
Abstract
Dormancy-associated MADS-box (DAM) genes serve as crucial regulators of the endodormancy cycle in rosaceous plants. Although pear DAM genes have been identified previously, the lack of a high-quality reference genome and techniques to study gene function have prevented accurate genome-wide analysis and functional verification of such genes. Additionally, the contribution of other genes to the regulation of endodormancy release remains poorly understood. In this study, a high-quality genome assembly for 'Cuiguan' pear (Pyrus pyrifolia), which is a leading cultivar with a low chilling requirement cultivated in China, was constructed using PacBio and Hi-C technologies. Using this genome sequence, we revealed that pear DAM genes were tandemly clustered on Chr8 and Chr15 and were differentially expressed in the buds between 'Cuiguan' and the high-chilling-requirement cultivar 'Suli' during the dormancy cycle. Using a virus-induced gene silencing system, we determined the repressive effects of DAM genes on bud break. Several novel genes potentially involved in the regulation of endodormancy release were identified by RNA sequencing and H3K4me3 chromatin immunoprecipitation sequencing analyses of 'Suli' buds during artificial chilling using the new reference genome. Our findings enrich the knowledge of the regulatory mechanism underlying endodormancy release and chilling requirements and provide a foundation for the practical regulation of dormancy release in fruit trees as an adaptation to climate change.
Similar content being viewed by others


Introduction
Bud endodormancy is a complex biological process necessary for the survival and development of perennial plants. During the endodormancy transition, buds undergo substantial internal changes, but there are no visible morphological changes1,2. Endodormancy release requires long-term exposure to cold, and the amount of cold needed to resume normal growth in the spring following the winter period is commonly referred to as the chilling requirement (CR). Insufficient chilling accumulation causes physiological disorders with adverse effects on bud sprouting, flowering, and fruit production. Global warming has resulted in insufficient winter chilling accumulation, leading to an unsatisfactory CR of some commercially grown deciduous fruit tree species in specific regions3,4. Therefore, elucidating the mechanism controlling the dormancy cycle may help researchers address the problems caused by climate change.
Several lines of evidence have shown that dormancy-associated MADS-box (DAM) genes are involved in the regulation of dormancy. DAM proteins belong to the AGAMOUS 24/SHORT VEGETATIVE PHASE (AGL24/SVP) subfamily of MADS-box proteins, and DAM genes were first described in Prunus spp., of which six tandemly repeated DAM genes (DAM1–6) are located on chromosomes 15,6,7. The large deletion fragment containing DAM1–4, which also eliminated the expression of DAM5 and DAM6, was shown to stop the growth cessation of buds of the evergrowing (evg) peach mutant5. Moreover, the ectopic expression of PmDAM6 in poplar promotes the formation of dormant terminal buds6, while DAM genes have also been identified in other rosaceous species8.
Three DAM genes have been characterized in pear, PpyMADS13-1, -2, and -3, also known as PpyDAM1/2/3, which were first identified via homologous cloning based on peach DAM genes9,10. After publication of the 'Suli' genome sequence, Niu et al. analyzed the pear MADS-box family and identified the same DAM genes10 (DAM1/2/3 or MADS13-1/-2/-3) that enhance endodormancy by inhibiting FT2 expression9,10,11. Further analysis showed that pear DAM genes are regulated by C-repeat binding factors and abscisic acid (ABA)-binding factors (ABFs), which mediate low-temperature-induced dormancy12,13. Although the biochemical functions of DAMs have been thoroughly studied in rosaceous fruit tree species, their roles in pear floral buds have not been characterized. In nonrosaceous species, DAM genes are usually named SVP or SVP-like (SVL). The poplar SVL protein enhances terminal bud endodormancy by directly promoting the biosynthesis of ABA14,15 and activating the expression of the callose synthetase gene CALLOSE SYNTHETASE 1. This causes plasmodesmata to close, preventing intercellular communication15. Thus, DAM genes mainly mediate growth repression during endodormancy.
Like FLCs during vernalization, DAM genes are thought to be regulated by epigenetic modifications during long-term exposure. Several lines of evidence indicate that H3K4me3 and H3ac are positively associated with DAM/SVP expression, whereas H3K27me3 is negatively associated with DAM/SVP expression in kiwifruit and Prunus spp16,17,18,19. Recently, a comprehensive analysis of peach DAM gene epigenetic regulation showed that multiple epigenetic events, including H3K27me3 and those involving 21-nucleotide small (s)RNAs and noncoding (nc)RNAs, were induced by chilling exposure and that such changes were inversely correlated with DAM downregulation19. Saito et al.20 observed H3K4me3 changes in 'Kosui' pear buds during the transition from endodormancy to ecodormancy, but they did not determine the association between MADS13-1 expression and H3K27me3. Similarly, SVL expression in poplar under chilling exposure was found to be unrelated to H3K27me3 levels14. Therefore, the association of epigenetic changes with the expression of DAM genes in pear is still unclear.
