
Abstract
A meta-analysis approach was used, to study the microbiomes of biofilms and planktonic communities underpinning microbial electrosynthesis (MES) cells. High-throughput DNA sequencing of 16S rRNA gene amplicons has been increasingly applied to understand MES systems. In this meta-analysis of 22 studies, we find that acetogenic and methanogenic MES cells share 80% of a cathodic core microbiome, and that different inoculum pre-treatments strongly affect community composition. Oxygen scavengers were more abundant in planktonic communities, and several key organisms were associated with operating parameters and good cell performance. We suggest Desulfovibrio sp. play a role in initiating early biofilm development and shaping microbial communities by catalysing H2 production, to sustain either Acetobacterium sp. or Methanobacterium sp. Microbial community assembly became more stochastic over time, causing diversification of the biofilm (cathodic) community in acetogenic cells and leading to re-establishment of methanogens, despite inoculum pre-treatments. This suggests that repeated interventions may be required to suppress methanogenesis.
Similar content being viewed by others


Introduction
Microbial electrosynthesis (MES) is an emerging electro-bioprocess for CO2 conversion to green chemicals, which promises to complement other nascent and established technologies aimed at decarbonizing industrial production1. In MES cells, specific microorganisms – electrotrophs – form a biofilm and catalyse CO2 reduction by accepting electrons from a solid cathode electrode, either directly, or by mediated pathways2. Bioelectrocatalytic CO2 reduction offers significant advantages over thermo- and electro-catalytic counterparts, since microorganisms act as inexpensive, self-regenerating catalysts active under mild temperature and pressure conditions, potentially achieving coulombic efficiencies exceeding 90%3,4. Furthermore, if mixed-species consortia are used as inoculum, flexible and resilient biofilm communities are formed, which can be adapted to synthesise a wide array of products from flue gas streams containing variable CO2 concentrations and various contaminants. However, MES is constrained by the low current density that can be supported by the biological catalyst, which remains one to two orders of magnitude lower than those achieved in state-of-art electrolysers5.
MES was first described by Nevin et al.6, who reported that the acetogenic microorganism Sporomusa ovata is capable of reducing CO2 by using electrons accepted from a cathode electrode. Subsequently, this function was proposed for several other microorganisms, including other Sporomusa sp. as well as Clostridium sp. and Moorella sp., and even for archaeal species such as Methanobacterium sp. and Methanococcus sp.7. However, discerning cathodic, direct electron transfer from the common H2-mediated CO2 reduction pathway is highly challenging. Indeed, our understanding of electron transfer mechanisms and metabolic pathways in MES is further complicated when complex, undefined microbial communities, rather than pure cultures, are used as inoculum. In such cases, the MES product spectrum, yields, production rates, and coulombic efficiency will be strictly linked to the distribution, abundance and networking of the microbial taxa underpinning bioelectrochemical conversion processes. Therefore, understanding and predicting the composition of MES microbial communities that develop from different inoculum sources could help to optimize the process.
Anaerobic and activated sludges are popular inocula for MES cells, and are typically pre-treated to eliminate methanogenic archaea when methane is not the target product8. Pretreatment is essential, even when using activated sludge as inoculum, since it will contain acetoclastic and hydrogenotrophic methanogenic species (albeit at concentrations 1–2 orders of magnitude lower than in anaerobic sludge) since the nuclei of the activated sludge flocs are typically anaerobic9. Heat shock8,10 and chemical pre-treatment with 2-bromoethanesulphonic acid (BESA)11,12 are the most commonly applied techniques to inhibit methanogens in MES cells. However, the possible impact of inoculum pre-treatment on other members of the electrosynthetic community has not yet been elucidated.
Microorganisms colonise MES cells either in planktonic form, or by forming a biofilm on the cathode surface, resulting in two different microbial communities11,13, of which the various roles and interactions have not been fully elucidated. By studying the metagenome of a mixed-species MES culture producing acetate from CO2, Marshall et al.14 developed a model in which three dominant microorganisms (i.e., the electrotroph Desulfovibrio, the acetogen Acetobacterium and the microaerophilic Sulfurospirillum) were implicated in the predominant metabolic pathways. Furthermore, Cabau-Painado et al.15 developed a model to predict microbial kinetics in MES, and link them to the cell performance. However, defining a general model to link microbial dynamics and productivity of the highly variable MES processes is arduous considering the scale of information currently available. A necessary first step in this direction is to better understand the microbial communities underpinning MES processes, including relevant microbial interactions; evolution in response to operating parameters (primarily pH, temperature, cathode potential); and how they affect production in MES cells.
High-throughput DNA sequencing, especially of 16S rRNA gene amplicons, using platforms such as Illumina Miseq, has provided a powerful tool to study microbial communities in biotechnologies16. Such microbiomics have also been increasingly adopted in MES research (Fig. 6). However, only 27.4% of MES-related studies had deposited their sequencing data into public databases. Nonetheless, several terabytes of data are available on online repositories such as Short Read Archive (SRA), from MES cells enriched using various inocula and operated under a variety of conditions. However, single-study results have low reproducibility due to the highly variable experimental set-ups used. Meta-analysis approaches, whereby results from different studies are merged to increase the statistical significance and to offer a global perspective, will help in elucidating the microbial dynamics of MES cells. Hence, in this study, publicly available sequencing data, along with data kindly provided by several authors, were collated to provide a meta-analysis of 16 S rRNA gene sequences from MES cells. The core microbiomes of the biofilm and planktonic communities in acetogenic and methanogenic cells were identified. Furthermore, the effects on microbial communities of common inoculation and operational strategies were assessed, providing valuable information for optimisation of MES cell performance.
