
Abstract
Supported nanoclusters (SNCs) with distinct geometric and electronic structures have garnered significant attention in the field of heterogeneous catalysis. However, their directed synthesis remains a challenge due to limited efficient approaches. This study presents a plasma-assisted treatment strategy to achieve supported metal oxide nanoclusters from a rapid transformation of monomeric dispersed metal oxides. As a case study, oligomeric vanadia-dominated surface sites were derived from the classic supported V2O5-WO3/TiO2 (VWT) catalyst and showed nearly an order of magnitude increase in turnover frequency (TOF) value via an H2-plasma treatment for selective catalytic reduction of NO with NH3. Such oligomeric surface VOx sites were not only successfully observed and firstly distinguished from WOx and TiO2 by advanced electron microscopy, but also facilitated the generation of surface amide and nitrates intermediates that enable barrier-less steps in the SCR reaction as observed by modulation excitation spectroscopy technologies and predicted DFT calculations.
Similar content being viewed by others
Introduction
Solid catalysts are employed in the production of over 80% of chemicals on a global scale1. Atomically dispersed supported solid catalysts, including single-atom catalysts (SACs) and multi-atom cluster catalysts, have recently garnered significant attention because of the maximum atom utilization, optimized charge distribution, and tuned coordination environment2,3,4,5. The SACs possess well-defined active centers and a unique confinement effect, while they may not be universally applicable to reactions that require multinuclear or adjacent active sites6,7,8. Beyond a simple combination of SACs, multi-atom cluster catalysts could result in enhanced activity because of the synergistic effects between adjacent atoms8,9,10. Such nanocatalysts have demonstrated remarkable catalytic performance in various reactions, such as CO oxidation, selective oxidation of hydrocarbons, selective catalytic reduction, selective hydrogenation, and electrochemical CO2 reduction8,9,10. However, achieving precise control over smaller oligomeric clusters (i.e., metal-oxo or metal cluster) is notably challenging, as they are highly susceptible to undergoing Oswald ripening, resulting in the gradual enlargement of these smaller clusters and formation of nanoparticles11,12,13,14.
Selective catalytic reduction of NOx with NH3 (i.e., NH3-SCR) to benign N2 and H2O reaction products by supported vanadia-based catalysts has been widely applied to control NOx emission from coal- and natural gas-fired power plants15,16,17,18,19. It is found that the active moieties of the supported vanadia-based catalysts are largely determined by the dispersed vanadyl surface sites20,21. Oligomeric surface vanadyl sites (dimers, trimers, etc.) have been proposed to hold higher intrinsic activity than isolated vanadyls sites at low temperatures for this bimolecular reaction22,23,24. Recently, the achievement of predominantly oligomeric vanadia surface sites for supported SCR catalysts was shown to be regulated through the loading amount of the active surface VOx or the surface WOx promoter prepared by incipient-wetness impregnation. With careful control of the vanadia loading below monolayer surface coverage on the anatase support (~0.1 wt% V2O5/m2 TiO2 or ~8 V atoms/nm2), the predominant surface VOx sites are found to be monomeric al low surface coverage (<0.03 wt% V2O5/m2 TiO2) and oligomeric at high surface coverage (0.05~0.1 wt% V2O5/m2 TiO2)20,22,25,26,27. However, as a case of the commercial TiO2 P25 support (56 m2/g), achieving the predominant formation of oligomeric vanadia surface sites requires a vanadia loading of ~3.5 wt% V (5 wt% V2O5), which would cause a serious degradation of the N2 selectivity and potential biological toxicity from volatilization of some vanadia27,28. Hence, it remains a significant challenge to precisely regulate a high concentration of vanadia clusters for commercially supported vanadia-based catalysts at low contents of active components. Despite extensive research recognizing the pivotal role of nitrates in the NH3-SCR reaction, the current study falls short in elucidating the correlation between nitrate formation and catalyst structure and lacks atomic-level insights into how nitrates participate in the reaction. There is a pressing need to address this issue and provide atomic-level insights into how nitrates engage in the NH3-SCR reaction29,30,31,32,33,34.
As illustrated in Fig. 1a, in the present study, a novel approach for the synthesis of supported vanadyl nanoclusters is presented through a secondary-level H2 plasma modification of conventional supported vanadia-based catalysts. Structural characterization revealed the transformation of isolated surface vanadyl sites into vanadia nanoclusters on the modified TiO2 surface during the plasma treatment. The resulting catalyst exhibited remarkable catalytic activity and stability in the NH3-SCR at low temperatures, with a tenfold increase in the turnover frequency (TOF). With the capabilities of advanced scanning transmission electron microscopy (STEM) and modulation excitation spectroscopy (MES) technologies as well as density functional theory (DFT) theoretical calculations, the distribution of supported vanadia clusters was identified at the atomic scale and the NH3-SCR reaction pathway was found to involve surface nitrate reaction intermediates.

a Schematic of the surface vanadyl species under plasma treatment over vanadia-based catalysts. HRTEM images of (b) OR and c PL. Selected high-angle annular dark field (HAADF) images of (d) OR and (e) PL. Enlarged view of the yellow region in d, (f) and e, g. 2D atomic maps of the EELS signals of (h) Ti, (i) O, and j V and W in combination with the simultaneously acquired HAADF image of the red region in (g).
Results and discussion
Preparation and identification of surface VOx sites on TiO2
The originally supported V2O5–WO3/TiO2 catalysts, “OR” for short, were synthesized by the incipient-wetness impregnation-drying-calcination method with a pure TiO2 (anatase) support. The OR powder was then subjected to an H2 plasma treatment using a radio-frequency discharge source within a Plasma Enhanced Chemical Vapor Deposition (PECVD) system. The plasma-modified catalysts, referred to as “PL” catalysts, were obtained after air calcination at 500 °C (Fig. S1). The loadings of V and W were quantified by inductively coupled plasma mass spectrometry (ICP-MS, Table S1). In the OR sample, the V content was found to be 0.81 wt% and the W content was 3.16 wt%. Similarly, in the PL sample, the V content was measured at 0.80 wt% and the W content at 3.18 wt%. X-ray diffraction (XRD) analysis of both the OR and PL catalysts exhibited diffraction peaks solely attributed to the anatase phase of the TiO2 support, with no discernible XRD peaks from crystalline V2O5 and WO3 nanoparticles (Fig. S2). The size distribution and morphology of the supported V2O5–WO3/TiO2 catalysts were examined using high-resolution transmission electron microscopy (HR-TEM) (Fig. S3). The plasma treatment did not result in significant changes in the average particle size (~21 nm) or interplanar spacing (0.32 nm) of the titania support compared to the pristine TiO2 (anatase) (Fig. 1b, c). For the PL catalyst (90 s plasma treatment), however, a notable lattice distortion resembling a core-shell structure of ~0.9 nm thickness of the shell as expected for amorphous V–W–O monolayer, larger than OR (~0.6 nm), along with blurred diffraction patterns at the edges is observed35. Prolonged plasma treatment (300 s) resulted in a further increased thickness of the distorted layer to 1.4 nm (Fig. S4).
The dispersion of the surface atoms was further examined by spherical aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). In the case of the OR catalyst (Fig. 1d, f), isolated atoms labeled within the indicated yellow circles are observed, indicating the presence of uniformly dispersed single atoms on the TiO2 support. In contrast, for the corresponding PL catalyst (Fig. 1e, g), fully exposed non-crystalline island-like nanoclusters, smaller than 0.8 nm in diameter, are observed. These agglomerated bright spots are attributed to W atoms given their significantly larger atomic number (Z = 74) compared to V (Z = 23) and Ti (Z = 22). To further differentiate between V and Ti in the PL catalyst, given their close Z numbers, a spectrum imaging technique was employed within a selected region (red region in Fig. 1g) to investigate V atoms within the nanoclusters. Electron energy loss spectroscopy (EELS) mapping within the electronic energy loss range of 350–850 eV was acquired (Fig. S5). The selected regions displayed a uniform distribution of the Ti element without any bright spots on the surface (Fig. 1h, i). Interestingly, the EELS mapping of V atoms precisely localized to the bright spots (Fig. 1j), indicating that V and W occupy the same positions within the nanoclusters. All the above confirmed that surface vanadyl sites on the surface of the TiO2 support are co-located with surface tungsten sites.