Pear cultivars with different CRs exhibit different endodormancy characteristics during chilling accumulation. The high-CR cultivar 'Suli' (with a CR of 800–1000 h)21 demonstrates a clear deep endodormancy stage (with bud breaking rates of almost 0 under forcing conditions) and obvious endodormancy release following chilling accumulation; conversely, the low-CR cultivar 'Cuiguan' (with a CR of <400 h)22 shows no deep endodormancy stage throughout chilling accumulation, and the lowest bud breaking rate under forcing conditions is ~20%. High-CR cultivars prefer low-CR cultivars to understand the biological changes that occur during endodormancy release because of the clear differentiation of endodormancy stages.
Since the first genome assembly of 'Suli' pear (Pyrus pyrifolia; white pear group) was published in 201323, the genomes of 'Bartlett' European pear (P. communis)24, Asian wild pear (P. betulifolia)25, and a select pear rootstock [(P. ussuriensis × P. communis) × spp.]26 have also been released. However, a reference genome for Chinese sand pear, the most widely cultivated Pyrus species27, is lacking. Additionally, the published genome of 'Suli', which is a representative Chinese white pear cultivar, is of relatively low quality because of technological limitations at the time of its assembly. Therefore, a high-quality genome for cultivated Chinese sand pear is needed.
In this study, a multiomics strategy was adopted to mine the essential genes regulating chilling-induced endodormancy release. We first assembled a high-quality genome of the low-CR pear cultivar 'Cuiguan', which is a popular cultivar in South China. Using this reference genome, we performed a genome-wide analysis of DAM genes and compared their expression patterns between pear cultivars with high and low CRs. We also verified DAM functions related to the inhibition of bud break in pear buds using a virus-induced gene silencing (VIGS) system. Because the high-CR cultivar 'Suli' has an obvious endodormancy stage and release process, using the high-quality 'Cuiguan' pear genome, we analyzed the transcriptome and genome-wide H3K4me3 modifications during the artificial chilling accumulation (ACA) period to identify candidate genes potentially involved in regulating endodormancy release. Overall, we obtained a new reference genome for cultivated Asian pear and identified several genes that are potentially involved in the regulation of the dormancy transition during the chilling accumulation period. These data will provide a basis for future investigations into the mechanism regulating endodormancy release in rosaceous fruit trees.
Results
De novo assembly of the 'Cuiguan' pear genome sequence
We assembled the P. pyrifolia 'Cuiguan' pear genome based on Illumina short reads, PacBio subreads, and Hi-C data using a hierarchical method. Approximately 36.85 Gb of Illumina paired-end short reads (270 bp) were produced, with an estimated heterozygosity ratio of 0.89%. Based on the 17-mer depth distribution of the Illumina reads, we estimated the genome to be 501.34 Mb in length. Next, we generated ~62.45 Gb of PacBio subreads (N50 = 19.07 kb) and used them to construct a genome comprising 694.68 Mb. Then, we used 74.03 Gb of clean Hi-C data to anchor the primary genome sequence. The final assembled genome (541.34 Mb; contig N50 = 1.28 Mb) included 17 pseudochromosomes (Table 1). We aligned the Illumina reads to the assembled genome, resulting in mapping of 98.58% of the reads. These 17 pseudochromosomes represented ~94% of the total assembled sequences and were denoted as the 'Suli' (P. bretschneideri) and P. betulifolia genomes (Supplementary Fig. S1).