Results and discussion
Statistical distribution of the samples
The statistical distribution of the samples across the parameters analysed is supplied in the Supplementary Fig. 1. All 261 samples were categorised as either inoculum sources (9%), planktonic (29%) or cathodic (62%) MES communities. The MES cells were mainly inoculated with anaerobic sludge (62%), or less commonly with activated sludge (22%) or other inocula (16%), and were in most cases enriched under H2:CO2 or in previously-operated MES cells. Inocula without any pre-treatment were used in 33% of samples, mainly in methanogenic MES cells, whereas pre-treatment using either 2-bromoethanesulfonic acid (BESA) (51%) or heat shock (16%) was applied to biomass used in most of the acetogenic MES cells.
Samples originated from MES cells operated at a range of temperatures: below 30 °C (36%), between 30 and 45 °C (52%), and above 45 °C (12%). Sampling timepoint (i.e. the number of operating days elapsed at the moment of sampling) was below 50 d (28%), between 50 and 100 d (17%), between 100 and 200 d (21%), and above 200 d (34%). The pH ranged between 5 and 8 in 69% of cases, and was <5 or >8 in 24% and 7% of cases, respectively. The cathodic potential applied, ranged from −0.7 to −1.1 V vs Ag/AgCl, but mostly at either −0.8 V (26%) or −1.0 V (44%). Only one study17 was performed in galvanostatic mode at an applied current of 10 A/m2, whereas one study18 controlled the cell voltage with a power supply. Acetate (68%) or methane (29%) were the main products of the MES cells included in this meta-analysis, with widely variable production rates and coulombic efficiencies. Low concentrations of other products such as ethanol, butanol, butyrate, propionate and caproate were seldomly reported.
Key differences between cathodic and planktonic communities in acetogenic and methanogenic MES cells
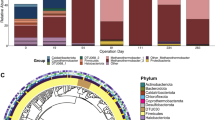
Samples were grouped by product (acetate or methane) and community type (planktonic or cathodic) for alpha diversity analysis, which indicated that species richness in the acetogenic planktonic community was only slightly higher than in the cathodic community but the difference was still deemed to be statistically significant (Fig. 1). The methanogenic planktonic community was significantly richer and more diverse than the cathodic community. However, the methanogenic planktonic group included only six samples and, therefore, this result is not conclusive. Cathodic and planktonic acetogenic communities clustered together in PCoA and were similar in both composition and phylogeny (Fig. 1). Samples from methanogenic MES systems clustered separately from the acetogenic MES samples in PCoA indicating that their microbial community compositions differed (Fig. 1). Indeed, PERMANOVA indicated that the main electrosynthesis product (acetate or methane) accounted for approximately 25% (p < 0.001) of the variance across all samples.

a Alpha Diversity Indices; Pielou’s Evenness, Rarefied Richness and Shannon Entropy for cathodic and planktonic communities in acetogenic and methanogenic cells. In the boxplots, center value lines indicate the median, boxes indicate the lower/upper quartiles (25%/75%) and lines extending parallel from the boxes (whiskers) show the variability outside the upper and lower quartiles. Lines of significance depict significant differences as follows: * (p < 0.05), ** (p < 0.01), or *** (p < 0.001) based on ANOVA. b Principal Component Analysis (Weighted Unifrac) of cathodic and planktonic communities in acetogenic and methanogenic cells, where ellipses were drawn using 95% confidence intervals based on standard deviation. c Heat Trees depicting differential abundances of taxa among the groups in question. The circle size and the colour intensity reflect the species abundance and the log2 median proportion between the two groups respectively.
Acetobacterium sp. were the most relatively abundant genus in the cathodic community (10–70% relative abundance; Supplementary Fig. 2). Exceptions included the two studies reporting thermophilic MES of acetate19,20, in which Moorella and Caloribacterium (at 50 °C), and Tepidiphilus and Coprothermobacter (at 70 °C), were enriched. Furthermore, Alqahtani et al.21 enriched a distinct community dominated by Proteobacteria, such as Marinobacter, and Firmicutes, such as Halanaerobiaceae, in MES cells inoculated with marine sediment using either a synthetic medium (10% salinity) or real brine (25% salinity) as electrolyte. Since the communities from these thermophilic and halophilic studies were so different from the other (mesophilic or ambient) studies, and only represented 31 samples, these have been excluded from all analysis other than the taxa plots (Supplementary Fig. 2). Methanobacterium sp. were dominant in methanogenic MES, with relative abundances often exceeding 50%. The exception was from Alqahtani et al.22, who enriched a distinct community with abundant Methanococcus in saline MES reactors (3.5% salinity) inoculated with salt marsh and mangrove sediments. Both Methanobacterium and Methanococcus are hydrogenotrophic archaea, which are generally recognised as the prevalent methane-producers in bioelectrochemical systems23.
We identified significant differences between the acetogenic and methanogenic cathodic communities using differential heat trees (Fig. 1). In particular, Firmicutes and Archaea (Fig. 1), which were more prominent in acetogenic and methanogenic systems respectively. The main difference between cathodic and planktonic acetogenic communities was that Acetobacterium sp. and Desulfovibrio sp. were significantly more abundant in the cathodic community (Fig. 1). Both genera were previously identified as key players in MES. Desulfovibrio sp. were hypothesised to accept electrons from the cathode through cytochromes, hydrogenases and/or formate dehydrogenase, and to subsequently reduce protons to molecular H2, and/or reduce CO2 to formate14. H2 and formate are then converted to acetate by Acetobacterium sp. through the Wood-Ljungdahl pathway14. Two more microbial genera (Arcobacteriaceae sp. and an uncultured Actinobacteria) were found upregulated within the cathodic community. It was suggested that certain Actinobacteria sp. can uptake electrons from the cathode24, whereas Arcobacter sp. is a sulphate reducer capable of CO2 fixation under autotrophic conditions, widely reported as member of the MES cathodic community25,26. Several microorganisms were more abundant in the planktonic community of acetogenic cells, including Gammaproteobacteria (including the aerobes Stenotrophomonas sp. and Pseudomonas sp.), Bacilli, Actinobacteria and Bacteroidia (Fig. 1). The growth of aerobic microorganisms is likely due to oxygen diffusion from the anodic compartment towards the cathodic compartment through the ion exchange membranes27.