The short-range structure of the reactive dehydrated surface VOx sites was further investigated with spectroscopic techniques. For the OR catalyst (Fig. 2c), characteristic Raman bands at 1005 and 1023 cm−1 correspond to the vibrations of terminal W=O and V=O bonds, respectively36,37,38,39. For the PL catalyst, the Raman band of the V=O vibration shifted from 1023 to 1030 cm−1, indicating an increase of oligomerized degree of the surface VOx sites24,25. The PL catalyst also exhibited a broad band centered at ~930 cm−1 assigned to the V–O vibration from the bridging V–O–Ti bond27. The lack of shift of the Raman band for the W=O bond for the PL catalyst suggests that the surface WOx species in the OR catalyst were minimally affected by the PL treatment. The absence of a crystalline V2O5 band at ~995 cm−1 confirmed that V2O5 NPs were not present39. Complementary supporting information about the states of the surface VOx species was provided by solid-state 51V MAS NMR spectroscopy in Fig. 2d. The 51V MAS NMR curves were deconvoluted and fitted into four sub-peaks of distinct vanadyl species with different extents of oligomerization (i.e., monomer, dimer, oligomer (trimer and longer) and crystalline V2O5 nanoparticles)40,41,42. The crystalline vanadyl sites were most probably related to surface VOx sites that had a similar structure because the crystalline V2O5 Raman band was not present at 995 cm−1 and would give a strong Raman band. An increase of oligomeric surface VOx sites (peak at −652 ppm) from 15 to 41% was found with the plasma treatment and the monomeric surface VOx sites (peak at −567 ppm) decreased from 57 to 24%. Therefore, a catalyst with predominantly oligomeric surface VOx species on the TiO2 support was successfully prepared through the applied “top–down” plasma treatment approach43.

a V2p and W4f XPS spectra, b H2-TPR profiles of PL (blue) and OR (red). c In situ dehydrated Raman spectra, and d deconvolution of the in situ solid-state 51V MAS NMR spectra of PL (blue) and OR (red).
The surface contents of V and W of the PL (1.25 at% and 6.21 at%) were significantly higher than the OR (0.91 at% and 4.0 at%) in the outer surface region (1–3 nm) from X-ray photoelectron spectroscopy (XPS) results, respectively (Fig. 2a, Table S1, and Fig. S6). The H2-TPR results reveal the easier reduction of surface V oxides while almost no change for W oxides. This was attributed to the stronger reducibility of surface-enriched V, indicating that the former was enriched and aggregated in the distorted layer (Fig. 2b)39. The above findings indicate that the active components, especially some of the dispersed VOx species, migrated to the topmost surface region (~1–3 nm) after the plasma treatment. The High Sensitivity-Low Energy Ion Scattering (HS-LEIS) results also supported the XPS findings (Figs. S7 and S8). For the OR catalyst, the V (3 atomic %) signal was in the minority compared to the W (39 atomic %) and Ti (58 atomic %) signals (Fig. S7). In contrast, the topmost surface region of the PL catalyst contains a much larger amount of V atoms, and the percentage of V reaches 28 atomic % with the contents of W and Ti decreasing to 52 atomic % and 28 atomic %, respectively (Fig. S8).
NO/NH3-SCR performance of supported VOx–WOx/TiO2 catalysts
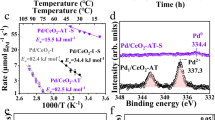
The effect of plasma treatment time on the NO/NH3-SCR activity of the supported V2O5–WO3/TiO2 catalysts was investigated under a high gas hourly space velocity (GHSV) of 375,000 cm3/(g h) in a fixed-bed reactor (Fig. S9). The NO reaction rate at 200 °C for the OR catalyst was 0.24 ± 0.02 × 10−6 mol g−1 s−1. After plasma treatment times of 60 s and 90 s, the reaction rate increased to 1.3 ± 0.03 × 10−6 mol g−1 s−1 and 2.8 ± 0.04 × 10−6 mol g−1 s−1, respectively (Fig. 3a). With longer treatment times, however, the reaction rate decreased to 2.3 ± 0.04 × 10−6 mol g−1 s−1 (120 s) to 1.1 ± 0.03 × 10−6 mol g−1 s−1 (300 s). The N2 selectivity, however, showed negligible change and was always around 99%. The increasing disparity in NO reaction rates between the OR and PL (90 s) catalysts as the reaction temperature rises is illustrated in Fig. 3b. The apparent activation energy (Ea) of the PL (90 s) catalyst was 27.9 ± 1.8 kJ/mol, which was significantly lower to the OR catalyst with an Ea of 41.6 ± 2.3 kJ/mol.

a Reaction rate and N2 selectivity of PL with different treatment time at 200 °C for NH3-SCR. b Reaction rate and apparent activating energy (Ea,inset) of PL and OR. c NO/NH3-SCR cycle stability of OR (orange) and PL (blue). Comparison of TOF values for the (d) reported NO/NH3-SCR catalysts and (e) different vanadia-based (PL and OR) NO/NH3-SCR catalysts at 200 °C. Error bars are standard deviations were calculated from triple activity testing.
Multi-cycle and long-term performance tests were conducted to assess the catalytic stability of the PL catalyst. Five consecutive cycles were performed, and the NO conversions and N2 selectivity remained unaltered throughout the cycles (Fig. 3c). Additionally, a 120-h catalytic stability test showed that the PL catalyst maintained a stable NO conversion of 97.1% at 270 °C. In contrast, the OR catalyst exhibited a lower NO conversion of 72.0% under similar conditions (Fig. S12). The TOF value of PL was superior to the commercial or reported SCR catalysts at the same or higher WHSV at 200 °C (Fig. 3d)23. Moreover, it was found that all supported V2O5/TiO2, V2O5–MoO3/TiO2, and V2O5–WO3/TiO2 catalysts with a different V loading (0.1–3.0 wt%) and stable W and Mo loading (3.22 ± 0.1) (Table S2) exhibited 4.5- to 9.5-fold increases in TOF values after a plasma treatment (Fig. 3e), indicating our strategy is universal for SCR activity improvement of supported vanadia-based catalysts.
Exploration of plasma effect on catalyst
In order to investigate the plasma effect on surface defects, we firstly prepared pure TiO2 samples with varying plasma treatment durations for reference. It was found that the Eg value of Ti slightly increased from 3.2 to 3.5 eV (Fig. S13 and Table S3) from UV–Vis spectroscopy, meanwhile the B1g, A1g, and Eg signals (at 399, 518, and 641 cm−1) belonging to TiO2 (anatase) decreased in intensity from Raman spectroscopy (Fig. S14) with increasing treatment duration. This indicates generation of defect sites on the TiO2 particles from the plasma treatment44. However, these signals corresponding to TiO2 defect sites disappear after calcination (Fig. S13 and Table S3), suggesting that defect sites could be filled by O2 at high temperatures that may diminish the enhanced catalytic activity for PL. To further substantiate this hypothesis, three controlled samples were also prepared by impregnation of vanadia precursors (ammonium metavanadate) on plasma treated TiO2 (V2O5–WO3(P)/TiO2), simultaneous impregnation of the vanadia and tungsta (aqueous ammonium tungstate) precursors on plasma treated TiO2 (V2O5–WO3/TiO2(P)), and impregnation of the aqueous ammonium metavanadate precursor on plasma treated WO3/TiO2 and dried at 100 °C and then calcined at 500 °C in air (V2O5–WO3/TiO2(RC)). The significant Raman bands associated with the TiO2 (anatase) and V=O vibrations of these catalysts remained almost unchanged (Fig. S14). However, all of these catalysts possessed poorer activity than untreated supported V2O5/TiO2, V2O5–WO3/TiO2, or PL catalysts (Fig. S15). This suggests that the improved performance of the PL catalysts is not related to surface defects on the support, but rather to the interaction of TiO2 with the surface vanadia species during the plasma chemical process.
