
Abstract
Pathogenic variants in the HIBCH gene cause HIBCH deficiency, leading to mitochondrial disorders associated with valine metabolism. Patients typically present with symptoms such as developmental regression/delay, encephalopathy, hypotonia and dystonia. Brain magnetic resonance imaging (MRI) shows bilateral lesions in the basal ganglia with/without brainstem involvement. Here, we report a case of a Japanese patient with Leigh-like syndrome caused by novel HIBCH variants. Long-term follow-up MRI revealed progressive cerebellar atrophy, which expands the phenotypic spectrum of HIBCH deficiency.
Similar content being viewed by others

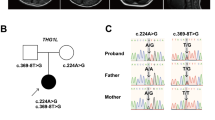
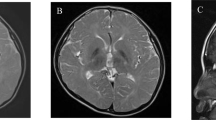
The patient was male with nonconsanguineous Japanese parents. He was born at 40 weeks of gestation via normal vaginal delivery following an uneventful pregnancy, and his birth measurements were within the normal ranges. The patient had no family history of neuromuscular disorders or delayed motor development. He gained head control at 4 months of age but was unable to sit up even at 10 months. He experienced poor weight gain from 7 months of age. At 10 months, he developed acute encephalopathy triggered by a viral infection, which led to neurological deterioration. The patient exhibited abnormal eye movements, dystonia of the left upper extremity, and paralysis of the left upper and lower extremities. The ketone body level was 11160 μmol/L. Lactate and pyruvate levels in the blood and cerebrospinal fluid were normal. 3-Hydroxy-isovaleric acid was slightly elevated in urinary organic acid analyses; however, it was classified as a nonspecific finding at that time. Brain magnetic resonance imaging (MRI) revealed bilateral symmetric signal abnormalities in the globus pallidus (Fig. 1A–D), suggesting possible metabolic encephalopathy. The patient was treated with a combination of a vitamin cocktail, carnitine, mannitol, and intravenous immunoglobulin therapy, causing temporary improvement of neurological symptoms. However, the patient subsequently developed various new symptoms, including nystagmus, athetosis, and spastic paraparesis. The abnormal signals in the globus pallidus resolved, but progressive cerebellar atrophy was observed (Fig. 1E–H). The patient acquired gross motor skills only to the point of a walking gait due to hypotonia. At 11 years of age, the patient’s developmental quotient, as assessed using the Kyoto Scale of Psychological Development 2020, was 40. Various tests, including mitochondrial gene point mutation screening, muscle biopsy, mitochondrial respiratory chain enzyme activity in muscle specimens, and ketone enzyme activity levels, yielded normal results.

A–D MRI at ten months showed bilateral symmetric signal abnormalities in the globus pallidus: axial T1-weighted (A), T2-weighted (B), fluid-attenuated inversion recovery (FLAIR) (C), and diffusion-weighted images (D). E FLAIR image at two years of age showed the disappearance of abnormal signals in the globus pallidus. F–H Sagittal T1-weighted MRI showing progressive cerebellar atrophy at one year (F), two years (G), and five years (H). I Schematic of the HIBCH gene with all reported variants labeled. Notes: Red boxes, variants identified in this study; yellow boxes, variants with cerebellar atrophy.
Written informed consent was obtained from the parents in accordance with the Review Board and Ethics Committee of Kyoto University. Whole-exome sequencing (WES) was performed when the patient was 11 years old. Trio-based WES was conducted using the xGen® Exome Research Panel v2 (IDT, Iowa, USA). The captured libraries were sequenced using DNBSEQ-G400 (MGI Tech, Shenzhen, China). WES analysis identified compound heterozygous variants of HIBCH that have not yet been reported as pathogenic variants. The first variant was identified in exon 10 [NM_014362.4:c.782 T > C, p.(Leu261Pro)] and was predicted to be deleterious by SIFT (score 0; https://sift.bii.a-star.edu.sg) and disease-causing by MutationTaster (prob 0.999; http://www.mutationtaster.org/). This variant was absent in gnomAD, HGVD, and 8.3KJPN and was moderately conserved across species. The second variant was located in exon 13 (NM_014362.4:c.1012-1 G > A) and was predicted to be deleterious by CADD (score 18.12; https://cadd.gs.washington.edu/) and disease-causing by MutationTaster (prob 0.999; http://www.mutationtaster.org/). This variant was absent in gnomAD, HGVD, and 8.3KJPN. Both variants were confirmed by Sanger sequencing; the c.782 T > C, p.(Leu261Pro) and c.1012-1 G > A variants were maternally and paternally inherited, respectively. Their pathogenicity was evaluated according to the 2015 American College of Medical Genetics and Genomics guidelines1. The c.782 T > C, p.(Leu261Pro) and c.1012-1 G > A variants were classified as likely pathogenic and pathogenic, respectively. In this patient, HIBCH deficiency was strongly considered because of the elevation of 3-hydroxy-isovaleric acid in urinary organic acid analyses and Leigh-like syndrome signs on MRI.