Core microbiome of acetogenic and methanogenic cells
Defining the core microbiome as including genera present in 85% of the samples considered, we identified five qualifying genera from the acetogenic and methanogenic cathodic communities, and twelve genera in the acetogenic planktonic community, which were plotted at different detection thresholds (numbers of reads) (Fig. 2). Given the few samples available, the core methanogenic planktonic community was not considered. Interestingly, the core microbiome of the acetogenic and methanogenic cathodic community was very similar and differed by only one genus. Four genera (Christensenellaceae_R − 7_group; Desulfovibrio sp.; Lentimicrobium sp.; and an uncultured genus in the Synergistaceae family) were present in the cathodic core microbiomes of both acetogenic and methanogenic MES cells. Acetobacterium sp. were additionally present in acetogenic samples and Methanobacterium sp. were present in methanogenic samples (Fig. 2). Therefore, four shared genera may be keystone members in MES. The analysis confirms the key role of Desulfovibrio sp., which is believed to be the main hydrogen-producer in MES, providing reducing equivalents to autotrophic microorganisms, such as Acetobacterium sp. or Methanobacterium sp. The role of the other core genera remains unclear, however, it may be the case that they have an unknown syntrophic association with either Acetobacterium sp or Methanobacterium sp. Synergistaceae were present in the core microbiome at thresholds up to 882 reads in the cathodic community (Fig. 2), likely acting as syntrophic partners of either acetogenic or methanogenic populations. In fact, the Synergistaceae were previously described, along with Christensenellaceae, as part of the “highly abundant core community” of a full-scale anaerobic digestion plant28, suggesting their importance in multiple anaerobic bioprocesses.

a Highly prevalent taxa in the core microbiomes of (a) acetogenic cathodic, b methanogenic cathodic and c acetogenic planktonic communities. Detection thresholds (number of reads) are shown on the y-axis.
The five members of the acetogenic cathodic core microbiome were also part of the planktonic core community (Fig. 2). However, the planktonic core community was more diverse, including seven more genera. The presence of aerobic microorganisms, such as Stenotrophomonas sp. and Pseudomonas sp., in the planktonic core microbiome suggests that oxygen intrusion occurred in most MES studies, highlighting the importance of oxygen scavengers in maintaining anaerobic conditions near the cathode.
Impact of inoculum source, and pre-treatment, on community development
The majority of cells were inoculated with either anaerobic or activated sludge (Supplementary Fig. 1), which had variable microbial community composition (Supplementary Fig. 3). For acetogenic MES, inocula were commonly pre-treated with BESA or by heat-shock. Significantly higher richness and evenness was observed in communities originating from heat-shock-pretreated inocula than in BESA-pretreated inocula (Supplementary Fig. 4). Pre-treated samples (BESA or heat shock) clustered together in PCoA (Fig. 3), and pre-treatment explained 33% of the total variability among samples (p = 0.001). Clustering based on pretreatment (Fig. 3) also aligned closely to clustering based on the product (acetate or methane), indicating that pre-treatment was an important factor influencing the MES microbiome and the metabolites produced.

a Principal Component Analysis (Weighted Unifrac) showing acetogenic and methanogenic samples clustered by pretreatment method where ellipses were drawn using 95% confidence intervals based on standard deviation. b Differential heat tree depicting differently abundant taxa between two groups based on seeding strategy (heat shocked activated sludge Vs BESA treated anaerobic sludge). The circle size and the colour intensity reflect the species abundance and the log2 median proportion between the two groups respectively. c Differential heat tree depicting differently abundant taxa between two groups based on seeding strategy (untreated anaerobic sludge Vs BESA treated anaerobic sludge). The circle size and the colour intensity reflect the species abundance and the log2 median proportion between the two groups respectively. Full size, high-resolution heat trees are provided in the Supplementary Information (Supplementary Figs. 6, 7).
Anaerobic sludge pre-treated with BESA and activated sludge pre-treated by heat-shock were the most common inocula used in acetogenic MES cells. Firmicutes were more abundant in heat-treated, activated sludge, including spore-forming microorganisms such as Lactobacillales, Sporomusaceae and several Clostridia (Fig. 3). Full-size, high-resolution heat trees are provided in the Supplementary Information (Supplementary Figs. 6, 7). The ability of these microorganisms to form spores29 infers an advantage that selects them in heat-shock pretreated inocula. Undesired methanogens, such as Methanobacterium sp. and Methanosaeta sp., were more abundant in BESA-pretreated anaerobic sludge (Fig. 3), which suggests that heat shock is a better pretreatment to supress methanogenesis. However, heat shock is less specific and can also affect other important organisms such as Acetobacterium sp., which were significantly less abundant in MES cells seeded with heat-shocked activated sludge (Fig. 3). Microorganisms involved in chain elongation and propionate production, such as Caproiciproducens sp. and Propionibacterium sp., were also less abundant when heat-shocked activated sludge was used as inoculum. Hence, heat shock should be avoided when targeting middle-chain fatty acids, and more specific strategies to inhibit methanogenic activity should then be considered.