To identify the function of H2, a series of extended experiments with different plasma treatment atmospheres (Ar and O2) and H2 thermal treatment without plasma were undertaken with the PL catalyst. The results suggested that only the Ar plasma treatment presented a slight activity enhancement (Fig. S10), while H2 thermal treatment had a negative effect on the SCR performance, especially at high temperatures (Fig. S11). These findings, therefore, further corroborate that an appropriate reductive atmosphere was significant for surface VOx cluster formation and activity improvement.
As plasma interacts with the catalyst surface, a considerable quantity of particles is projected onto the material. During this interaction, involving ions, neutral particles, and the material surface, the kinetic energy of the incident particles is transferred to surface atoms via collision cascades44,45,46,47,48,49,50. Given that the bond strength of Ti–O (dissociation enthalpy of 662 kJ/mol) is higher than that of V–O (644 kJ/mol)51. Additionally, the high coordination number (CN = 6) and the octahedral structure of titanium dioxide contribute to the enhanced stability of the crystal structure. When the absorption energy of the V–O bonds in the plasma exceeds their inherent bond energy, the bonds become increasingly prone to disruption, leading to the migration of V atoms to the catalyst’s surface. Here, V atoms aggregate, bonding to form polymeric vanadium oxide. This aggregation effectively lowers the system’s energy52, highlighting a crucial aspect of the catalyst’s interaction with plasma.
Investigation on the SCR reaction mechanism
Operando DRIFTS spectra during temperature-programmed measurements were initially undertaken with an online MS detector at the reaction cell outlet under the NO–NH3–O2–Ar reaction mixture from 100 to 300 °C. The OR catalyst showed characteristic IR peaks attributed to the surface NH4+ species adsorbed on Brønsted acid sites (B–NH4+: 1400 and 1670 cm−1), surface NH3 coordinated at Lewis acid sites (L–NH3: 1230 and 1604 cm−1) and adsorbed NO2 at 1340 cm−1 as shown in Fig. S16a16,21,22. In the case of the PL catalyst, at 100 °C, L–NH3 (at 1604 cm−1) and adsorbed NO2 (at 1340 cm−1) are also observed. As the temperature increased to 150–250 °C, additional infrared (IR) peaks appeared, including from surface bridging nitrate (νs (N–O)2 at 1288 cm−1 and νas (N–O)2 at 1599 cm−1), bidentate nitrate (νas (N–O)2 at 1579 cm−1), and NH2NO (vs (N–H) at 1330 cm−1, v (N=O) at 1490 cm−1) as shown in Fig. S16b18,53,54,55,56,57. For the PL catalyst, the temperature for formation of N2 in the outlet initiated at 115 °C that was much lower than the initiation temperature of 180 °C for the OR catalyst reflecting the greater activity for the PL catalyst (Fig. S17).
The MES-DRIFTS measurements were conducted at 150 °C in order to determine the participating surface species in the SCR reaction. The MES studies employed alternating pulses of NH3 and NO while maintaining a constant O2 concentration (5 vol%) in a flowing Ar environment (Details given in Figs. S18, S20, and S22). In Fig. 4a and b, the PL catalyst exhibited the MES-DRIFTS peaks from NH3 related peaks (1190, 1370, 1460, 1542, 1618, 3260, 3406 cm−1) and adsorbed H2O (1618 cm−1). The V=O (2035 cm−1) at overtone region showed an opposite sign to NH3 introduction, indicating NH3 adsorption on it (Fig. S19). The bridging nitrates (1288 vs(N–O)2 and 1599 cm−1 vas(N–O)2), bidentate nitrates (1260 vs(N–O)2 and 1579 cm−1 vas(N–O)2) and bridging M–O(H)–M (3653 cm−1) respond to NO introduction21,58,59. The phase delay for bridging M–(OH)–M is opposite in phase to the surface NH3*, NH4+*, and NH2*, suggesting that N–H cleavage does form terminal V–OH hydroxyls rather than bridging hydroxyls.

a Schematic of surface species and their corresponding IR vibrations (The yellow shading represents the positions of IR vibrations). MES DRIFT spectra of PL during (b) NO + O2/NH3 + O2 and (c) NO + O2/ND3 + O2 modulation experiment. d In situ Raman spectra of OR and PL under different reaction conditions (5% O2/Ar, 2000 ppm of NH3/Ar, 2000 ppm of NO + 2000 ppm of NH3/Ar, 2000 ppm of NO + 2000 ppm of NH3 + 5% O2/Ar in sequence). e MES-Raman spectra of PL during NO + O2/NH3 + O2 modulation experiment. The above experiments were carried out at 150 °C.
Due to the overlap of IR peaks associated with bidentate nitrate, bridging nitrate, L–NH3, and H2O around 1600 cm−1, there is mutual interference among the IR peaks of these species. To address this challenge, MES-DRIFTS studies utilized isotopically labeled reactant ND3 to prevent overlapping60,61,62,63. As shown in Fig. 4c, the IR peaks of adsorbed ND2 (1124 cm−1), nitrate intermediates (1260, 1288, 1579, and 1599 cm−1), and D2O (1385 cm−1) were still observed for the PL catalyst. In contrast to Marberger et al.’s study, isotope experiment confirmed that the band at 1599 cm−1 should be attributed to nitrate rather than NH3, suggesting the involvement of nitrate in the reaction54. The assignment for the aforementioned peaks has been reinforced based on Marberger et al., with a detailed comparison provided in Section 7 of the Supplementary Information54. The coverage of bridging bidentate NO3* increases substantially in this experiment compared to the NH3 experiment, by using the hydroxyl mode as an internal reference. It is proposed that surface NH3* (ND3*) undergoes N–H (N–D) cleavage to yield surface NH2* (ND2*) that then associatively couples with surface NO3* to yield the surface NH2NO2(OH) intermediate. Given that the N–D cleavage is kinetically slower than N–H cleavage, this increases the surface coverage of NO3* that is not consumed as quickly. This might close the reduction half cycle if further N–H bond breaking occurs to release H2O and N2. In comparison, the characteristic bands belonging to adsorbed nitrate and amide species were not found for the OR catalyst in the MES-DRIFTS (Figs. S21a and S23).
The evolution of the normalized signal intensity of surface intermediate species appearing in Fig. 4b as a function of phase angle were examined (Fig. S24) to further discern the chronological order of the species’ appearance. The phase angle of L–NH3 precedes that of B–NH4+ by 10°, indicating faster reactivity of L–NH3 than B–NH4+. Previously, in situ time-resolved IR spectroscopy demonstrated that L–NH3 would not convert to B–NH4+ at below 200 °C during NH3-SCR64. Therefore, surface L–NH3 appears to be the primary active site and species involved in the SCR reaction in this study. In contrast, the peaks of surface NH2 shifted by 20° with respect to the L–NH3 peaks. Simultaneously, the IR peak from surface NH2NO species displays a phase shift of 10° relative to the peaks from the surface Bi–NO3 and Bri–NO3 species, suggesting a temporal delay in the formation of surface NH2NO species with respect to the generation of surface nitrate species. The appearance of NH2* is delayed even more (20°) with respect to the introduction of NH3 than the appearance of surface NH2NO* (10°) upon the introduction of NO*, this indicates that N–H cleavage as rate-determining step. Hence, it appears that the PL catalyst exhibits a distinct reaction pathway involving surface L–NH3 and adsorbed nitrate species, potentially leading to the formation of a surface NH2NO intermediate for SCR65,66,67,68,69,70.