Annotation and phylogenetic analysis of the 'Cuiguan' pear genome
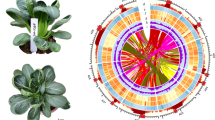
We identified a total of ~314.99 Mb of repetitive elements, occupying ~58.19% of the assembled sequence. We detected LTR copia and LTR gypsy as two of the repetitive elements, which accounted for a large proportion of the total repetitive sequences in the assembled genome (Fig. 1a, Supplementary Table S1). The LTR assembly index (LAI) of the 'Cuiguan' pear genome was 17.62. We combined three gene prediction strategies [ab initio, homology-based search, and RNA sequencing (RNA-seq)] to predict 42,622 protein-coding genes with an average length of 3151 bp. The number of annotated genes was similar to that in the 'Suli' pear genome (Fig. 1a, Table 1). Moreover, 39,929 genes (93.7%) were detected on the pseudochromosomes. We used the BUSCO 3 program28 to evaluate the predicted genes based on 1440 highly conserved plant genes. Specifically, 94.8% complete BUSCOs were identified, reflecting the integrity of our genome and the successful prediction of protein-coding genes. These predicted genes were further annotated using the NR, Gene Ontology (GO), EuKaryotic Orthologous Groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG), and TrEMBL databases, and 99.14% (42,257) of the genes were successfully annotated (Supplementary Table S2).

a 'Cuiguan' pear genome profile. (1) pseudochromosomes; (2) LTR copia density; (3) LTR gypsy density; (4) gene density; (5) GC content distribution. The colored lines connect gene pairs within syntenic blocks. b Phylogenetic tree of 'Cuiguan' pear and eight other rosaceous species. The orange clade includes four Pyrus species whose genome has been published
We used 510 single-copy gene families from the following species to construct a phylogenetic tree according to the maximum likelihood method (Fig. 1b): 'Cuiguan' pear (P. pyrifolia), 'Suli' pear (P. bretschneideri)23, P. betulifolia25, P. communis24, Malus domestica29, P. persica30, Rosa chinensis31, Fragaria vesca32, Rubus occidentalis33, and Solanum lycopersicum34 (outgroup; not shown in the Figure). 'Cuiguan' pear and 'Suli' pear were closely clustered, indicative of a close genetic relationship. We also calculated the synonymous substitution rates (Ks) of collinear gene pairs using the 'Cuiguan' pear, apple, and peach genomes. The Ks density distribution revealed a peak shared by pear and apple (Supplementary Fig. S2), consistent with the recent whole-genome duplication event that occurred before the species differentiation between pear and apple occurred.
Identification and analysis of DAM genes in the 'Cuiguan' and 'Suli' pear genomes
DAM genes encode key bud endodormancy regulators in rosaceous plant species. Genome-wide analysis revealed five intact DAM genes in the 'Cuiguan' pear genome, which we renamed according to their chromosomal positions. Three and two DAM genes were tandemly arrayed on chromosomes 8 (DAM1–3) and 15, respectively. We named the two DAM genes on chromosome 15, which encode identical proteins, DAM4-1 and DAM4-2 (Fig. 2). Collinearity analysis revealed a strong collinear relationship between the peach EVG locus and loci on pear chromosomes 8 and 15 (Fig. 2a, b). The alignment of DAM proteins showed that the peach and pear DAM proteins are highly similar. However, the similarity between sequences from the same species was greater than those between different species (Supplementary Fig. S3). In the phylogenetic tree (Fig. 2c), the five pear DAMs clustered together with four apple SVP proteins and were grouped with six peach DAM proteins.

a Collinear blocks between peach chromosome 1 and 'Cuiguan' pear chromosomes 8 and 15. A 10 Mb partial chromosome region is shown around each collinear block. The detailed collinearity relationship of the green blocks is shown in (b). b Detailed collinear blocks between the peach EVG locus and pear DAM genes. Chromosomal segments are indicated by the horizontal bars. Genes in the positive and negative strands are shown in the green and blue boxes, respectively. The peach DAM genes are marked in red, and the pear DAM genes are marked in orange. The collinear DAM gene pairs are linked by orange curves, whereas the other genes are linked by blue curves. c Phylogenetic analysis of rosaceous DAM proteins. Ppe Prunus persica, Ppy Pyrus pyrifolia, Md Malus domestica. The pear DAM genes are shown in blue text. d DAM gene expression patterns in 'Suli' and 'Cuiguan' pears during a natural endodormancy cycle. The error bars indicate the standard deviations of three biological replicates. The dashed lines indicate the date of endodormancy release
We next compared DAM gene expression patterns during the endodormancy cycle between 'Suli' pear and 'Cuiguan' pear (Fig. 2d). Because the DAM4-1 and DAM4-2 sequences are identical, their PCR products were indistinguishable. Consequently, we used DAM4 to represent the expression of DAM4-1 and DAM4-2. For 'Suli' pear, two expression patterns were observed for the four DAM genes. The DAM3 expression level decreased continuously from September, when we started sampling, until endodormancy release. DAM1, DAM2, and DAM4 expression levels were upregulated from mid-October, when endodormancy started, and peaked prior to endodormancy release, after which they decreased substantially. The DAM3 expression pattern in 'Cuiguan' pear was similar to that in 'Suli' pear. DAM1, DAM2, and DAM4 expression levels decreased shortly after endodormancy release, which was in contrast to the corresponding expression patterns in 'Suli’ pear.