As expected, methanogenic archaea were less abundant in (typically acetogenic) MES cells seeded with BESA pre-treated anaerobic sludge than in (typically methanogenic) MES cells seeded with untreated sludge (Fig. 3). However, methanogens such as Methanobrevibacter sp. and Methanobacterium sp. correlated positively with the sampling timepoint in acetogenic MES cells (Fig. 4), suggesting that methanogens are only temporarily inhibited by BESA pretreatment. This could be due to either adaptation of the community, or degradation of the inhibitor. It was indeed reported that sulphate-reducing bacteria, including Desulfovibrio sp., can reduce BESA to H2S12. Therefore, pre-treatment needs to be periodically repeated to reliably inhibit methanogenesis. However, in this study, Desulfovibrio sp. was lower in abundance in samples treated with BESA, which disagrees with its possible role in BESA reduction in MES cells. Conversely, several acetogenic Firmicutes (including Acetobacterium sp. and Sporomusa sp.), and potential chain-elongating microorganisms (Caproiciproducens sp. and Clostridium_sensu_stricto_12), were more abundant in BESA-treated MES cells, potentially favoured when competition from methanogens is removed. Once again, this result indicates that BESA may offer a better pre-treatment when targeting the production of middle-chain fatty acids.

Correlation heatmap of the 25 most abundant genera from (a) acetogenic, and (b) methanogenic cells with environmental variables. Kendall correlations between the taxa and the environmental variables were calculated. Significance levels are indicated by asterisks as *(p < 0.05), **(p < 0.01) or ***(p < 0.001).
Response of microbial communities to MES operating conditions
Several significant correlations were identified between genera in acetogenic, and methanogenic, MES cells and operating, or performance, parameters (pH, temperature, potential applied, coulombic efficiency, production rate and product concentration) (Fig. 4). Sporomusa sp. correlated positively with temperature (in the range 10–35 °C, since thermophilic samples were excluded) in acetogenic cells and were more abundant under mesophilic conditions. However, Sporomusa sp. have also been observed to grow autotrophically in MES cells at lower temperatures30. Other key genera, such as Clostridium sp., Acetobacterium sp. and Desulfovibrio sp., did not correlate with temperature. Desulfovibrio sp. did, however, correlate positively with pH. This could explain the higher abundance of Desulfovibrio in the cathodic rather than planktonic community, as the local pH at the surface of the cathode is higher due to alkalization phenomena31. Clostridium sp. and Oscillibacter sp. correlated negatively with pH, indicating that they prefer mildly acidic conditions. Interestingly, Sporomusa sp. correlated negatively with acetate concentration in the catholyte, suggesting lower tolerance for product accumulation than other acetogens, such as Acetobacterium sp. (Fig. 4).
Significant correlations were also found between several organisms and the cathodic potential applied to the MES cell (ranging from −0.8 to −1.1 V vs Ag/AgCl) (Fig. 4). Sporomusa sp. correlated positively with potential, suggesting its ability to perform electrosynthesis at relatively higher potentials than other acetogenic microorganisms. Acetogenesis with Sporomusa ovata was observed at potentials as high as −0.4 vs SHE (−0.6 vs Ag/AgCl)6, although the electron transfer mechanism for CO2 reduction at such potential is still debated. While direct electron transfer from the cathode to microorganisms appeared as the most reasonable hypothesis, due to the insufficient potential to generate hydrogen abiotically, it was recently argued that microbially-secreted metabolites/cell-components might enable H2 evolution at higher potentials32. This is particularly true in mixed-culture MES, wherein several microorganisms can participate in H2 catalysis at the cathode, thus making H2-mediated electron transfer the prevalent acetogenic route. Furthermore, acetogenic microorganisms can increase the onset potential and evolution rate of H2 by promptly consuming it and maintaining low partial pressure near the cathode33.
The abundance of key microorganisms, such Desulfovibrio sp. and Acetobacterium sp., did not correlate with the applied potential, suggesting their flexibility, whereas Sulfurospirillum sp. preferred low potentials (Fig. 4). Sulfurospirillum sp. are capable of carbon fixation using H2 as reducing agent, and were suggested to protect the strictly anaerobic members of the community by consuming traces of oxygen often present in MES cathodes, albeit at the cost of acetate consumption14. Thus, Sulfurospirillum sp. are favoured at low potentials both directly, due to the faster H2 evolution, and indirectly, due to the fast accumulation of acetic acid resulting from the abundance of reducing equivalents. Similarly, the preference of Oscillibacter sp., suspected chain-elongating microorganisms11, for low potential can be attributed to the observed higher acetate production rates. This hypothesis is supported by the fact that both Sulfurospirillum and Oscillibacter sp. correlated positively with acetate concentration in the catholyte (Fig. 4).
In methanogenic MES cells, several microorganisms, including acetoclastic Methanosaeta sp., correlated positively with pH (ranging between 6.8 and 8.5 among the samples). This was not the case for Methanobacterium sp., which are often dominant in methanogenic MES under both neutral and slightly alkaline pH conditions. Overall, the methanogenic community appears to be less associated with temperature and applied potential than the acetogenic community (Fig. 4).
Cathodic microbial community assembly
To assess microbial community assembly processes in the cathodic community over time, samples were grouped into four ranges based on the sampling timepoint (i.e. on the duration of cell operation). Those were: 0–50 days, 51–100 days, 101–200 days and 200+ days. Only acetogenic MES samples were considered due to the lack of methanogenic MES samples older than 100 d. Shannon diversity and species richness increased significantly over time from young (<100 d) to old (>100 d) biofilms (Fig. 5). This is an interesting finding, since MES cells are typically operated as semi-closed systems, allowing the entry only of gas, thus excluding immigration as a source of de novo richness and diversity.

a Alpha Diversity Indices; Pielou’s Evenness, Rarefied Richness and Shannon Entropy for cathodic acetogenic communities. In the boxplots, center value lines indicate the median, boxes indicate the lower/upper quartiles (25%/75%) and lines extending parallel from the boxes (whiskers) show the variability outside the upper and lower quartiles. Lines of significance depict significant differences as follows: * (p < 0.05), ** (p < 0.01), or *** (p < 0.001) based on ANOVA. b Normalized stochasticity ratio (NST) using Ružička metric and Taxa-Richness constraints of proportional-fixed (P-F) and proportional-proportional (P-P) which stipulates that the probabilities of taxa occurrence are proportional to the observed occurrence frequencies, and taxon richness in each sample is either fixed or proportional. c Scatter plot indicating winner diversity and winner prevelance for genera exhibiting lottery model-like behaviour (a genera member makes up >90% of the genera’s abundance). Samples in each panel are grouped into four time periods 0–50 d, 51–100 d, 101–200 d and 200 + d.
