
Abstract
The X-linked human glutamate receptor subunit 3 (GRIA3) gene (MIM *305915, Xq25) encodes ionotropic α amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)-type glutamate receptor subunit 3, which mediates postsynaptic neurotransmission. Variants in this gene can cause a variety of neurological disorders, primarily reported in male patients. Here, we report a female patient with developmental and epileptic encephalopathy who carries the novel de novo GRIA3 variant NM_007325.5: c.1982T > C: p.Met661Thr.
Similar content being viewed by others


Human glutamate receptor subunit 3 (GRIA3) encodes subunit 3 (GluA3) of the α-3-hydroxy-5-methyl-4-isoxazole propionic receptor (AMPAR). Four types of isoforms, i.e., GluA1–4, produce diverse combinations of pore-forming transmembrane complexes and play important roles in mediating excitatory synaptic transmission via glutamate and synaptic plasticity, which are closely regulated by alternative splicing of flip and flop isoforms1,2. AMPARs, including complexes with GluA3, are broadly distributed in the brain, particularly in the hippocampus, cerebral cortex, and thalamus, regions associated with epileptic activity3. GRIA3 variants cause many neurodevelopmental disorders of various degrees of severity, such as cognitive impairment (MIM *300699)4, movement disorders5, and epilepsy6. Approximately 20 variants have been reported, including balanced translocation, deletion, duplication, and missense variants. Most patients have been men whose mothers were carriers, with only five females reported7,8,9,10,11.
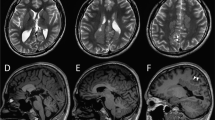
The patient was a 13-year-old Japanese girl, the second child of healthy, nonconsanguineous parents with no family history of neurological disorders. Her mother had three miscarriages and one stillbirth. She was delivered via scheduled cesarean section at 38 weeks and 3 days of gestation due to fetal distress, acidemia, and limb hypertonia. Her birth weight was 2648 g (−0.86 SD); her head circumference was 33.5 cm (0.43 SD). She exhibited hypertonia and increased deep tendon reflexes. Epilepsy onset occurred at 3 months of age with tonic and clonic seizures. An electroencephalogram (EEG) showed frequent spikes in the central area, which readily developed into generalized spikes and waves (Fig. 1c). Brain magnetic resonance imaging revealed mild frontal lobe atrophy and slight ventricular enlargement (Fig. 1d). Treatment with carbamazepine and ethosuximide was ineffective. The seizures, initially resistant to drug treatment, were gradually brought under control using lamotrigine, clobazam, levetiracetam, and lacosamide. Seizures occurred several times per month, sometimes in clusters. The patient could control her neck at 4 months and roll over at 9 months of age. However, she was bedridden and had poor speaking ability at one year of age. She now presents with severe developmental delay, scoliosis and hip dislocation. Characteristic facial features are not apparent (Fig. 1a, b).

a, b The patient had no specific facies. She was bedridden, with hypertonia and severe psychomotor delay. c Electroencephalogram of the first seizure at 3 months of age. Spikes in the central area that easily developed into generalized waves were noted. d Brain magnetic resonance image at 13 years of age. Slight frontal lobe atrophy and ventricular enlargement were not aggravated.
The FOXG1 variant, analyzed by Sanger sequencing, was not detected during her infancy. Written informed consent for genetic analysis was obtained from her parents; the Ethical Committee of the Asahikawa Habilitation Center for Children (No. R04-03) approved the study. Microarray analysis did not show any pathogenic copy number variants. The Initiative on Rare and Undiagnosed Disease, a nationwide consortium of the Japan Agency for Medical Research and Development, performed whole-exome sequencing and variant filtering for pathogenicity in the patient and her parents. We identified a de novo heterozygous variant in the patient: NM_007325.5: c.1982T>C: p.Met661Thr of GRIA3. Currently, this variant is not listed in public databases, including the Exome Variant Server (http://evs.gs.washington.edu/EVS/), the 1000 Genome Project database (http://www.internationalgenome.org/), dbSNP (http://www.nebi.nlm.gov/SNP), the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/), and the Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp/). According to the American College of Medical Genetics and Genomics guidelines12, the variant was classified as likely pathogenic (PS2, PM2, PP2, PP3). It was confirmed as deleterious by SIFT and probably damaging by Polyphen-2 analyses. No additional variants associated with the patient’s clinical features were identified.
This variant is located in the linker region between the third transmembrane domain (M3) and the S2 extracellular domain of the glutamate binding domain (Fig. 2), next to a previously reported variant, p.Arg660Thr10, affecting a highly conserved amino acid. The patient in that case was a female with clinical manifestations similar to those of our patient: hypertonia, epileptic encephalopathy, and global developmental delay. Sun et al. revealed by functional analysis that p.Arg660Thr was a gain-of-function variant that slowed deactivation and desensitization kinetics10. A male patient with a gain-of-function variant also exhibited muscle hypertonia13. Although we did not conduct a functional study, hypertonia may be indicate a gain of function, as AMPAR is also expressed in the lower motor neurons14. Neurodevelopmental disorder with or without seizures and gait abnormalities (#617864), an AMPAR-related disorder, features hypertonia or increased startle reflex. However, epilepsy was reported regardless of the patients’ sex. GluA3-knockout mice reportedly exhibit marked EEG changes, suggesting a critical role in the generation of slow cortical oscillation15.

