
Abstract
Familial renal glucosuria (FRG) is characterized by persistent glucosuria despite normal blood glucose levels in the absence of overt tubular dysfunction. SGLT2 is a sodium-glucose cotransporter expressed in the proximal tubule; loss-of-function variants in SLC5A2 are the primary cause of FRG. Heterozygous variants have rarely been reported in Japanese individuals. Here, we identified a novel SLC5A2 heterozygous variant, c.1348G>T: p.Gly450Trp, in a Japanese family comprising two children and their father.
Similar content being viewed by others


Familial renal glucosuria (FRG) is characterized by persistent glucosuria despite normal blood glucose levels in the absence of overt tubular dysfunction1. Mutations in sodium/glucose cotransporter 2 (SGLT2, SLC5A2, OMIM: 182381) have been identified and were recently reported to be involved in FRG. SGLT2 is critical for glucose reabsorption in the proximal convoluted tubules2. The SLC5A2 gene [7.7 kb, on chromosome 16p11.2] contains 14 exons and encodes 672 amino acids. FRG is generally caused by homozygous or compound heterozygous variants of SLC5A23,4. Here, we report a case of a Japanese family with a novel heterozygous variant (NM_003041.4: c.1348G>T: p.Gly450Trp) of SLC5A2.
The probands were two Japanese sisters: a 9-year-old girl (Patient 1) and a 7-year-old girl (Patient 2). They underwent a health checkup and exhibited urinary glucose positivity. Patient 1 presented with persistent urinary glucose excretion and was diagnosed with renal glucosuria when she was five years old. However, her condition was not critical. Family history (Fig. 1a) revealed that her father and her great-grandfather also exhibited urinary glucose excretion but had not been diagnosed with diabetes or renal disease. Her mother did not present with glucosuria or hyperglycemia. The medical and developmental histories of both patients were insignificant. They exhibited normal body mass index (BMI) and did not have cataracts or a Kayser-Fleischer ring. Clinical findings (Table 1) showed no signs of hyperglycemia or elevated levels of HbA1c, creatinine, or liver enzymes. The probands were diagnosed with renal glucosuria since there were few characteristic findings suggestive of Fanconi syndrome, Lowe syndrome, or Wilson disease.

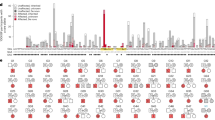
a Family pedigree. Patients 1 and 2 and their father and grandfather were diagnosed with FRG. The grandfather’s detailed medical history was unknown, and he was an SLC5A2 carrier. b,c. Mutation analysis in family members affected by FRG. b Genomic DNA sequence analysis. The G → T transition at SLC5A2 cDNA position 1348 results in a glycine to tryptophan substitution at position 450 of the SLC5A2 protein (Gly450Trp). The variant was carried in a heterozygous state by Patient 1, Patient 2, and their father. The unaffected mother displayed a wild-type (WT) sequence. c Protein alignment. The G450 residue (highlighted with red color) is conserved among the human SGLT subtypes and across SGLTs of various other species.
Based on the family history, the probands were suspected of having FRG. Genomic DNA was extracted from blood samples from both patients for diagnosis and sequenced using next-generation sequencing (NGS) with a custom panel named SPEEDI-KID5 after obtaining written informed consent from the children’s parents. This study was conducted in accordance with the Declaration of Helsinki. Permission was obtained from the institution’s ethics committee. Pooled barcoded libraries for NGS were prepared using SureSelect QXT kits (Agilent Technologies, Inc.) according to the manufacturer’s protocol. The prepared libraries were sequenced via SE 150-bp reads using Illumina MiSeq sequencers. SPEEDI-KID covers genetic variants causing FRG, such as SLC5A1, SLC5A2 and SLC2A2 (GLUT2). We also confirmed the presence of variants by Sanger sequencing. PCR primers were designed to flank approximately 200 bp of the final candidate variants, and the target sequences were amplified by PCR. They were then sequenced using Sanger sequencing on a 3130 DNA Analyzer (Applied Biosystems, Tokyo, Japan)5.
We found a missense variant, NM_003041.4 (SLC5A2): c.1348G>T: p.Gly450Trp, in exon 11. Single-base substitutions were confirmed by Sanger sequencing (Fig. 1b). There were three carriers of the variant, namely, Patient 1, Patient 2, and their father, and no variants were identified in their mother. This variant was present in the children and their father in a heterozygous state. Thus, a correlation between genotype and phenotype was confirmed in the family. Therefore, the genetic form of this variant was suggested to be autosomal dominant and is not registered in gnomAD v3.1.2 (https://gnomad.broadinstitute.org/), ToMMo 8.3 KJPN (https://jmorp.megabank.tohoku.ac.jp/202102/variants/statistics/), the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). All databases were accessed in June 2022. In silico evaluation tools of SIFT and PROVEAN predicted p.Gly450Trp of SLC5A2 as deleterious, and Polyphen-2 predicted it as probably damaging. These programs revealed a high pathological significance prediction score, whereas the 2015 ACMG criteria indicated that the variant was of uncertain significance. Variants of uncertain significance are difficult to interpret; thus, their pathological significance cannot be denied6. The G450 residue is conserved among human SGLT family members and across SGLT homologs in other species (Fig. 1c). Based on the molecular diagnosis and clinical features of the family members, it is plausible that this novel missense variant of SLC5A2 contributes to FRG pathogenesis in a compound heterozygous state.