The study of patterns of microbial community assembly has gained popularity in recent years and may be useful in understanding, and controlling, microbial communities in engineered systems, such as MES cells. This facet of microbial ecology describes the forces governing community development to determine mechanisms and factors controlling microbial diversity34. Community assembly mechanisms broadly fit two categories: deterministic community assembly, which indicates that interspecies interactions, or environmental gradients, create niches, influencing patterns of diversity35,36; and stochastic community assembly, which implies that community assembly is governed by more-neutral processes, such as ecological drift, birth and death dynamics, speciation, and immigration35,36.
NST analysis was applied to consider whether deterministic or stochastic mechanisms dominated cathodic community assembly. A value of >0.50 indicates a community is more influenced by stochastic factors, whereas a value <0.50 indicates deterministic forces are prominent. Early biofilm development was more influenced by deterministic than stochastic processes (Fig. 5). This was likely caused by adaptation of the community to the new environment (i.e., MES cathode and electrolyte) or effects of BESA or heat-shock pre-treatments. In older samples (>100 d), stochasticity in cathode community increased significantly, leading to higher richness and diversity (Fig. 5). Since the relative abundance of acetogen competitors, such as Methanobacterium sp., increased over time (Fig. 4), the stochasticity observed in older biofilms may be detrimental to cell performance. Therefore, a management strategy may be required to reduce stochasticity and maintain acetogenic populations. This may include periodic inhibition of the methanogenic population coupled with acetate extraction to limit the toxic effect of product accumulation on the acetogenic community.
Lottery model analysis identified Methanobacterium sp. as ‘lottery winners’, indicating that in the Methanobacterium genus a single ASV often made up 90% of its abundance. In early biofilm samples (0–50 d) Methanobacterium sp. had high winner prevalence but very low winner diversity, meaning that in most (>70%) samples a single Methanobacterium ASV was dominant. However, in older biofilms (100 + d) Methanobacterium lottery winners were more diverse. Since it is unlikely that new Methanobacterium ASVs were introduced by immigration, stochasticity may have caused them to proliferate through acclimatisation, adaptation or even speciation, providing more potential winning ASVs.
The second lottery winner was Desulfovibrio sp., which had high winner prevalence and high winner diversity in the young biofilms, strongly suggesting that several members of this genus can quickly colonise the cathode. Winner prevalence and diversity lessened over time, indicating that fewer, though highly competitive, Desulfovibrio ASVs dominanted older biofilms. Furthermore, winner behaviour was apparent in fewer samples in aged biofilms, indicating Desulfovibrio was gradually outcompeted, with only a few highly competitive ASVs remaining. This result is in agreement with the negative correlation found between Desulfovibrio and sampling timepoint (Fig. 4). It is interesting that only two genera were identified as displaying lottery winner behaviour, as this indicates that none of the other genera were dominated by single ASVs. This result suggests that MES cells strongly select for specific Desulfovibrio and Methanobacterium ASVs, whereas the composition of other genera was more random and varied between studies.
Correlation between key microorganisms and MES cell performance
Sporomusa sp abundance correlated strongly with coulombic efficiency (CE) (Fig. 4). The Sporomusa genus includes several highly efficient species, such as S. ovata and S. malonica, capable of acetogenesis in MES with >90% CE30. Such high CE can be attributed to the direct electron uptake capability37 and/or to the efficient metabolism of Sporomusa sp. using H2 as electron donor30. Acetobacterium sp. abundance also correlated positively with CE, whereas neither Acetobacterium nor Sporomusa sp. correlated with production rates. Production rates are likely more dependent on the operating parameters (e.g. the concentration of reducing equivalents) than the organism performing acetogenesis. No correlation was found between Sulfurospirillum sp. abundance and CE. However, it correlated positively with production rate, confirming the importance of its oxygen scavenging role in MES. As expected, methanogens such as Methanobacterium and Methanobrevibacter sp. were associated with low CE and/or low acetate production rates. More surprisingly, Clostridium sensu stricto 12 correlated negatively with CE, suggesting its role in chain elongation pathways, rather than acetogenesis, as previously hypothesised38.
In methanogenic MES cells, neither CE nor methane production rates correlated with Methanobacterium sp., which dominates the microbial community of methanogenic MES cells regardless of their productivity. In fact, Methanobacterium sp. constituted around 40% of the community of the granular sludge-based electromethanogenic reactor that achieved the highest methane production rate so far, with 85% CE18, but was also highly abundant in poorly performing cells (Supplementary Fig. 2). Methanobacterium is a highly resilient genus of hydrogenotrophic microorganisms, which prevails in methanogenesis under stressed conditions28. In MES cells, such conditions are promoted by local pH gradients at the electrodes; ion migration; and oxygen intrusion from the anode chamber. Interestingly, Methanosarcina, sp. which are capable of both hydrogenotrophic and acetoclastic metabolism, were associated with low CE and methane production rates (Fig. 3). Cytochrome-containing archaea, such as some Methanosarcina sp., show restricted growth on H2 and CO2, as well as at least a ten-fold higher H2 partial pressure threshold than archaea lacking cytochromes, such as Methanobacterium39. Thus, it is plausible that in well-performing cells, Methanobacterium keeps the H2 partial pressure at the cathode low and thus outcompetes Methanosarcina, which is abundant in poorly-performing cells due to H2 accumulation.