Establishment of VIGS and DAM gene silencing in pear buds
To further clarify DAM protein functions during the endodormancy cycle, we established a VIGS system in pear buds to study gene function during bud endodormancy. The experiment was conducted using excised 'Suli' pear shoots infected by Agrobacterium harboring pTRV1 and pTRV2-DAM (Fig. 3a). To silence DAM genes only, the selected fragment was taken from a relatively conserved region of DAM genes (half of the K-box domain) but did not include the conserved MADS-box region (Supplementary Fig. S4).

a Agroinfiltration of cut pear shoots. b VIGS of pear DAM genes increased the floral bud break rate after 21 days of cultivation under forcing conditions. The error bars indicate the standard deviations of three biological replicates. The asterisk indicates a significant difference compared with the empty vector control according to Student’s t-test (*P < 0.05). c DAM1, DAM2, and DAM4 expression levels in buds at 5 days after VIGS treatment. The error bars indicate the standard deviations of three biological replicates. The asterisks indicate significant differences compared with the empty vector control according to Student’s t-test (**P < 0.01; ***P < 0.001). d EXPA1 expression in buds at 5 days after VIGS treatment. The error bars indicate the standard deviations of three biological replicates. The asterisk indicates a significant difference compared with the empty vector control according to Student’s t-test (*P < 0.05)
The bud break percentage was significantly higher in DAM-silenced dormant 'Suli' pear buds than in control buds after a 10-day forcing treatment (Fig. 3b), indicating that silencing DAM genes in pear buds promoted endodormancy release. At 5 days after agroinfiltration, quantitative real-time (qRT)-PCR analysis indicated that DAM gene (DAM1, DAM2, and DAM4) expression levels were significantly downregulated (Fig. 3c). Furthermore, the expression of EXPANSIN A1 (EXPA1), which is related to endodormancy release35, was significantly upregulated after the DAM genes were silenced (Fig. 3d). The silencing of DAM genes may have promoted endodormancy release because of the associated induced expression of growth-related genes (e.g., EXPA1). Thus, we confirmed that high DAM gene expression levels are correlated with the maintenance of pear bud endodormancy.
Artificial chilling accumulation and phytohormone contents
To determine the effects of long-term chilling on the regulation of endodormancy release, 1-year-old detached shoots of 'Suli' pear plants were collected on October 15, 2019, for ACA treatment (Fig. 4a). This date was selected because the 'Suli' pear buds were in the endodormancy stage and because the temperature was still greater than 10 °C (Fig. 4b). After a 30-day chilling accumulation period, the bud break percentage began to increase, exceeding 50% at 50 days, which is when bud endodormancy was released (Fig. 4c).

a Outline of ACA treatment and sample harvesting. b Endodormancy process (bud break percentage) of 'Suli' pear buds and daily temperature during the endodormancy transition. Orange and blue lines represent the maximum and minimum daily temperatures, respectively. The black dots indicate the bud break percentage on different sampling dates. The red arrow represents the sample used for ACA treatment. c Floral bud break rate during ACA treatment. d–g Changes in ABA, ACC, and JA contents in bud samples during ACA treatment. Phytohormone contents were measured by UPLC-MS/MS. The error bars indicate the standard deviations of three biological replicates, whereas the dots indicate individual values. The different letters above the bars indicate significant differences between samples according to one-way ANOVA (Duncan’s test, P < 0.05)
We also measured the abundance of several phytohormones in buds subjected to ACA treatment and those under natural conditions to confirm the effects of chilling accumulation. The ABA content was relatively high during the first 20 days of the ACA treatment, after which it decreased gradually, which was consistent with changes under natural conditions (Fig. 4d; Supplementary Fig. S5a). Interestingly, changes in the abundance of the ethylene precursor ACC were the opposite of those observed for the ABA content (Fig. 4e; Supplementary Fig. S5b), implying that ethylene influences endodormancy induction and release. Additionally, the jasmonate (JA and JA-Ile) contents tended to gradually decrease during the chilling accumulation period (Fig. 4f, g; Supplementary Fig. S5c).