In situ, Raman experiments were performed to investigate the molecular structures of the catalytic active sites involved in the nitrate route. For both the OR and PL catalysts under an O2 environment, terminal V=O (1023–1030 cm−1), W=O (1005 cm−1), and bridging V–O–Ti (~930 cm−1) vibrational bands are present with the bridging V–O–Ti vibration very strong for the PL catalyst (Fig. 4d)27,36. Upon introduction of NH3, the V=O band selectively diminishes reflecting the interaction of ammonia with this bond. Upon the addition of NO to the NH3 stream, the bridging V–O–Ti band selectively decreases, especially pronounced for the PL catalyst, suggesting a correlation between the changes in the bridging V–O–Ti bond and the reaction of NO. When O2 is added to the NH3 + NO stream, the V=O, and V–O–Ti bands increase in intensity because of the oxidation of the reduced surface VOx sites and consumption of the surface ammonia species that broaden the IR bands. The intensity of the W=O band is less dramatically affected by the changing environmental conditions reflecting its inability to undergo efficient redox and ammonia coordination compared to the surface VOx site.
In situ MES-Raman spectra were also collected to further investigate the structure of the responsive surface metal oxide sites of the catalyst (Fig. S25). Phase-sensitive detection (PSD) of the PL catalyst revealed a strong correlation between the ν(V=O) and ν(W=O) vibrational modes of the VOx and WOx surface sites and the introduction of NH3 (Fig. 4e)54. Moreover, it was found that the two bands at 902 cm−1 and 930 cm−1 were indeed related to bridging V–O–Ti vibrations from two inequivalent adsorption sites (terminal Ti–O and Ti–(OH)–Ti), and not to W–O–Ti vibrations, which was confirmed by conducting similar experiments with the VOx/TiO2 (PL) catalyst (Fig. S26)71,72,73. The terminal V=O phase angle peak (80°) lagged behind that of bridging V–O–Ti vibrations (60°), indicating NH3 activation on V=O would be the RDS in agreement with IR-MES (Fig. S27). Furthermore, the IR phase angle of nitrate (Fig. S24) being out-of-phase with the diminishment of V–O–Ti suggests NH3 adsorption perturbs bridging V–O–Ti and not NO. With aid of theoretical calculations, it was found that the adsorption of nitrate on dimer VOx resulted in the charge redistribution and decrease in the covalence of V–O bonds, which caused the formation of weak IR bands from the bridging V–O–Ti vibrations (Fig. S28).
Upon integrating the findings from in situ spectroscopic studies, the introduction of NO was observed to prompt the formation of both bridging and bidentate nitrates, as illustrated in Fig. 4a, b. Intriguingly, the adsorption center coincides precisely with the dimeric V sites, a phenomenon distinctly highlighted in Raman spectroscopy by the attenuation of the V=O signal, as shown in Fig. 4e. This particular adsorption pattern highlights the indispensable role of dimeric V sites within PL catalysts, where each V site collaboratively participates in the formation of the nitrate intermediate. Following this, NH3 adsorption leads to the generation of NH2NO species, as captured in the MES-DRIFTS data presented in Fig. 4a. This adsorption event is coupled with the simultaneous formation of bridging hydroxyl groups, revealing the concurrent dehydration process, as delineated in Fig. 4a, b. Literature suggests that the decomposition of NH2NO transpires swiftly22. This proposed reaction mechanism accentuates the critical importance of dimeric V sites as the central activation sites for NO adsorption, underlining their significance in the catalytic process.
DFT calculations were conducted to further elucidate the mechanism underlying the superior activity of the PL catalyst. The models of monomeric and dimeric surface vanadia sites on the TiO2(101) anatase surface were constructed and optimized for comparison of the reaction pathway (Fig. S29). The four stages of NH3 and NO-assisted vanadyl reduction (referred to as Red) and one stage of O2 involved in re-oxidation of the reduced surface vanadyl site (referred to as Ox), constituting the NO/NH3-SCR reaction cycle is illustrated in Fig. 5. We compared the relative energy profiles of the nitrate-first generation pathway and the NH2-first generation pathway, revealing that the relative energy for NH3 cleavage to form NH2 is higher than that for the nitrate pathway (Fig. S30). Therefore, on dimeric surface vanadia sites, the reaction is more inclined to proceed via the nitrate pathway. With the introduction of NO, as observed by MES-DRIFTS, nitrate species and the disappearance of V=O and V–O–Ti species detected by Raman spectroscopy were noted. This suggests that NO was adsorbed on distinct sites (i.e., bridging V=O with V=O (A→B), V–OH (D→E), or V–OOH (I→J)) and coordinating with bidentate V–O2 (L→M) together with NH3 adsorption. DFT calculations indicate that NH3 has a higher affinity for adsorption on the catalyst surface (Fig. S31), forming lower energy structures, as illustrated by the B configuration in Fig. 5a. Additionally, NH2NO species was observed on the MES-DRIFTS, corresponding to the assistance of surface nitrates for NH3 dehydrogenation into the surface nitroso intermediate (B→C, E→F, J→K and M→N), and subsequently decomposition into N2 and H2O (C→D, F→G, K→L and N→O). MES-DRIFTS observations also revealed M–(OH)–M and M–(OD)–M, corresponding to steps (C→D, F→G, K→L, N→O) involving the dehydration process forming V–OH. Furthermore, adsorbed H2O and D2O were observed, corresponding to step (C→D, O→A, F→G, K→L, N→O) involving the dehydration to form H2O. Additionally, two sub-reaction pathways were involved in the Ox stage: (I) H2O desorption from a reduced vanadia (V*) site (G→H), and (II) gas-phase O2 replenishment on the reduced vanadia (H→I) site. Finally, after the H2O desorbed from V=O groups (O→A), the catalytic cycle was completed. The calculated formation energy of nitrate species on monomeric and dimeric surface vanadia sites are −0.34 eV and −0.47 eV (Fig. S32), respectively. Consequently, comparatively, nitrate formation is less favorable on monomeric vanadia sites. Additionally, MES-DRIFTS did not detect any nitrate species. Therefore, the monomeric surface vanadia site followed a reaction pathway without nitrate species as shown in Fig. 5b. The rate-determining step for the monomeric surface vanadia site was the V–OOH dehydration (G→H) with 1.44 eV in the Red 2 stage, which is in agreement with previous reported monomeric vanadia/TiO2 surfaces22. However, the rate-determining step changed to the transition state of NH3 dehydrogenation on the V=O bond with V–OH (E→F) with 1.17 eV over dimeric surface vanadia sites, which was also in the stage of vanadyl reduction. To further investigate the dehydration behavior for dimeric surface vanadia, the transition states for the generation of H2O (G→H and N→O processes) are calculated in Fig. S33. It is shown that their energy barriers for G→H (TS5) and N→O (TS6) are 0.98 eV and 1.02 eV, respectively, which is indeed lower than the relative energy of 1.17 eV for TS2 (Figs. 5 and S33.) The results indicated that dimeric surface vanadia also significantly reduced the energy barrier for H2O desorption.

The optimized molecular structures for the reactant, transition states, intermediates, product, and reaction energies were determined using DFT theory for each elementary step in the NH3-SCR mechanism over the surfaces of (a) dimeric surface vanadia site and (b) monomeric surface vanadia site. Red, cyan, green, blue, and white circles denote O, Ti, V, N, and H atoms, respectively.