To conclude, meta-analysis revealed the key role of H2-producing Desulfovibrio electrotrophs in early-stage MES biofilms, suggesting that both acetogenesis and methanogenesis are H2-mediated. Heat shock is a more effective pre-treatment than BESA to prevent the onset of methanogenesis, but may hamper product diversification, and should be avoided when targeting middle chain carboxylic acid production. Oxygen-scavenging microorganisms were important members of the core planktonic MES community. Their positive correlation with CE confirmed their important role in efficient MES cells, and that oxygen intrusion from the anode chamber is an unresolved challenge. Richness, diversity and stochasticity in the acetogenic cathodic community increase over time, mainly due to increasing competition from methanogens, suggesting that pre-treatment of the microbial community should be periodically repeated.
Methods
Data collection
Research articles on MES were retrieved using the Google Scholar, Scopus and Elsevier search engines using the keywords “microbial electrosynthesis”, “MES”, “biocathode”, and “bioelectro”. The resulting articles were screened to select the studies that included 16 S rRNA gene sequence analysis on the Illumina Miseq or Hiseq platforms, and which used universal bacterial and archaeal oligonucelotide primers. The reason for curtailing the studies to just those that utilised Illumina platform is based on authors’ recent work where it was shown, based on mock communities, that Illumina platforms are more quantitative with less systematic biases40. After initial screening, 61 articles were considered suitable for the study, although sequencing data were available in public databases from only 16 (26.2%) of the studies. Sequencing data from an additional 16 studies were requested, and obtained, from the respective authors, while the remaining 29 articles (46.8%) were regrettably excluded from the study due to the unavailability of the data (Fig. 6).

Suitable publicly available data, along with data requested and obtained from the Authors, was included to the meta-data table. Data from 2021 includes only studies published by June.
Ten more articles were excluded in a second screening due to poor sequencing quality and/or unavailability of metadata, resulting in a final total of 22 studies in the meta-analysis. The studies included are listed in the Supplementary Table 1. Publicly available data were downloaded using the Sequence Read Archive (SRA) toolkit provided by the NCBI. Metadata were collected from the NCBI and directly from the respective published articles. A total of 261 samples were included in a meta-data table and categorised based on parameters, including: primers used; region of the 16 S rRNA gene targeted; inoculum and pre-treatment used; microbial community type (cathodic, planktonic or inoculum); sampling timepoint (i.e. in the MES trial); potential/current applied; operating parameters (e.g., pH, temperature); product spectrum, concentration and production rates; and coulombic efficiency. When the required data were not explicitly provided in the text, it was obtained by extrapolating an approximate value from the figures in the manuscript, where possible. When necessary, units of measurement were converted to allow inter-study comparisons.
Sequence processing
Sequences from each study were processed individually using Qiime2. Since multiple V regions of the 16 S rRNA gene were considered in this study, the DEBLUR algorithm was used to generate amplicon sequence variants (ASVs). DEBLUR was used over DADA2 because, for longer amplicon regions (e.g, V3-4), sometimes DADA2 is unable to give the optimum number of ASVs as the denoising is done separately for forward and reverse reads before finding sufficient overlap to assemble them together. Taxonomy was assigned using a Bayesian Least Common Ancestor (BLCA) approach (using the SILVA138 database) as recommended by Keating et al.41, to obtain more sequence-level resolution. Since multiple V-regions were used, and some do not overlap, the idea was to collate together as many ASVs as possible to facilitate the generation of phylogentic tree based on full 16 S rRNA sequences. ASVs from each study (matched to the reference database) were collated in a single biom file and sequences without sequence-level assignments were removed. Our collation strategy is already evaluated to cause minimal loss in beta diversity and is mentioned in detail in Thom et al.42, with the code already released as part of Keating et al.41. Our analysis suggested that the beta diversity patterns between samples (both visual and statistical cues) were preserved when using a minimal set of well-resolved ASVs at the sequence level, as justified in Figures Supplementary Figs. 8–15. Finally, a phylogenetic tree and a biom file with taxonomy were generated using MAFFT and QIIME2, respectively.
Statistical analysis
All further statistical analysis was done in R (version 3.6.2) using the collated biom file, taxonomy file and phylogenetic tree. Samples were grouped based on relevant metadata parameters, such as ‘community type’ or the main product synthesised by the MES cell. Alpha and beta diversity analyses were done with the Vegan package. Alpha diversity indices used included: rarefied richness, Shannon entropy and Pielou’s evenness. Beta diversity was observed using Principal Coordinate Analysis (PCoA) with the Weighted Unifrac distance metric. Analysis of variance in relation to explanatory variables in the metadata was done using Vegan’s Adonis() function with the Weighted Unifrac distance metric. This function, referred to as permutational analysis of variance (PERMANOVA), fits linear models to distance matrices and used a permutation test with pseudo-F ratios. The core microbiome was determined by identifying taxa with a minimum prevalence of 85% across all samples considered. Whilst there is no single threshold to describe what is core microbiome, particularly when there can be inter-study variations due to biases accumulated by study design, choice of V region, and extraction protocol, we have followed the authors recommendations in terms of absolute read counts43. Correlation analysis was used to identify potential relationships between abundant genera and selected environmental variables using Kendall correlation, a nonlinear measure that is more robust. The p-values for correlations were adjusted for multiple testing using the Benjamini and Hochberg procedure to give significant associations. Taxa with significantly differential abundance between groups were visualised using heat trees with a Wilcoxon p-value test adjusted for multiple comparisons on the proportional microbiome data, as recommended by Foster et al.44.
