epigenetic program. Cancer Res. 78, 3926–3937, https://doi.org/10.1158/0008-5472.CAN-17-3719 (2018).
Pradhan, S. & Slater, J. H. Tunable hydrogels for controlling phenotypic cancer cell states to model breast cancer dormancy and reactivation. Biomaterials 215, 119177, https://doi.org/10.1016/j.biomaterials.2019.04.022 (2019).
Guiro, K., Patel, S. A., Greco, S. J., Rameshwar, P. & Arinzeh, T. L. Investigating breast cancer cell behavior using tissue engineering scaffolds. PLoS ONE 10, e0118724, https://doi.org/10.1371/journal.pone.0118724 (2015).
Marlow, R. et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 73, 6886–6899, https://doi.org/10.1158/0008-5472.CAN-13-0991 (2013).
Cavnar, S. P. et al. Modeling selective elimination of quiescent cancer cells from bone marrow. Neoplasia 17, 625–633, https://doi.org/10.1016/j.neo.2015.08.001 (2015).
Montagner, M. & Sahai, E. In vitro models of breast cancer metastatic dormancy. Front Cell Dev. Biol. 8, 37, https://doi.org/10.3389/fcell.2020.00037 (2020).
Montagner, M. et al. Crosstalk with lung epithelial cells regulates Sfrp2-mediated latency in breast cancer dissemination. Nat. Cell Biol. 22, 289–296, https://doi.org/10.1038/s41556-020-0474-3 (2020).
Clark, A. M. et al. A liver microphysiological system of tumor cell dormancy and inflammatory responsiveness is affected by scaffold properties. Lab Chip 17, 156–168, https://doi.org/10.1039/c6lc01171c (2016).
Clark, A. M., Ma, B., Taylor, D. L., Griffith, L. & Wells, A. Liver metastases: Microenvironments and ex-vivo models. Exp. Biol. Med (Maywood) 241, 1639–1652, https://doi.org/10.1177/1535370216658144 (2016).
Roelofs, C., Hollande, F., Redvers, R., Anderson, R. L. & Merino, D. Breast tumour organoids: promising models for the genomic and functional characterisation of breast cancer. Biochemical Soc. Trans. 47, 109–117 (2019).
Neal, J. T. et al. Organoid modeling of the tumor immune microenvironment. Cell 175, 1972–1988. e1916 (2018).
Marlow, R. & Dontu, G. in Mammary Stem Cells 213–220 (Springer, 2015).
Morris, A. H. et al. Engineered niches to analyze mechanisms of metastasis and guide precision medicine. Cancer Res. 80, 3786–3794 (2020).
Aguado, B. A., Bushnell, G. G., Rao, S. S., Jeruss, J. S. & Shea, L. D. Engineering the pre-metastatic niche. Nat. Biomed. Eng. 1, 0077 (2017).
Bushnell, G. G., Wicha, M. S. & Jeruss, J. S. Precision health for breast cancer metastasis: biomaterial scaffolds as an engineered metastatic niche to define, study, and monitor metastatic progression. Oncoscience 6, 380 (2019).
Bushnell, G. G. et al. Biomaterial scaffolds recruit an aggressive population of metastatic tumor cells in vivo. Cancer Res., canres.2502.2018, https://doi.org/10.1158/0008-5472.can-18-2502 (2019).
Bushnell, G. G. et al. Microporous scaffolds loaded with immunomodulatory lentivirus to study the contribution of immune cell populations to tumor cell recruitment in vivo. Biotechnol. Bioeng. 117, 210–222 (2020).
Carpenter, R. A., Kwak, J.-G., Peyton, S. R. & Lee, J. Implantable pre-metastatic niches for the study of the microenvironmental regulation of disseminated human tumour cells. Nat. Biomed. Eng. 2, 915 (2018).
Goss, P. E. et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N. Engl. J. Med. 375, 209–219 (2016).
Keydar, I. et al. Establishment and characterization of a cell line of human breast carcinoma origin. Eur. J. Cancer (1965) 15, 659–670 (1979).
Puchalapalli, M. et al. NSG mice provide a better spontaneous model of breast cancer metastasis than athymic (nude) mice. PLoS ONE 11, e0163521 (2016).
Lefley, D. et al. Development of clinically relevant in vivo metastasis models using human bone discs and breast cancer patient-derived xenografts. Breast Cancer Res. 21, 1–21 (2019).
Gawrzak, S. et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol. 20, 211–221 (2018).
Soule, H., Vazquez, J., Long, A., Albert, S. & Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl Cancer Inst. 51, 1409–1416 (1973).
Wada, M. et al. P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 36, 6649–6657 (2017).
Barkan, D. et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 68, 6241–6250 (2008).
Rucci, N. et al. In vivo bone metastases, osteoclastogenic ability, and phenotypic characterization of human breast cancer cells. Bone 34, 697–709 (2004).
Sowder, M. E. & Johnson, R. W. Enrichment and detection of bone disseminated tumor cells in models of low tumor burden. Sci. Rep. 8, 1–13 (2018).
Jessen, K. A. et al. Molecular analysis of metastasis in a polyomavirus middle T mouse model: the role of osteopontin. Breast Cancer Res. 6, 1–13 (2004).
Borowsky, A. D. et al. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin. Exp. metastasis 22, 47–59 (2005).
Thies, K. A. et al. Stromal platelet-derived growth factor receptor-β signaling promotes breast cancer metastasis in the brain. Cancer Res 81, 606–618, https://doi.org/10.1158/0008-5472.Can-19-3731 (2021).
Simmons, J. et al. Animal models of bone metastasis. Vet. Pathol. 52, 827–841 (2015).
Pei, X. F. et al. Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. Vitr. Cell. Developmental Biol.-Anim. 40, 14–21 (2004).