In situ UV–Vis time-resolved spectroscopy was further employed to compare the V=O reduction step for PL and OR catalysts19,74. The kinetics of V5+ reduction was determined by monitoring the percentage of reduced V5+ sites under NH3 and NH3–NO exposure conditions at 150 °C, using the d-d transition band of reduced vanadia at 799 nm as a reference (Figs. S34, S35, and Table S6). It was observed that the specific reduction rates of V5+ for the PL catalyst (0.76 × 10−2 min−1) were approximately twice as high as those for the OR catalyst (0.40 × 10−2 min−1). This finding further suggests that the improved kinetics of the surface V5+ reduction step is highly correlated with the enhanced catalytic activity of the PL catalyst.
To uncover the factors contributing to the decline in activity following prolonged plasma treatment, we analyzed the spectral characteristics and reducibility of the PL-300s catalyst. Raman spectra (Fig. S36a) revealed a significant decrease in terminal V=O bonds in the PL-300s sample. V 2p XPS analysis (Fig. S36b) indicated a shift in the valence state of V from 5+ to 4+. H2-TPR profiles (Fig. S36c) showed a reduction in hydrogen consumption for the V reduction peak, suggesting a suppression of V’s redox ability from 1.16 cm3/g in PL to 0.63 cm3/g in PL-300s. In-situ UV–Vis experiments (Fig. S36d) demonstrated a decrease in reducible V5+ content to 11.8% in PL-300s after introducing NH3 and NO, indicating fewer active V sites. The deterioration of the NH3-SCR activity of the catalyst is exacerbated by the prolonged plasma treatment, as the destruction of V=O is pivotal for activating NH3 and promoting the generation of NH2NO (steps B→C, E→F, J→K, M→N)19,40,75.
In summary, supported vanadia nanocluster catalysts were successfully fabricated by transformation from monomeric surface VOx sites from a classic supported V2O5–WO3/TiO2 catalyst via a H2 plasma treatment. The plasma treatment resulted in the generation of a distorted lattice shell overlayer on the surface of the TiO2 support. This facilitated surface migration and reconstruction of surface VOx and WOx sites on the titania support under an H2-reducing atmosphere. The atomic-scale distribution of oligomeric surface vanadia sites on TiO2 were identified by the combination of HAADF-STEM and EELS microscopy. The plasma-treated supported oligomeric surface vanadia sites exhibited superior SCR activity, selectivity, and long-term stability in comparison to the conventional supported SCR VWTi catalyst. Moreover, the novel plasma-assisted method also significantly enhanced the activity of other supported vanadia-based catalysts. The structural investigations indicate that the oligomerization of the surface vanadyl sites was not caused by just the plasma treatment of the TiO2 support, but required the coexistence of vanadia and titania in the plasma reduction atmosphere to trigger aggregation of monomeric surface VOx sites at the TiO2 surface defects to generate strong interactions between the active components and oxide support. The MES investigations revealed that oligomeric surface VOx sites provide exclusive centers for adsorbed bridging and bidentate nitrates, and assisted in NH3 activation to generate amide intermediates. DFT calculations revealed that the enhanced activity from oligomerization of surface vanadyl sites is related to the barrier-less steps of the V5+ reduction from NH3 dehydrogenation with the assistance of adsorbed nitrates. This research contributes to a deeper understanding of structure-activity relationship and reaction mechanism of the widely used supported vanadia-based/TiO2 catalysts for NH3-SCR.
Methods
Catalyst preparation
Tungsten and vanadium were deposited on anatase-TiO2 (4.75 g, >99.8% anatase, Maklin Co.) using tungsten oxalate (0.2325 g, Maklin Co.) and ammonium metavanadate (0.0645 g, Maklin Co.) dissolved in an aqueous oxalic acid solution (1 mol/l). Typically, water was removed slowly by using a rotary evaporator, and the obtained solid was dried at 100 °C overnight and calcined at 500 °C for 3 h in air. Plasma treatment was performed in a PECVD system (KeJing co. Anhui, China) equipped with a high-frequency generator operating at 13.56 MHz and a power of up to 500 W. 100 mg of the OR catalyst powder was evenly spread on the quartz plate and introduced into the plasma chamber for x s, the H2/Ar/O2 was introduced as a pulse into the chamber. H2 was slowly introduced into the chamber before plasma treatment until atmospheric pressure was reached. Before commencing the plasma treatment procedure, the chamber pressure was reduced to 10 Pa employing a vacuum pump. Subsequently, the power supply was incrementally raised to 500 w, employing a slow and controlled approach. At this stage, the timing protocol was initiated. After 10-s plasma treatment, hydrogen gas was reintroduced into the chamber. The aforementioned steps were iterated multiple times to attain the intended duration for modification. Subsequently, the samples were calcined at 500 °C for 3 h in air.
Catalyst characterization
HAADF images of the samples were obtained using a ThermoFisher Themis Z transmission electron microscope with a convergence angle of 25 mrad and inner and outer collection angles of 59 and 200 mrad, respectively. To acquire the spectroscopic data needed for EELS elemental mapping, the electron probe (in our setups the probe has a diameter of ∼1.0 Å) was scanned in cluster regions and an EELS spectrum (350–850 eV) was acquired at each point together with HAADF image as reference. After using the average spectra as individual components in a linear combination, the spectra were fitted, and 2D atomic maps of the spectral weights were generated in combination with the simultaneously acquired HAADF image. XPS was performed with an XPS spectrometer (Thermo, Escalab 250Xi, USA) with Al Kα radiation. The temperature-programmed reduction with H2 (H2-TPR) experiments was carried out on a chemisorption instrument. Before conducting the testing, the catalyst samples were subjected to a pre-treatment at 300 °C for 60 min using helium as carrier gas. This pre-treatment was performed to remove moisture and impurities from the samples. (Micromeritics, AutoChem II 2920, USA). Quasi in situ HS-LEIS spectra were obtained using the Qtac100 HS-LEIS spectrometer (ION-TOF) equipped with a highly sensitive double toroidal analyzer. Using a Bruker Avance III 500 spectrometer with a resonance frequency of 131.6 MHz, the 51V solid-state NMR tests were performed at 11.7 T. A 1.9-mm HX double-resonance probe was utilized at a spinning rate of 40 kHz. The in situ 51V NMR spectra of the dehydrated samples were carried out employing a Hahn−echo pulse sequence, with a π/2 pulse width of 1.5 μs. For the present samples, a total of 60,000 scans were conducted, with a recycle delay of 0.3 s between each scan.
Measurement of NH3-SCR activity and kinetics
The NH3-SCR activity and kinetic data were measured with a tubular quartz reactor system, TOF are calculated by dividing the amount of NO molecules converted per second at low NO conversion (<15%) by the per V atoms on the surface of catalysts. (Additional details are provided in the supplementary information Section 2) 60. Outlet NO, NO2, NH3, N2O, SO2, and H2O concentrations were monitored by a Fourier-transformed infrared spectrometer (MBGAS-3000; ABB Co.)75.