Screening of dormancy transition-related genes by RNA-seq
To characterize temporal gene expression changes that occur during the dormancy transition, we analyzed samples treated with ACA and those under natural conditions by RNA-seq. A total of 21 libraries from seven time points of the ACA treatment and 15 libraries from five time points of the natural dormancy cycle were sequenced. Sequencing generated ~252.16 Gb of clean reads, which we mapped to the previously released 'Suli' pear genome, with average mapping rates of 82.15% and 71.59% for the samples treated with ACA and those under natural conditions, respectively (Supplementary Fig. S6); however, when the new 'Cuiguan' pear genome was used as the reference sequence, the mapping rates were as high as 89.84% and 77.95%, respectively (Supplementary Fig. S6). Therefore, we analyzed the RNA-seq data based on the 'Cuiguan' pear genome sequence. The HISAT2 featureCounts-DESeq2 pipeline revealed 9392 differentially expressed genes (DEGs) in the ACA-treated samples and 14,673 DEGs in those under natural conditions (Supplementary Dataset).
Our analysis of the pear DAM gene expression patterns indicated that all the genes were differentially expressed when the results under the ACA treatment and natural conditions were compared. The DAM1, DAM2, and DAM4 expression patterns were similar, with high expression levels during the early-ACA period (0–10 days) and decreasing levels toward endodormancy release (10–50 days, Supplementary Fig. S7a). In contrast, DAM3 expression was continuously downregulated during the ACA period (Supplementary Fig. S7). These changes were in accordance with the expression patterns under natural conditions (Fig. 2c; Supplementary Fig. S7b). qRT-PCR analysis of functional DAM genes confirmed their expression patterns in the ACA-treated samples (Supplementary Fig. S7c).
To further identify the genes involved in the regulation of the dormancy transition, using Mfuzz analysis. we independently categorized the DEGs in ACA-treated samples and samples under natural conditions into clusters based on their expression patterns36. Nine clusters were obtained for each sample type (Supplementary Fig. S8). Clusters with linear changes during the ACA period (clusters 2, 4, 5, 6, and 9) as well as clusters 3, 5, 7, 8, and 9 for samples under natural conditions were further analyzed (Supplementary Fig. S8a, b). Specifically, the 1,001 overlapping genes between these two DEG pools were selected for further analyses (Fig. 5a).

a Venn diagram of genes in specific clusters for ACA-treated samples and samples under natural conditions. The 1001 overlapping genes were considered candidate genes regulating endodormancy. The blue circle shows DEGs in samples under natural conditions (clusters 3, 5, 7, 8, and 9), whereas the yellow circle shows DEGs in ACA-treated samples (clusters 2, 4, 5, 6, and 9). b GO analysis of the 1001 putative candidate genes. The orange, blue, and green text correspond to the molecular function, cellular component, and biological process GO categories, respectively. The dot color represents the −log10 corrected p value (Benjamini–Hochberg corrected), whereas the dot size reflects the number of enriched genes. c Heatmaps of TPM values for ACA clusters 2/4 and (d) the expression of selected genes as determined by qRT-PCR. e Heatmaps of the TPM values for ACA cluster 5 and (f) the expression of selected genes as determined by qRT-PCR. g Heatmaps of the TPM values for ACA cluster 9 and (h) the expression of selected genes as determined by qRT-PCR. The line graphs in c, e, and g show the expression trends of the correlated clusters
GO analysis indicated that the 1001 genes were mainly associated with enzyme activities, plastoglobules, and phenylpropanoid metabolic processes (Fig. 5b), suggesting that these genes contribute to multiple biological activities. We analyzed the expression patterns of ACA-treated samples and identified genes in clusters 2/4 (ACA-clusters 2/4, Supplementary Table S3) and cluster 5 (ACA-cluster 5, Supplementary Table S4) with downregulated expression toward endodormancy release as candidate genes inhibiting endodormancy release. Cluster 9 genes for ACA-treated samples (ACA cluster 9, Supplementary Table S5) were considered candidate genes that promote endodormancy release.
Genes encoding transcription factors including NACs (NAC10, NAC40, and NAC88), MYB108, TCP12, ZAT12, ABRE-BINDING FACTOR 2 (ABF2), and DAM4 (Fig. 5c; Supplementary Table S3) and enzymes such as protein phosphatase 2C, sorbitol dehydrogenase, and E3 ubiquitin-protein ligase were identified in ACA clusters 2/4. The enriched GO terms among the DEGs in clusters 2/4 were porphyrin-containing compound metabolic process, tetrapyrrole metabolic process, and pigment metabolic process (Supplementary Fig. S9a). The expression levels of these genes were upregulated during the early-chilling accumulation period (0–20 days; October 15–December 15) and were downregulated toward endodormancy release (20–50 days; December 15–January 20) (Fig. 5c; Supplementary Fig. S9b). We further validated the expression patterns of these genes during the dormancy transition via qRT-PCR (Fig. 5d).