3-Hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency is a rare mitochondrial disorder associated with valine metabolism. Most patients experience delayed motor milestone development in early infancy and develop Leigh/Leigh-like syndrome after a febrile infection or metabolic crisis, leading to secondary regression and a range of neurological symptoms2. The movement disorders in this patient are characteristic symptoms of the disease3,4. Among them, dystonia, observed from an early onset, is a common symptom, occurring in 71% of patients3. Neuroradiological features are similar to those of other causes of Leigh/Leigh-like syndrome, characterized by signal abnormalities in the basal ganglia. In previous cases, the probability of cerebellar atrophy was low at 17%3. Table 1 summarizes the characteristics of six reported cases of HIBCH deficiency with cerebellar atrophy, including the present case3,5. Five of these six patients had Leigh/Leigh-like syndrome, with dystonia being a typical complication in 5 of 6 patients. Ataxia was present in 3 of 6 patients. All patients except one were unable to walk independently. Patient 2 was considered to have a relatively mild phenotype without Leigh/Leigh-like syndromes, which might be related to residual HIBCH enzyme activity. No deaths occurred during the observation period. Figure 1I shows a schematic of the HIBCH gene labeled with all the reported pathogenic variants6. There is no hot spot mutation site in the HIBCH gene. The variants identified in this study are shown in red boxes. Previously reported variants with cerebellar atrophy are indicated by yellow boxes. The mutation sites of the HIBCH gene varied, and there was no correlation between the specific mutation sites and cerebellar atrophy.
To our knowledge, this is the first report of progressive cerebellar atrophy in patients with HIBCH deficiency evaluated using long-term MRI follow-up. Our patient showed no cerebellar atrophy at disease onset; however, progressive atrophy of the cerebellum was revealed over five years. Although there have been only a few reports of HIBCH deficiency with cerebellar atrophy, there may be more cases with follow-up over time that were not detected at the initial diagnosis.
There have been several reports on the usefulness of metabolic analysis7,8,9. Increased C-4 carnitine in the acylcarnitine profile or increased urinary excretion of 2-methyl-2,3-dihydroxybutyrate and 3-hydroxy-isovaleric acid are suggestive findings3,10. In our case, C-4 carnitine in the acylcarnitine profile and 2-methyl-2,3-dihydroxybutyrate in urinary organic acid analyses were not elevated, but elevated 3-hydroxy-isovaleric acid levels were detected. In another report, all metabolic analyses yielded negative results despite the severity of the disease11. Therefore, in the case of Leigh/Leigh-like syndrome, where movement disorders are the primary symptom and the globus pallidus is involved, a genetic diagnosis using next-generation sequencing should be considered, even if metabolic analyses are negative.
Regarding treatment, some reports have shown improvement in symptoms and imaging findings with a valine-restricted diet12,13,14. In this patient, after the initial rapid worsening of the neurological symptoms, the patient’s manifestations were not exacerbated in the chronic phase, leading the parents to decline dietary treatment after the diagnosis.
In conclusion, HIBCH deficiency is a rare disease with diagnostic challenges due to limited abnormal metabolic studies and various nonspecific movement disorders. However, the presence of bilateral symmetrical signal abnormalities in the globus pallidus during the acute stage and progressive cerebellar atrophy during the chronic stage should raise suspicion of HIBCH deficiency. Considering the potential effect of a valine-restricted diet in the early phase, aggressive genetic analysis should be performed promptly when this syndrome is suspected.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3315. https://doi.org/10.6084/m9.figshare.hgv.3318.
References
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Gene Med. 17, 405–424 (2018).
Tan, H. et al. Truncating mutations of HIBCH tend to cause severe phenotypes in cases with HIBCH deficiency: a case report and brief literature review. J. Hum. Genet. 63, 851–855 (2018).
François-Heude, M. C. et al. Movement disorders in valine metabolism diseases caused by HIBCH and ECHS1 deficiencies. Eur. J. Neurol. 29, 3229–3242 (2022).
Schottmann, G. et al. A movement disorder with dystonia and ataxia caused by a mutation in the HIBCH gene. Mov. Disord. 31, 1733–1739 (2016).
Marti-Sanchez, L. et al. Delineating the neurological phenotype in children with defects in the ECHS1 or HIBCH gene. J. Inherit. Metab. Dis. 44, 401–414 (2021).
Wang, J. et al. Cinical, metabolic, and genetic analysis and follow-up of eight patients with HIBCH mutations presenting with Leigh/Leigh-like syndrome. Front. Pharm. 12, 605803 (2021).
Ferdinandusse, S. et al. HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet J. Rare Dis. 8, 188 (2013).
Peters, H. et al. Metabolite studies in HIBCH and ECHS1 defects: implications for screening. Mol. Genet. Metab. 115, 168–173 (2015).
Stiles, A. R. et al. Successful diagnosis of HIBCH deficiency from exome sequencing and positive retrospective analysis of newborn screening cards in two siblings presenting with Leigh’s disease. Mol. Genet. Metab. 115, 161–167 (2015).
Çaker, N. E. & Görükmez, O. 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency cases diagnosed by only HIBCH gene analysis and novel pathogenic mutation. Ann. Indian Acad. Neurol. 24, 372–378 (2021).
D’Gama, A. M. et al. A phenotypically severe, biochemically “silent” case of HIBCH deficiency in a newborn diagnosed by rapid whole exome sequencing and enzymatic testing. Am. J. Med. Genet. A 182, 780–784 (2020).
Abdenur, J. E. et al. Medical nutrition therapy in patients with HIBCH and ECHS1 defects: clinical and biochemical response to low valine diet. Mol. Genet. Metab. Rep. 24, 100617 (2020).
Soler-Alfonso, C. et al. Identification of HIBCH gene mutations causing autosomal recessive Leigh syndrome: a gene involved in valine metabolism. Pediatr. Neurol. 52, 361–365 (2015).
Yamada, K. et al. Clinical and biochemical characterization of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency that causes Leigh-like disease and ketoacidosis. Mol. Genet. Metab. Rep. 1, 455–460 (2014).
Acknowledgements
We thank the patient and his family members for their cooperation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taura, Y., Tozawa, T., Isoda, K. et al. Leigh-like syndrome with progressive cerebellar atrophy caused by novel HIBCH variants. Hum Genome Var 10, 23 (2023). https://doi.org/10.1038/s41439-023-00251-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00251-y