MES (modulation excitation spectroscopy) experiments
In situ DRIFTS was performed using a FT-IR spectrometer (Thermo Fisher Scientific, Nicolet 6700) equipped with a mercury–cadmium telluride detector and a low void volume cell (Jiaxing Puxiang Tech. Ltd, RC-DRS -K01). The thermocouple was directly placed into the catalyst powder for temperature measurement. For the concentration modulation excitation experiments, the solenoid valves were used to automatically switch between gases. The pulse sequence according to Fig. S17 (NO + O2/NH3 + O2 modulation: 2000 ppm NO/Ar vs. 2000 ppm NH3/Ar, constant 5% O2/Ar) was introduced into the reaction cell. The set of time-resolved spectra obtained from the modulation experiments was converted into MES spectra using PSD:
where I(t) is the set of time-resolved data, ω the stimulation frequency, k the demodulation index (k = 1 is the fundamental harmonic and was used in this work), T the modulation period, and ϕPSD the phase angle. Python was used to process the time-resolved data using PSD. The modulation period (T = 240 s) is defined as the time required to conclude one full sequence. A single modulation period typically consisted of 240 consecutive time-resolved FTIR spectra, identical modulation sequences were applied and consisted of 12 consecutive. The FTIR spectra were recorded as 8 scans at a resolution of 4 cm−1 62,63,76.
In situ Raman spectroscopic characterization
Raman spectra were carried on a Senterra II Raman spectrometer (Bruker Optic), with an excitation wavelength of 532 nm and a low void volume cell (Jiaxing Puxiang Tech. Ltd, RC-RAMAN-K01). For the time-resolved experiments, the sample was first treated in a 10% O2/Ar (50 ml/min) flow at 500 °C for 30 min. Then the reaction cell was cooled to 150 °C (200 °C) in an Ar flow (50 ml/min) and collected as background spectra. Then the catalyst was sequentially exposed to 5% O2 (50 ml/min), 2000 ppm of NH3 (50 ml/min), 2000 ppm of NH3 + 2000 ppm of NO (50 ml/min), and 2000 ppm of NH3 + 2000 ppm of NO (50 ml/min) + 5% O2 (50 ml/min). The Raman spectra were recorded every 2 min at a resolution of 2 cm−1. The same pulse sequence and data processing methods as FTIR were employed for the Raman concentration modulation excitation experiments. The Raman spectra were recorded every 30 s at a resolution of 4 cm−1.
Computational details
First-principles calculations were performed using the DFT framework within the Vienna ab initio simulation package (VASP 5.4.4)77,78,79. A (3 × 1) supercell of the anatase (101) surface with double layer was employed as substrate for the commercial SCR catalyst surface80,81,82. The thickness of vacuum layer of anatase (101) surface was set over 15 Å. For relaxation of vanadia-loaded anatase surface and gases absorbed models, atoms at bottom eight layers were fixed, which means the upper vanadia clusters were allowed to relax and interact with gas molecules. After geometric optimization with generalized gradient approximation Perdew–Burke–Ernzerhof (GGA-PBE) functionals, the lattice parameters became 11.28 Å × 9.94 Å × 20.31 Å, which is in good agreement with input experimental lattice parameters (11.33 Å × 10.2 Å × 20.84 Å) of anatase (101) surface shown in Fig. S29. PBE functionals, based on the GGA, were widely used to account for exchange-correlation of V2O5/TiO2 catalyst for selective catalytic reduction with ammonia22,82,83,84,85. The interaction between the ions and the electrons was described by projector-augmented wave methods85. The pseudopotentials used for the present models were constructed by the electron configurations as V 3s2sp63d44s1 states, Ti 3s2sp63d24s2 states, N 2s22p3 states, H 1s states, and O 2s22p4 states. The energy cut-off value was set at 600 eV86. The convergence criteria of total energies and forces were 10−6 eV/atom and 0.05 eV/Å. The first Brillouin zone was sampled by a Monkhorst–Pack 2 × 2 × 1 K-point mesh87. The adsorption energies and electron density difference were calculated according to the adsorption or interfacial models88,89,90,91. We used dimeric vanadyl species as the model for our DFT calculations because they are the basic structural unit of various polymeric vanadia structures and can reasonably represent the coupling effect in them. The coupling effect between two adjacent vanadyl species (i.e., within a dimer unit of vanadia) at the reaction site was common in dimeric and higher-order polymeric vanadia structures. It sped up the whole catalytic cycle during the NH3-SCR of NO over the polymeric vanadyl species, and thus, we expected that dimeric and higher-order polymeric vanadia would have similar effects on the SCR reaction. Free energy correction was performed by including the zero-point energy and enthalpic and entropic contributions from vibrational degrees of freedom, with the substrate fixed. Climbing Image Nudged Elastic Band method was employed to find the minimum energy path connecting the reactants and products91,92,93. The fast inertial relaxation engine was used as optimizer in CI-NEB.
Data availability
The data generated within the paper and its Supplementary Information file are available from the corresponding authors upon request. Source data of Figs. 2–4 are provided in a Source Data file. Source data are provided with this paper.
Code availability
All code used in the simulations supporting this paper is available from the respective authors upon request.
References
Weckhuysen, B. M. Solid catalysts under the spotlight. Nat. Catal. 1, 101–102 (2018).
Liu, L. C. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981–5079 (2018).
Cui, X. J., Li, W., Ryabchuk, P., Junge, K. & Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 1, 385–397 (2018).
Li, X. et al. Functional CeOx nanoglues for robust atomically dispersed catalysts. Nature 611, 284–288 (2022).
Wang, A. Q., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65–81 (2018).
Yang, X. F. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Tyo, E. C. & Vajda, S. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 10, 577–588 (2015).
Liu, J. C., Xiao, H. & Li, J. Constructing high-loading single-atom/cluster catalysts via an electrochemical potential window strategy. J. Am. Chem. Soc. 142, 3375–3383 (2020).
Wang, X. N. et al. Atomic-precision Pt6 nanoclusters for enhanced hydrogen electro-oxidation. Nat. Commun. 13, 1596 (2022).
Li, X. Z. et al. Atomically precise single metal oxide cluster catalyst with oxygen-controlled activity. Adv. Funct. Mater. 32, 2200933 (2022).
Dong, C. Y. et al. Supported metal clusters: fabrication and application in heterogeneous catalysis. ACS Catal. 10, 11011–11045 (2020).
Li, R. T. et al. In situ identification of the metallic state of Ag nanoclusters in oxidative dispersion. Nat. Commun. 12, 1406 (2021).
Piotrowski, M. J. et al. Theoretical study of the structural, energetic, and electronic properties of 55-atom metal nanoclusters: a DFT investigation within van der Waals corrections, spin-orbit coupling, and PBE+U of 42 metal systems. J. Phys. Chem. C 120, 28844–28856 (2016).
Baghdasaryan, A. & Bürgi, T. Copper nanoclusters: designed synthesis, structural diversity, and multiplatform applications. Nanoscale 13, 6283–6340 (2021).
Paolucci, C. et al. Dynamic multinuclear sites formed by mobilized copper ions in NO selective catalytic reduction. Science 357, 898–903 (2017).
Topsøe, N. Y. Mechanism of the selective catalytic reduction of nitric-oxide by ammonia elucidated by in-situ online Fourier-transform infrared-spectroscopy. Science 265, 1217–1219 (1994).
Ma, Z. X. et al. Oxide catalysts with ultrastrong resistance to SO2 deactivation for removing nitric oxide at low temperature. Adv. Mater. 31, 1903719 (2019).
Smirniotis, P. G., Peña, D. A. & Uphade, B. S. Low-temperature selective catalytic reduction (SCR) of NO with NH3 by using Mn, Cr, and Cu oxides supported on hombikat TiO2. Angew. Chem. Int. Ed. 40, 2479–2482 (2001).
Inomata, Y. et al. Bulk tungsten-substituted vanadium oxide for low-temperature NOx removal in the presence of water. Nat. Commun. 12, 557 (2021).
Lian, Z. H. et al. Adsorption-induced active vanadium species facilitate excellent performance in low-temperature catalytic NOx abatement. J. Am. Chem. Soc. 143, 10454–10461 (2021).
Inomata, Y. et al. Bulk vanadium oxide versus conventional V2O5/TiO2: NH3-SCR catalysts working at a low temperature below 150 °C. ACS Catal. 9, 9327–9331 (2019).