The expression of genes in ACA cluster 5 continuously decreased during the chilling accumulation period (Fig. 5e; Supplementary Fig. S10a). Several genes encoding transcription factors (e.g., ethylene response factor [ERF], basic helix–loop–helix [bHLH], and MADS-box types) were identified. Additionally, most pear DAM genes, including DAM1, DAM2, and DAM3, were categorized in this cluster. The expression of DAM3 decreased more rapidly than did that of DAM1 and DAM2. Moreover, the strigolactone esterase DAD2 gene and several genes encoding antioxidant-related enzymes, such as glutathione S-transferase and peroxidase, were also included in this cluster. GO enrichment analysis suggested that the genes in this cluster participate in transcriptional regulation and protein dimerization (Supplementary Fig. S10b). The expression profiles of selected genes in the samples under natural conditions were similar to those in the ACA-treated samples (Fig. 5f).
In ACA cluster 9, the expression levels of genes encoding transcription factors (e.g., bHLH, TCP, ERF, G-box-binding factor, auxin response factor, and zinc finger protein types) increased throughout the chilling accumulation period (Fig. 5g). Additionally, the expression levels of some structural genes, including YUCCA, cyclin-J18 (CYCJ18), fructokinase, and sucrose synthase genes, were significantly upregulated during the chilling accumulation period (Fig. 5g; Supplementary Fig. S11a; Supplementary Table S5). Interestingly, the expression of some genes related to epigenetic regulation, including those encoding histone H2AX, methyltransferase, histone-lysine N-methyltransferase SUVR2-like, and acyltransferase, was also upregulated during prolonged chilling accumulation (Supplementary Table S5), suggesting that epigenetic modifications might be involved in chilling-induced endodormancy release. The enriched GO terms among the DEGs in cluster 9 were pyrimidine-containing compound process, ribose phosphate biosynthetic process, RNA processing, and reproductive system development (Supplementary Fig. S11b). In addition, the expression levels of candidate genes [TCP4 and GH3 (encoding glucan endo-1,3-beta-glucosidase 3)] were upregulated during the chilling accumulation period (Fig. 5h), as confirmed via qRT-PCR.
H3K4me3 chromatin immunoprecipitation and sequencing (ChIP-seq) analysis of dormant 'Suli' pear buds during the chilling accumulation period
To investigate the possible epigenetic regulation of endodormancy release, we determined the genome-wide distribution of H3K4me3 modifications in dormant pear buds. The ChIP-seq data revealed that H3K4me3 histone marks were mainly associated with transcription start sites (TSSs) (Supplementary Fig. S12a) in all ACA-treated samples. Moreover, gene expression was closely related to the presence of H3K4me3 at the TSS (Supplementary Fig. S12b–f).
During the ACA period, we found that the expression of DAM genes in ACA clusters 4 and 5 decreased toward endodormancy release (Supplementary Fig. S7; Supplementary Tables S3, S4). We also showed that H3K4me3 modification was clearly enriched around the TSS of all DAM genes. For DAM1 and DAM3, the enrichment of H3K4me3 decreased beginning at the middle of the ACA period and was lowest after 50 days (Fig. 6a). However, for DAM4-1 and DAM4-2, H3K4me3 enrichment increased soon after initiating ACA treatment and then decreased until the end of the treatment period (Fig. 6b).

a, b Enrichment of H3K4me3 in DAM loci on 'Cuiguan' pear chromosomes 8 and 15. The peaks represent the normalized H3K4me3 signals. The exons are indicated by blue bars, whereas the introns are indicated by gray horizontal lines. The transcription orientation is marked above the gene structures. c H3K4me3 level at the CYCJ18 (EVM0000777.1) locus. The arrow indicates the coding direction. The exons are indicated by blue boxes, whereas the introns are indicated by lines. The yellow bars represent the relative expression of CYCJ18 during ACA treatment. The error bars indicate the standard deviations of three biological replicates. d H3K4me3 level at the NAC88 (EVM0012159.1) locus. The arrow indicates the coding direction. The exons are indicated by blue boxes, whereas the introns are indicated by lines. The yellow bars represent the relative expression of NAC88 during ACA treatment
Finally, our RNA-seq data indicated that the expression of a cell division-related gene, CYCJ18 (cluster 9), of the cyclin family steadily increased during the chilling accumulation period (Supplementary Table S5). The ChIP-seq data revealed that H3K4me3 was enriched at the CYCJ18 locus during the chilling accumulation period (Fig. 6c). Additionally, the expression of the cold-induced transcription factor-encoding gene NAC88, whose H3K4me3 level increased and then decreased during the chilling accumulation period, was upregulated during endodormancy but downregulated during endodormancy release (Fig. 6d).