Alikhani, N. et al. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene 32, 961–967, https://doi.org/10.1038/onc.2012.113 (2013).
Faraji, F. et al. Cadm1 is a metastasis susceptibility gene that suppresses metastasis by modifying tumor interaction with the cell-mediated immunity. PLoS Genet. 8, e1002926 (2012).
Holzer, R. G. et al. Development and characterization of a progressive series of mammary adenocarcinoma cell lines derived from the C3 (1)/SV40 Large T-antigen transgenic mouse model. Breast Cancer Res. Treat. 77, 65–76 (2003).
Liu, P. et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway–dependent and PI3K pathway–independent mechanisms. Nat. Med. 17, 1116–1120 (2011).
Jallow, F. et al. Dynamic interactions between the extracellular matrix and estrogen activity in progression of ER+ breast cancer. Oncogene 38, 6913–6925 (2019).
Ramasamy, K. et al. (AACR, 2018).
Ewens, A., Mihich, E. & Ehrke, M. J. Distant metastasis from subcutaneously grown E0771 medullary breast adenocarcinoma. Anticancer Res. 25, 3905–3915 (2005).
Hiraga, T. & Ninomiya, T. Establishment and characterization of a C57BL/6 mouse model of bone metastasis of breast cancer. J. Bone Miner. Metab. 37, 235–242 (2019).
Aslakson, C. J. & Miller, F. R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 52, 1399–1405 (1992).
de Souza Garcia, C. M., de Araújo, M. R., Lopes, M. T. P., Ferreira, M. & Cassali, G. D. Morphological and immunophenotipical characterization of murine mammary carcinoma 4t1. Braz. J. Vet. Pathol. 7, 158–165 (2014).
Johnstone, C. N. et al. Functional and molecular characterisation of EO771. LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis. Models Mechanisms 8, 237–251 (2015).
Lincoln, D. W., Phillips, P. G. & Bove, K. Estrogen-induced Ets-1 promotes capillary formation in an in vitro tumor angiogenesis model. Breast cancer Res. Treat. 78, 167–178 (2003).
Aslakson, C. J., Rak, J. W., Miller, B. E. & Miller, F. R. Differential influence of organ site on three subpopulations of a single mouse mammary tumor at two distinct steps in metastasis. Int. J. Cancer 47, 466–472 (1991).
Miller, B., Miller, F., Wilburn, D. & Heppner, G. Analysis of tumour cell composition in tumours composed of paired mixtures of mammary tumour cell lines. Br. J. Cancer 56, 561–569 (1987).
Carlson, P. et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 21, 238–250 (2019).
Rockwell, S. C., Kallman, R. F. & Fajardo, L. F. Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J. Natl Cancer Inst. 49, 735–749 (1972).
Morris, V. L., Tuck, A. B., Wilson, S. M., Percy, D. & Chambers, A. F. Tumor progression and metastasis in murine D2 hyperplastic alveolar nodule mammary tumor cell lines. Clin. Exp. Metastasis 11, 103–112 (1993).
Alonso, D. F. et al. Characterization of F3II, a sarcomatoid mammary carcinoma cell line originated from a clonal subpopulation of a mouse adenocarcinoma. J. Surgical Oncol. 62, 288–297 (1996). 10.1002/(SICI)1096-9098(199608)62:4<288::AID-JSO14>3.0.CO;2-1.
Lollini, P.-L. et al. High-metastatic clones selected in vitro from a recent spontaneous BALB/c mammary adenocarcinoma cell line. Clin. Exp. Metastasis 2, 251–259 (1984).
Chen, Y. & Rittling, S. R. Novel murine mammary epithelial cell lines that form osteolytic bone metastases: effect of strain background on tumor homing. Clin. Exp. Metastasis 20, 111–120 (2003).
Acknowledgements
S.A.M. is supported by the National Institutes of health (NIH) 2R01CA155243 and R01CA200970 and Cancer Research and Prevention Institutes of Texas CPRIT RP170172. H.L is supported by the National Science Foundation (NSF) Center for Theoretical Biological Physics (NSF PHY-2019745). H.L./S.A.M. are supported by NSF grant PHY-19357672. D.J. is supported by a training fellowship from the Gulf Coast Consortia in the Computational Cancer Biology Training Program (CPRIT grant no. RP170593). M.S.W. is supported by the Breast Cancer Research Foundation (BCRF) BCRF-18–173 and NIH R35CA197585. We thank Jordan Pietz and the Creative Communications Department at MD Anderson for help with graphic design of figures.
Author information
Authors and Affiliations
Contributions
All authors wrote, edited, and approved the final version of the manuscript. H.L., S.A.M., and M.S.W. also supervised and provided funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bushnell, G.G., Deshmukh, A.P., den Hollander, P. et al. Breast cancer dormancy: need for clinically relevant models to address current gaps in knowledge. npj Breast Cancer 7, 66 (2021). https://doi.org/10.1038/s41523-021-00269-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-021-00269-x
This article is cited by
-
Glucocorticoid receptor: a harmonizer of cellular plasticity in breast cancer—directs the road towards therapy resistance, metastatic progression and recurrence
Cancer and Metastasis Reviews (2024)
-
A synthetic metastatic niche reveals antitumor neutrophils drive breast cancer metastatic dormancy in the lungs
Nature Communications (2023)
-
Chromatin profile-based identification of a novel ER-positive breast cancer subgroup with reduced ER-responsive element accessibility
British Journal of Cancer (2023)
-
The genomic regulation of metastatic dormancy
Cancer and Metastasis Reviews (2023)
-
Breast cancer: emerging principles of metastasis, adjuvant and neoadjuvant treatment from cancer registry data
Journal of Cancer Research and Clinical Oncology (2023)