Approximately 90 variants in the SLC5A2 gene have been reported to be associated with FRG from various countries, including the United States and China3,7. In most cases of these reports, the variants are homozygous or compound heterozygous for FRG, and obvious racial differences are not reported. In Japan, a heterozygous variant has been reported, and there is no case that carries the same variants as our patients8. Missense variants account for loss of function, as with other variants. It has been reported that patients with heterozygous SLC5A2 variants tend to exhibit relatively milder glucosuria than those with homozygous or compound heterozygous variants4. The patients’ urinary glucose levels were mild, which is consistent with the results of previous studies. SGLT2 is expressed in the S1 segment of the early proximal convoluted tubule and plays a critical role in the process of glucose reabsorption with a 1:1 Na + -to-glucose coupling ratio2. It has been reported that variants, even on the C-terminal side of SGLT2, could cause a reduction in SGLT2 expression on the cell membrane of renal tubules. The cubilin-amnionless complex is a receptor protein expressed on the luminal cell membrane of proximal tubules, and a deficit of cubilin causes Imerslund–Gräsbeck syndrome. Imerslund–Gräsbeck syndrome characterized by anemia attributed to selective intestinal malabsorption of cobalamin and low-molecular weight proteinuria is thought to develop in homozygotes. However, it has also been reported that heterozygous variants inhibit the glycosylation of cubilin and prevent membrane expression9. Similarly, in these cases, this heterozygous variant might cause a decrease in the membrane expression of proximal tubular cells10. FRG is an asymptomatic disorder except for glucosuria. Patients with FRG are not affected by severe clinical consequences, and the disorder is considered to be benign. However, by confirming a diagnosis using genetic testing and distinguishing FRG from diabetes, unnecessary testing or treatments can be prevented. In addition, patients with FRG provide an ideal model for identifying and investigating the pathogenic variants of SLC5A2. SGLT2 inhibitors have attracted attention as therapeutic agents for type 2 diabetes in recent years1, and the analysis of the SLC5A2 gene and examination of the pathophysiology of FRG could provide useful clues to the progress of diabetes and its treatment.
There are certain limitations to this report. A quantitative test for urinary glucose excretion was not evaluated, especially for the father who did not undergo examinations in our hospital. However, he does undergo these examinations at another hospital every year. Moreover, histological analysis of the kidneys in the probands was not performed. In conclusion, a novel SLC5A2 variant was identified in a family with familial renal glucosuria. Further investigations are required to clarify the mechanism of SGLT2.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3258.
References
René, S. & Joaquim, C. Familial renal glucosuria and SGLT2: from a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 5, 133–141 (2010).
Yoshikatsu, K., Wen-Sen, L., Guofeng, Y., Dennis, B. & Matthias, A. H. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J. Clin. Invest. 93, 397–404 (1994).
Shentang, L., Yeyi, Y., Lihua, H., Min, K. & Zuocheng, Y. A novel compound heterozygous mutation in SLC5A2 contributes to familial renal glucosuria in a Chinese family, and a review of the relevant literature. Mol. Med. Rep. 19, 4364–4376 (2019).
Lei, Y., Ping, H., Guo-Ping, L. & Hong, Z. Novel SLC5A2 mutation contributes to familial renal glucosuria: Abnormal expression in renal tissues. Exp. Ther. Med. 12, 649–652 (2016).
Takayasu, M. et al. Comprehensive genetic testing approach for major inherited kidney diseases, using next-generation sequencing with a custom panel. Clin. Exp. Nephrol. 21, 63–75 (2017).
Ma, N. et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human induced pluripotent stem cells. Circulation 138, 2666 (2018).
Santer, R. et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J. Am. Soc. Nephrol. 14, 2873–2882 (2003).
Sada, K. et al. Clinical and genetic analysis in a family with familial renal glucosuria: Identification of an N101K mutation in the sodium–glucose cotransporter 2 encoded by a solute carrier family 5 member 2 gene. J. Diabetes Investig. 11, 573–577 (2020).
Udagawa, T. et al. Amnionless-mediated glycosylation is crucial for cell surface targeting of cubilin in renal and intestinal cells. Sci. Rep. 8, 1–12 (2018).
Yu, L., Wu, M., Hou, P. & Zhang, H. SLC5A2 mutations, including two novel mutations, responsible for renal glucosuria in Chinese families. BMC Nephrol. 21, 1–8 (2020).
Acknowledgements
The authors express our gratitude to the patients and their parents for their cooperation. We would like to thank Editage (www.editage.com) for their English language editing services. This work was partially supported by JSPS KAKENHI Grant Number 20K16848 and the Yukiko Ishibashi Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hatano, M., Udagawa, T., Kanamori, T. et al. A novel SLC5A2 heterozygous variant in a family with familial renal glucosuria. Hum Genome Var 9, 42 (2022). https://doi.org/10.1038/s41439-022-00221-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00221-w