Discussion
The 'Cuiguan' pear genome is a new reference genome for cultivated asian pear
PacBio long reads can enhance the accuracy and integrity of genome assemblies, whereas Hi-C sequencing enables pseudochromosomes to be correctly assembled. Compared with the 'Suli' pear (P. bretschneideri) genome sequence released by Wu et al.23, the 'Cuiguan' pear (P. pyrifolia) genome sequence constructed in this study had a higher contig N50 value. The LAI represents genome assembly continuity because LTR retrotransposons are poorly assembled in draft genomes. In the current study, the LAI of the 'Cuiguan' pear genome was 17.62 (Table 1), which is similar to that of the Arabidopsis thaliana TAIR10 reference genome37, while BUSCO evaluation confirmed that the 'Cuiguan' pear genome was relatively complete. Moreover, its RNA-seq read mapping rate was relatively high (Supplementary Fig. S6). These results imply that this is a high-quality reference genome that could be useful for genome-based research on Asian pear. 'Cuiguan' and 'Suli' are the main pear cultivars grown in China and are usually considered to be P. pyrifolia and P. bretschneideri, respectively. In this study, they were clustered together, revealing a very close genetic relationship (Fig. 1b) and supporting our earlier conclusion that Chinese white pear cultivars such as 'Suli' should be treated as P. pyrifolia cultivars (i.e., the P. pyrifolia white pear group)38,39,40.
Characterization and function of pear DAM genes
Three MADS-box proteins (DAM1/2/3 or MADS13-1/-2/-3) from 'Suli' and 'Kosui' pear cultivars were previously identified in studies involving homology-based cloning9,10,11. Only one intact DAM gene and two partial DAM genes have been annotated in the released 'Suli' pear genome10. To unify the naming of pear DAM genes, the five DAM genes detected in the 'Cuiguan' pear genome were renamed according to their chromosomal locations (Supplementary Table S6). Phylogenetic analysis (Fig. 2) implied that apple and pear DAM genes evolved before the separation of Malus and Pyrus and that they share the same origin as Prunus DAM genes. Specifically, peach DAM genes were tandemly arrayed at the EVG locus5, and pear DAM1/2/3 were tandemly arrayed on chromosome 8. We also detected a strong collinear relationship between the DAM locus on peach chromosome 1 and that on pear chromosome 8 (Fig. 2a). However, peach and pear DAM genes clustered separately in the phylogenetic tree, suggesting that these clusters were derived from segmental duplication events that occurred after the separation of the tribes Amygdaleae and Maleae.
A previous study proved that the regulated DAM1, DAM2, and DAM4 (originally named MADS13-1, MADS13-2, and MADS13-3, respectively) expression patterns in 'Kosui' Japanese pear leaf buds are consistent with endodormancy establishment and release9. In the present study, we identified DAM3 and DAM4-2 as additional pear DAM genes. Two expression patterns were detected for these DAM genes, especially in 'Suli' pear, which has a high CR. DAM1, DAM2, and DAM4 expression levels increased during endodormancy but decreased rapidly after entering the ecodormancy stage, consistent with the results of our previous studies10,13 and with reported DAM6 expression patterns in peach and Japanese apricot6,41. In contrast, DAM3 expression decreased continuously beginning in September, reaching its lowest level upon endodormancy release. Although the DAM3 expression pattern was similar in 'Cuiguan' pear and 'Suli' pear, the expression level was much lower and decreased earlier in 'Cuiguan' pear. Moreover, DAM1, DAM2, and DAM4 expression changed more in 'Suli' pear than in 'Cuiguan' pear. Because endodormancy maintenance requires high DAM gene expression levels, our results suggest that the limited changes in DAM1, DAM2, and DAM4 expression during endodormancy might be responsible for the relatively weak dormancy of 'Cuiguan' pear. Previous studies also concluded that differences in the expression of these DAM genes might be related to CR diversity9,13.