He, G. Z. et al. Polymeric vanadyl species determine the low-temperature activity of V-based catalysts for the SCR of NOx with NH3. Sci. Adv. 4, eaau4637 (2018).
Qu, W. Y. et al. An atom-pair design strategy for optimizing the synergistic electron effects of catalytic sites in NO selective reduction. Angew. Chem. Int. Ed. 61, e202212703 (2022).
Lai, J. K. et al. Structure-activity relationships of hydrothermally aged titania-supported vanadium-tungsten oxide catalysts for SCR of NOx emissions with NH3. ACS Catal. 11, 12096–12111 (2021).
Lai, J. K. & Wachs, I. E. A perspective on the selective catalytic reduction (SCR) of NO with NH3 by supported V2O5-WO3/TiO2 catalysts. ACS Catal. 8, 6537–6551 (2018).
Haber, J., Machej, T., Serwicka, E. M. & Wachs, I. E. Mechanism of surface spreading in vanadia-titania system. Catal. Lett. 32, 101–114 (1995).
Chen, S. et al. Coverage-dependent behaviors of vanadium oxides for chemical looping oxidative dehydrogenation. Angew. Chem. Int. Ed. 59, 22072–22079 (2020).
Janssens, T. V. W. et al. A consistent reaction scheme for the selective catalytic reduction of nitrogen oxides with ammonia. ACS Catal. 5, 2832–2845 (2015).
Greenaway, A. G. et al. Detection of key transient Cu intermediates in SSZ-13 during NH3-SCR deNOx by modulation excitation IR spectroscopy. Chem. Sci. 11, 447–455 (2020).
Negri, C. et al. Evidence of mixed‐ligand complexes in Cu−CHA by reaction of Cu nitrates with NO/NH3 at low temperature. ChemCatChem 11, 3828–3838 (2019).
Yao, L. et al. Promotional effects of nitrogen doping on catalytic performance over manganese-containing semi-coke catalysts for the NH3-SCR at low temperatures. J. Hazard. Mater. 387, 121704 (2022).
Chen, L., Li, J. H. & Ge, M. F. Promotional effect of Ce-doped V2O5-WO3/TiO2 with low vanadium loadings for selective catalytic reduction of NOx by NH3. J. Phys. Chem. C 113, 21177–21184 (2009).
Liu, Z. M., Zhang, S. X., Li, J. H., Zhu, J. Z. & Ma, L. L. Novel V2O5-CeO2/TiO2 catalyst with low vanadium loading for the selective catalytic reduction of NOx by NH3. Appl. Catal. B 158, 11–19 (2014).
Cheng, J., Xu, R. N., Song, L. Y., He, H. & Chen, B. H. Unveiling the role of microwave induction on V2O5@AC catalysts with enhanced activity for low-temperature NH3-SCR reaction: an experimental and DFT study. Environ. Sci. Nano 10, 1313–1328 (2023).
Dong, K. et al. Plasma-induced defective TiO2-x with oxygen vacancies: a high-active and robust bifunctional catalyst toward H2O2 electrosynthesis. Chem. Catal. 16, 1437–1448 (2021).
Chen, S. et al. Modulating lattice oxygen in dual-functional Mo–V–O mixed oxides for chemical looping oxidative dehydrogenation. J. Am. Chem. Soc. 141, 18653–18657 (2019).
Guo, M. Y., Lis, B. M., Ford, M. E. & Wachs, I. E. Effect of redox promoters (CeOx and CuOx) and surface sulfates on the selective catalytic reduction (SCR) of NO with NH3 by supported V2O5-WO3/TiO2 catalysts. Appl. Catal. B 306, 121108 (2022).
Ruan, C. Y. et al. Selective catalytic oxidation of ammonia to nitric oxide via chemical looping. Nat. Commun. 13, 718 (2022).
Went, G. T., Leu, L. J. & Bell, A. T. Quantitative structural analysis of dispersed vanadia species in TiO2(Anatase)-supported V2O5. J. Catal. 134, 479–491 (1992).
Jaegers, N. R. et al. Mechanism by which tungsten oxide promotes the activity of supported V2O5/TiO2 catalysts for NOx abatement: structural effects revealed by 51V MAS NMR spectroscopy. Angew. Chem. Int. Ed. 131, 12739–12746 (2019).
Hu, J. Z. et al. Investigation of the structure and active sites of TiO2 nanorod supported VOx catalysts by high-field and fast-spinning 51V MAS NMR. ACS Catal. 5, 3945–3952 (2015).
Borovkov, V. Y., Mikheeva, E. P., Zhidomirov, G. M. & Lapina, O. B. Theoretical and experimental studies of the nature of the catalytic activity of VOx/TiO2 systems. Kinet. Catal. 44, 710–717 (2003).
Gao, X. T. & Wachs, I. E. Investigation of surface structures of supported vanadium oxide catalysts by UV-vis-NIR diffuse reflectance spectroscopy. J. Phys. Chem. B 104, 1261–1268 (2000).
Ye, Z. P. et al. A review of the advances in catalyst modification using nonthermal plasma: process, mechanism and applications. Adv. Colloid Interface Sci. 308, 102755 (2022).
Santos, A. M., Catapan, R. C. & Duarte, D. A. The potential of non-thermal plasmas in the preparation of supported metal catalysts for fuel conversion in automotive systems: a literature overview. Front. Mech. Eng. 6, 42 (2020).
Liu, X. Z., Long, H. Y., Hu, S. H. & Wen, K. Photocatalytic TiO2 nanoparticles activated by dielectric barrier discharge plasma assisted ball milling. J. Nanosci. Nanotechnol. 20, 1773–1779 (2020).
Dou, S. et al. Plasma‐assisted synthesis and surface modification of electrode materials for renewable energy. Adv. Mater. 30, 1705850 (2018).
Zou, J. J., Liu, C. J. & Zhang, Y. P. Control of the metal−support interface of NiO-loaded photocatalysts via cold plasma treatment. Langmuir 22, 2334–2339 (2006).
Li, K. et al. Research on manganese oxide catalysts surface pretreated with non-thermal plasma for NO catalytic oxidation capacity enhancement. Appl. Surf. Sci. 264, 557–562 (2013).
Mistry, H. et al. Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123 (2016).
Dean, J. A. & Lange, N. Lange’s Handbook of Chemistry 16th ed. 1, 1–331, New York (McGraw-Hill, 2005).
El-Roz, M. et al. High-visible-light photoactivity of plasma-promoted vanadium clusters on nanozeolites for partial photooxidation of methanol. ACS Appl. Mater. Interfaces 9, 17846–17855 (2017).
Wang, H., Sun, Y. J. & Dong, F. Insight into the overlooked photochemical decomposition of atmospheric surface nitrates triggered by visible light. Angew. Chem. Int. Ed. 61, e202209201 (2022).
Marberger, A., Ferri, D., Elsener, M. & Kröcher, O. The significance of Lewis acid sites for the selective catalytic reduction of nitric oxide on vanadium-based catalysts. Angew. Chem. Int. Ed. 55, 11989–11994 (2016).
Nuguid, R. J. G., Ferri, D., Marberger, A., Nachtegaal, M. & Kröcher, O. Modulated excitation Raman spectroscopy of V2O5/TiO2: mechanistic insights into the selective catalytic reduction of NO with NH3. ACS Catal. 9, 6814–6820 (2019).
Marberger, A. et al. Time-resolved copper speciation during selective catalytic reduction of NO on Cu-SSZ-13. Nat. Catal. 1, 221–227 (2018).
Nasir, J. A. et al. Influence of solvent on selective catalytic reduction of nitrogen oxides with ammonia over Cu-CHA zeolite. J. Am. Chem. Soc. 145, 247–259 (2022).