Methionine at position 661 is highly conserved among different species and GluA subunits.
Why GRIA3 mutations impact female patients remains unknown. Among developmental epileptic encephalopathies, female patients with WDR45- or SMC1A-related disorders exhibit equal or worse clinical manifestations than male patients11. Neither X-inactivation nor mosaicism can entirely account for the range of symptoms or severity. In addition, the association between genotype and phenotype has not been defined, except for the localization of movement disorder variants in the TM4 region4,5. Carbamazepine has been reported to be effective for a patient with a gain-of-function variant13. In our case, carbamazepine was ineffective, possibly because it does not act directly on AMPAR but inhibits glutamate release from presynaptic neurons16. In addition, variants tend not to act in isolation but elicit adaptive changes in other systems, complicating patients’ responsiveness to treatment17. Antiepileptic medications that are currently in use may be effective to some extent by manipulating neuronal networks in the brain18. We intend to classify antiepileptic medications while taking into account the use of perampanel, a noncompetitive AMPAR antagonist, as a first-line treatment. From the perspective of treatment decisions, the accumulation of case data is crucial.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3276.
References
Kamalova A., Nakagawa T. AMPA receptor structure and auxiliary subunits. J. Physiol. 599, 453–469 (2021).
Renner, M. C. et al. Synaptic plasticity through activation of GluA3-containing AMPA-receptors. Elife 6, e25462 (2017).
Schwenk, J. et al. Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron 84, 41–54 (2014).
Wu, Y. et al. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc. Natl Acad. Sci. USA 104, 18163–18168 (2007).
Piard, J. et al. The GRIA3 c.2477G >A Variant Causes an Exaggerated Startle Reflex, Chorea, and Multifocal Myoclonus. Mov Disord 35, 1224–1232 (2020).
Trivisano, M. et al. GRIA3 missense mutation is cause of an x-linked developmental and epileptic encephalopathy. Seizure 82, 1–6 (2020).
Gécz, J. et al. Characterization of the human glutamate receptor subunit 3 gene (GRIA3), a candidate for bipolar disorder and nonspecific X-linked mental retardation. Genomics 62, 356–368 (1999).
Allen, N. M. et al. Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia 57, e12-7 (2016).
Hesse, A. N., Bevilacqua, J., Shankar, K., & Reddi, H. V. Retrospective genotype-phenotype analysis in a 305 patient cohort referred for testing of a targeted epilepsy panel. Epilepsy Res. 144, 53–61 (2018).
Sun, J. H. et al. X-linked neonatal-onset epileptic encephalopathy associated with a gain-of-function variant p.R660T in GRIA3. PLoS Genet. 17, e1009608 (2021).
Martinez-Esteve Melnikova, A., et al. The p.Glu787Lys variant in the GRIA3 gene causes developmental and epileptic encephalopathy mimicking structural epilepsy in a female patient. Eur. J. Med. Genet. 65, 104442 (2022).
Richards, S. et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Hamanaka, K. et al. Amelioration of a neurodevelopmental disorder by carbamazepine in a case having a gain-of-function GRIA3 variant. Hum. Genet. 141, 283–293 (2022).
Gregory, J. M. et al. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 250, 67–78 (2020).
Kryger, M. H., Roth, T., & Goldstein, C. A. Kryger’s Principles and Practice of Sleep Medicine (Elsevier Health Sciences, 2021).
Sitges, M., Chiu, L. M., & Reed, R. C. Effects of levetiracetam, carbamazepine, phenytoin, valproate, lamotrigine, ocarbamazepine, topiramate, vinpocetine and sertraline on presynaptic hippocampal Na(+) and Ca(2+) channels permeability. Neurochem. Res. 41, 758–769 (2015).
Balestrini, S. & Sisodiya, S. M. Pharmacogenomics in epilepsy. Neurosci. Lett. 667, 27–39 (2018).
Meisel, C. Antiepileptic drugs induce subcritical dynamics in human cortical networks. Proc. Natl Acad. Sci. USA 117, 11118–11125 (2020).
Acknowledgements
The authors sincerely appreciate the cooperation of the patient, her family, and all the doctors who participated in her treatment.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okano, S., Makita, Y., Miyamoto, A. et al. GRIA3 p.Met661Thr variant in a female with developmental epileptic encephalopathy. Hum Genome Var 10, 4 (2023). https://doi.org/10.1038/s41439-023-00232-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00232-1