Stochasticity in community assembly was assessed using a Normalized Stochasticity Ratio (NST) with various distance measures (incidence-based: Jaccard, Kulczynski and abundance-based: Ruzcika, Kulczynski) using the NST package. A 50% stochasticity ratio was used to identify more deterministic (<50%) and more stochastic (>50%) assembly45. For the calculations, taxa occurrence frequency and the sample richness were constrained as proportional (P) or fixed (F) in the combinations PF or PP, and 1,000 randomizations were performed for each model. We have adopted this approach after original authors’ detailed benchmarking of different choices available. Permutational multivariate ANOVA (PANOVA) was then used to assess statistical significance.
A competitive lottery model was used to identify ASVs that dominated their clade (i.e. genera)46,47. The model assumes competition among phylogenetically similar species, which will occasionally result in one species, or ASV, dominating by making up >90% of the abundance within that clade (based on a stickbreaking model used on simulated communities)46. Here, the lottery ‘winners’ were identified as ASVs dominating a particular genus. Genera with lottery-type behaviour (i.e. containing lottery winners) were then plotted in terms of winner prevalence (proportion of samples having a ‘winner ASV’ among that genus) and winner diversity (the diversity of winning ASVs in samples from which winners were observed). Diversity values closer to 0 suggest that a single ASV consistently dominates that genus across all samples, whilst high diversity values suggest more evenly spread ASVs as winners in that group.
Data availability
E- supplementary data of this work can be found in online version of the paper. A list of all studies used in this meta-analysis has been provided in the supplemental material. Publically available sequencing data has been obtained from the NCBI sequence Read Archive. Data from studies which are not publically available were provided upon request by the authors.
Code availability
All code used in this manuscript has been made available at https://userweb.eng.gla.ac.uk/umer.ijaz/#bioinformatics
References
Grim, R. G. et al. Transforming the carbon economy: challenges and opportunities in the convergence of low-cost electricity and reductive CO2 utilization. Energy Environ. Sci. 13, 472–494 (2020).
Karthikeyan, R., Singh, R. & Bose, A. Microbial electron uptake in microbial electrosynthesis: a mini-review. J. Ind. Microbiol. Biotechnol. 46, 1419–1426 (2019).
Dessì, P. et al. Microbial electrosynthesis: Towards sustainable biorefineries for production of green chemicals from CO2 emissions. Biotechnol. Adv. 46, 107675 (2021).
Jourdin, L., Freguia, S., Flexer, V. & Keller, J. Bringing high-rate, CO2‑based microbial electrosynthesis closer to practical implementation through improved electrode design and operating conditions. Environ. Sci. Technol. 50, 1982–1989 (2016).
Claassens, N. J., Cotton, C. A. R., Kopljar, D. & Bar-Even, A. Making quantitative sense of electromicrobial production. Nat. Catal. 2, 437–447 (2019).
Nevin, K. P., Woodard, T. L., Franks, A. E., Summers, Z. M. & Lovley, D. R. Microbial Electrosynthesis: Feeding Microbes Electricity To Convert Carbon Dioxide and Water to Multicarbon Extracellular Organic Compounds. MBio 1, 2 (2010).
Logan, B. E., Rossi, R., Ragab, A. & Saikaly, P. E. Electroactive microorganisms in bioelectrochemical systems. Nat. Rev. Microbiol. 17, 307–319 (2019).
Izadi, P., Fontmorin, J. M., Godain, A., Yu, E. H. & Head, I. M. Parameters influencing the development of highly conductive and efficient biofilm during microbial electrosynthesis: the importance of applied potential and inorganic carbon source. npj Biofilms Microbiomes 6, 1–15 (2020).
Wu, W. et al. Cultivation of anaerobic granular sludge in UASB reactors with aerobic activated sludge as seed. Water Res. 21, 789–799 (1987).
Izadi, P., Fontmorin, J. M., Virdis, B., Head, I. M. & Yu, E. H. The effect of the polarised cathode, formate and ethanol on chain elongation of acetate in microbial electrosynthesis. Appl. Energy 283, 116310 (2021).
Dessì, P. et al. Carboxylic acids production and electrosynthetic microbial community evolution under different CO2 feeding regimens. Bioelectrochemistry 137, 107686 (2021).
Bian, B., Xu, J., Katuri, K. P. & Saikaly, P. E. Resistance assessment of microbial electrosynthesis for biochemical production to changes in delivery methods and CO2 flow rates. Bioresour. Technol. 319, 124177 (2021).
Song, Y. E. et al. Biofilm matrix and artificial mediator for efficient electron transport in CO2 microbial electrosynthesis. Chem. Eng. J. 427, 131885 (2022).
Marshall, C. W. et al. Metabolic Reconstruction and Modeling Microbial Electrosynthesis. Sci. Rep. 7, 8391 (2017).
Cabau-Peinado, O., Straathof, A. J. J. & Jourdin, L. A General Model for Biofilm-Driven Microbial Electrosynthesis of Carboxylates From CO2. Front. Microbiol. 12, 1–17 (2021).
Thompson, L. R. et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017).
Chu, N. et al. Waste C1 Gases as Alternatives to Pure CO2 Improved the Microbial Electrosynthesis of C4 and C6 Carboxylates. ACS Sustain. Chem. Eng. 8, 8773–8782 (2020).
Zhou, H. et al. Optimization of a newly developed electromethanogenesis for the highest record of methane production. J. Hazard. Mater. 407, 124363 (2021).
Rovira-Alsina, L. et al. Thermophilic bio-electro CO2 recycling into organic compounds. Green. Chem. 22, 2947–2955 (2020).
Yang, H. Y. et al. Mixed-culture biocathodes for acetate production from CO2 reduction in the microbial electrosynthesis: Impact of temperature. Sci. Total Environ. 790, 148128 (2021).
Alqahtani, M. F. et al. Enrichment of Marinobacter sp. and Halophilic Homoacetogens at the Biocathode of Microbial Electrosynthesis System Inoculated With Red Sea Brine Pool. Front. Microbiol. 10, 1–18 (2019).