Bahrami, B. et al. In situ FTIR characterization of NH3 adsorption and reaction with O2 and CO on Pd-based FCC emission control additives. Appl. Catal. A 39, 11–21 (2011).
Topsøe, N. Y., Dumesic, J. A. & Topsøe, H. Vanadia-titania catalysts for selective catalytic reduction of nitric-oxide by ammonia: I.I. Studies of active sites and formulation of catalytic cycles. J. Catal. 151, 241–252 (1995).
Chang, C. H. & Nesbitt, D. J. Sub-doppler slit jet infrared spectroscopy of astrochemically relevant cations: symmetric (v1) and antisymmetric (v6) NH stretching modes in ND2H2+. J. Chem. Phys. 148, 014304 (2018).
Hadjiivanov, K. I. Identification of neutral and charged NxOy surface species by IR spectroscopy. Catal. Rev. 42, 71–144 (2000).
Centeno, M. A., Carrizosa, I. & Odriozola, J. A. In situ DRIFTS study of the SCR reaction of NO with NH3 in the presence of O2 over lanthanide doped V2O5/Al2O3 catalysts. Appl. Catal. B 19, 67–73 (1998).
Ramis, G., Busca, G., Lorenzelli, V. & Forzatti, P. Fourier transform infrared study of the adsorption and coadsorption of nitric oxide, nitrogen dioxide and ammonia on TiO2 anatase. Appl. Catal. 64, 243–257 (1990).
Zhu, M. H., Lai, J. K., Tumuluri, U., Wu, Z. I., & Wachs, I. E. Nature of active sites and surface intermediates during SCR of NO with NH3 by supported V2O5-WO3/TiO2 catalysts. J. Am. Chem. Soc. 139, 15624–15627 (2017).
Li, S. Y., Song, L. Y., Li, J. & He, H. Promotional mechanisms of activity and SO2 tolerance of NdVOx/TiO2 catalysts for selective catalytic reduction of NOx with NH3. ACS Catal. 13, 2867–2884 (2023).
Song, K. L. et al. Insight into the origin of excellent SO2 tolerance and de-NOx performance of quasi-Mn-BTC in the low-temperature catalytic reduction of nitrogen oxide. ACS Catal. 13, 5020–5032 (2023).
Xiong, S. C., Liao, Y., Xiao, X., Dang, H. & Yang, S. J. Novel effect of H2O on the low temperature selective catalytic reduction of NO with NH3 over MnOx-CeO2: mechanism and kinetic study. J. Phys. Chem. C 119, 4180–4187 (2015).
Xiong, S. C. et al. Global kinetic study of NO reduction by NH3 over V2O5-WO3/TiO2: relationship between the SCR performance and the key factors. Ind. Eng. Chem. Res. 54, 11011–11023 (2015).
Daya, R. et al. Alternate pathway for standard SCR on Cu-zeolites with gas-phase ammonia. React. Chem. Eng. 6, 1042–1052 (2021).
Chen, L. et al. A complete multisite reaction mechanism for low-temperature NH3-SCR over Cu-CHA. ACS Catal. 10, 5646–5656 (2020).
Guo, M. Y., Lis, B. M., Ford, M. E. & Wachs, I. E. The effect of non-redox promoters (AlOx, POx, SiOx and ZrOx) and surface sulfates on supported V2O5-WO3/TiO2 catalysts in selective catalytic reduction of NO with NH3. Appl. Catal. B 306, 121128 (2022).
Luca, V., Thomson, S. & Howe, R. F. Spectroscopic investigation of vanadium speciation in vanadium-doped nanocrystalline anatase. J. Chem. Soc. Faraday Trans. 93, 2195–2202 (1997).
Magg, N. et al. Vibrational spectra of alumina- and silica-supported vanadia revisited: an experimental and theoretical model catalyst study. J. Catal. 226, 88–100 (2004).
Zhu, M. H. et al. Reaction pathways and kinetics for selective catalytic reduction (SCR) of acidic NOx emissions from power plants with NH3. ACS Catal. 7, 8358–8361 (2017).
Yan, T. et al. Promoter rather than Inhibitor: phosphorus incorporation accelerates the activity of V2O5-WO3/TiO2 catalyst for selective catalytic reduction of NOx by NH3. ACS Catal. 10, 2747–2753 (2020).
Ferri, D. et al. Revealing the dynamic structure of complex solid catalysts using modulated excitation X-ray diffraction. Angew. Chem. Int. Ed. 53, 8890–8894 (2014).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Luo, B. C. et al. Structural and electronic properties of cubic KNbO3 (001) surfaces: a first-principles study. Appl. Surf. Sci. 351, 558–564 (2015).
Mino, L., Cazzaniga, M., Moriggi, F. & Ceotto, M. Elucidating NOx surface chemistry at the anatase (101) surface in TiO2 nanoparticles. J. Phys. Chem. C 127, 437–449 (2022).
Langhammer, D., Kullgren, J. & Österlund, L. Photoinduced adsorption and oxidation of SO2 on anatase TiO2 (101). J. Am. Chem. Soc. 142, 21767–21774 (2020).
Weirich, T. E., Winterer, M., Seifried, S., Hahn, H. & Fuess, H. Rietveld analysis of electron powder diffraction data from nanocrystalline anatase, TiO2. Ultramicroscopy 81, 263–270 (2000).
Song, I. et al. Simple physical mixing of zeolite prevents sulfur deactivation of vanadia catalysts for NOx removal. Nat. Commun. 12, 901 (2021).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Luo, B. C. et al. Superhierarchical inorganic/organic nanocomposites exhibiting simultaneous ultrahigh dielectric energy density and high efficiency. Adv. Funct. Mater. 31, 2007994 (2020).
Luo, B. C. et al. Interfacial bonding and electronic structure between copper thiocyanate and hybrid organohalide lead perovskites for photovoltaic application. J. Phys. Chem. Lett. 10, 5609–5616 (2019).
Luo, B. C. et al. Interfacial electronic properties of ferroelectric nanocomposites for energy storage application. Mater. Today Energy 12, 136–145 (2019).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22176008, 21906004, and 52202154). Bingcheng Luo acknowledges support from the High-performance Computing Platform of China Agricultural University. We thank Si Jiang for the scientific discussion and valuable suggestions. We thank Didi Li at East China University of Science and Technology for their help with X-ray absorption spectroscopy characterization. We thank Yuan Xu (Bruker Co.) for assistance with collecting Raman spectra. We acknowledge support from National Supercomputer Center in Tianjin, and the energy calculations were performed on Tianhe new generation supercomputer. We thank Xiumei Wang (Bruker NMR Facility) for assistance with collecting NMR spectra. The work at Lehigh University was supported as part of Understanding & Control of Acid Gas-Induced Evolution of Materials for Energy (UNCAGE-ME), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0012577.
Author information
Authors and Affiliations
Contributions
X.L. and Y.Y. proposed and designed the research plan and experimental scheme. Y.Y., K.L., M.Z., I.E.W. and X.L. performed the characterization of the materials. Y.Y. carried out the synthesis and the activity tests of the materials and data analysis. Y.Y., X.L., K.L., M.Z., B.M.L., Y.S. and I.E.W. conducted mechanistic investigation experiments and analysis. B.L. performed the first-principles calculations. X.L., M.Z., I.E.W., B.L., K.L., T.Z. and Y.Y. co-wrote the manuscript. All authors discussed the results and provided comments on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jamal Abdul Nasir, Hong He and Toru Murayama for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yin, Y., Luo, B., Li, K. et al. Plasma-assisted manipulation of vanadia nanoclusters for efficient selective catalytic reduction of NOx. Nat Commun 15, 3592 (2024). https://doi.org/10.1038/s41467-024-47878-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47878-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