Alqahtani, M. F. et al. Enrichment of salt-tolerant CO2–fixing communities in microbial electrosynthesis systems using porous ceramic hollow tube wrapped with carbon cloth as cathode and for CO2 supply. Sci. Total Environ. 766, 142668 (2021).
Siegert, M., Li, X. F., Yates, M. D. & Logan, B. E. The presence of hydrogenotrophic methanogens in the inoculum improves methane gas production in microbial electrolysis cells. Front. Microbiol. 5, 1–12 (2014).
Kobayashi, H. et al. Analysis of a Methanogen and an Actinobacterium Dominating the Thermophilic Microbial Community of an Electromethanogenic Biocathode. Archaea 4, 6 (2021).
Izadi, P., Gey, M. N., Schlüter, N. & Schröder, U. Bidirectional electroactive microbial biofilms and the role of biogenic sulfurin charge storage and release. iScience 24, 102822 (2021).
Mateos, R., Sotres, A., Alonso, R. M., Moran, A. & Escapa, A. Enhanced CO2 Conversion to Acetate through Microbial Electrosynthesis (MES) by Continuous Headspace Gas Recirculation. Energies 12, 3297 (2019).
Huang, D., Song, B. Y., Li, M. J. & Li, X. Y. Oxygen diffusion in cation-form Nafion membrane of microbial fuel cells. Electrochim. Acta 276, 268–283 (2018).
Trego, A. C. et al. First proof of concept for full-scale, direct, low-temperature anaerobic treatment of municipal wastewater. Bioresour. Technol. 341, 125786 (2021).
Galperin, M. Y. Genome Diversity of Spore-Forming. Firmicutes. Microbiol. Spectr. 1, 1–27 (2013).
Aryal, N., Tremblay, P. L., Lizak, D. M. & Zhang, T. Performance of different Sporomusa species for the microbial electrosynthesis of acetate from carbon dioxide. Bioresour. Technol. 233, 184–190 (2017).
Zeppilli, M., Paiano, P., Torres, C. & Pant, D. A critical evaluation of the pH split and associated effects in bioelectrochemical processes. Chem. Eng. J. 422, 130155 (2021).
Tremblay, P. L., Faraghiparapari, N. & Zhang, T. Accelerated h2 evolution during microbial electrosynthesis with sporomusa ovata. Catalysts 9, 1–11 (2019).
Philips, J. Extracellular electron uptake by acetogenic bacteria: Does H2 consumption favor the H2 evolution reaction on a cathode or metallic iron? Front. Microbiol. 10, 2997 (2020).
Zhou, J. & Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 81, e00002–e00017 (2017).
Chase, J. M. Stochastic Community Assembly Causes Higher Biodiversity in More Productive Environments. Sci. (80-.). 328, 1388–1391 (2010).
Ning, D. et al. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 11, 4717 (2020).
Nevin, K. P. et al. Electrosynthesis of organic compounds from carbon dioxide is catalyzed by a diversity of acetogenic microorganisms. Appl. Environ. Microbiol. 77, 2882–2886 (2011).
Liu, B., Kleinsteuber, S., Centler, F., Harms, H. & Sträuber, H. Competition Between Butyrate Fermenters and Chain-Elongating Bacteria Limits the Efficiency of Medium-Chain Carboxylate Production. Front. Microbiol. 11, 1–13 (2020).
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W. & Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591 (2008).
D’Amore, R. et al. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics 17, 55 (2016).
Keating, C., Trego, A. C., Sloan, W., O\textquoterightFlaherty, V. & Ijaz, U. Z. Circular Economy of Anaerobic Biofilm Microbiomes: A Meta-Analysis Framework for Re-exploration of Amplicon Sequencing Data. bioRxiv https://doi.org/10.1101/2020.12.23.424166 (2020).
Thom, C., Smith, C. J., Moore, G., Weir, P. & Ijaz, U. Z. Microbiomes in drinking water treatment and distribution: A meta-analysis from source to tap. Water Res. 212, 118106 (2022).
Lahti, L., Shetty, S., Blake, T. & Salojarvi, J. Tools for microbiome analysis in R. Microbiome package. (2019).
Foster, Z. S. L., Sharpton, T. J. & Grünwald, N. J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLOS Comput. Biol. 13, e1005404 (2017).
Ning, D., Deng, Y., Tiedje, J. M. & Zhou, J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl Acad. Sci. 116, 16892–16898 (2019).
Verster, A. J. & Borenstein, E. Competitive lottery-based assembly of selected clades in the human gut microbiome. Microbiome 6, 186 (2018).
Trego, A. C. et al. Combined Stochastic and Deterministic Processes Drive Community Assembly of Anaerobic Microbiomes During Granule Flotation. Front. Microbiol. 12, 1165 (2021).
Acknowledgements
PD received funding from Science Foundation Ireland (award nr. 19/FIP/ZE/7572PF). UZI was funded by NERC IRF NE/L011956/1 and both UZI, WTS are additionally funded by EPSRC (EP/P029329/1 and EP/V030515/1). GC and SM were supported by a European Research Council Starting Grant (3C-BIOTECH 261330) and by a Science Foundation Ireland Career Development Award (17/CDA/4658) to GC.
Author information
Authors and Affiliations
Contributions
S.M. and P.D. conceptualised the study, collected the data, performed the analyses and drafted the manuscript and are therefore co-first authors of the study. D.P. contributed in data collection; U.Z.I. wrote the code for data collation and analysis; G.C. and U.Z.I. contributed to interpretation of the data; D.P., P.F., W.T.S., G.C. and U.Z.I. thoroughly revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mills, S., Dessì, P., Pant, D. et al. A meta-analysis of acetogenic and methanogenic microbiomes in microbial electrosynthesis. npj Biofilms Microbiomes 8, 73 (2022). https://doi.org/10.1038/s41522-022-00337-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-022-00337-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