Ectopic expression of PmDAM6 in poplar reportedly inhibits plant growth and delays bud break6. Overexpression of PpyDAM4 (previously designated PpyDAM3) in pear calli was also shown to inhibit growth and downregulate CYCLIN D and EXPASIN 1 expression13. Except for the peach evg mutant5, there has been no direct genetic evidence of DAM functions related to the floral bud endodormancy of Amygdaloideae plant species, likely because of a lack of effective transformation techniques and a long juvenile period. In this study, we established a VIGS technique to functionally characterize genes in pear floral buds. Previous investigations have confirmed that tobacco rattle virus (TRV) can infect apple and pear42,43,44. Therefore, TRV was used to silence DAM genes in dormant pear buds to further clarify DAM functions during bud dormancy transition. Because we were unable to identify the buds in which DAM genes were silenced, we collected all the buds. Our subsequent analysis confirmed that the silencing, or at least downregulation, of DAM1, DAM2, and DAM4 expression led to an increase in bud break percentage under forcing conditions (Fig. 3). As expected, the expression of the downstream gene EXPA1 was significantly upregulated in pear buds when DAM1, DAM2, and DAM4 were silenced. Thus, our results confirmed that EXPA1, which is closely associated with bud break, is located downstream of the DAM genes in pear buds. Future studies involving the overexpression of these genes in transgenic plants may help elucidate the functions of encoded proteins during the dormancy transition.
Candidate genes potentially regulating pear endodormancy release
Analyses of RNA-seq data for buds collected under natural conditions and those of plants treated with specific chemicals have revealed many candidate genes encoding key dormancy regulators3,17,43,44,45, but the precise functions of these genes remain unclear. However, under natural conditions, bud status can be affected by disease, insect pests, and other dynamic environmental cues, which might affect the identification of candidate genes. ACA treatment may enable a more accurate gene expression analysis. Gabay et al.45 recently used ACA-treated vegetative buds to quantify the metabolite contents in European pear, revealing that fatty acids including α-linolenic acid influence the dormancy transition. In the present study, we comprehensively analyzed the RNA-seq data for ACA-treated floral buds and the floral buds under natural conditions.
Mfuzz analysis allowed us to cluster DEGs in ACA-treated samples and samples under natural conditions into nine groups. Because DAM1, DAM2, and DAM3 were grouped in ACA cluster 5 and encode proteins that usually form functional dimers, the transcription factors-encoding genes (e.g., ZAT10, RAP2.3, TIFY10A, and ERF5) in cluster 5 might be closely related to DAM genes with respect to expression and function. GO analysis indicated that ACA cluster 9 genes are associated with reproduction-related processes and cell division (Supplementary Fig. S11), implying that cell division is activated and floral organs develop during the progression of chilling accumulation in preparation for endodormancy release and flowering. This finding is consistent with our recent findings35. Among these genes, CYCJ18 is responsible for nuclear division46, whereas GH3, belonging to the glucan hydrolase superfamily, encodes an enzyme that hydrolyzes 1,3-β-glucan, which regulates dormancy in poplar15,47. GH3 expression was upregulated during the chilling accumulation period, similar to the expression of GH17 members in poplar47, suggesting that GH3 might be crucial for callose degradation in pear buds and the promotion of endodormancy release. However, the functions of these genes need to be thoroughly characterized in future studies.
Among the transcription factor-encoding genes, NAC10, NAC40, and NAC88 expression levels were continuously downregulated toward endodormancy release, similar to DAM gene expression (Figs. 5, 6; Supplementary Fig. S9b). Earlier research indicated that NAC10 and NAC88 may be involved in plant responses to low temperature as well as the regulation of dormancy transition48. Any potential relationships among NAC10, NAC88, and DAM4 need to be experimentally verified, as do the functions of NAC10 and NAC88 related to bud dormancy. The expression of several genes encoding bHLH transcription factors (e.g., bHLH10-like, bHLH90, and bHLH91-like) was also upregulated during the chilling accumulation period, which is indicative of a potential role in endodormancy release (Fig. 5g). The expression of an identified TCP family gene, TCP4, was upregulated during the chilling accumulation period, with peak levels at endodormancy release. Interestingly, in A. thaliana, TCP family proteins interact with the bHLH protein PIF4, target the gibberellin (GA) biosynthesis-related gene GA20ox1 and the atypical HLH transcription factor-encoding gene PRE6, thereby regulating thermomorphogenic growth49. The expression of PRE1, which positively regulates GA signaling and cell elongation in A. thaliana50, was also upregulated in pear buds during the chilling accumulation period. Thus, TCP4 may interact with bHLH proteins to activate PRE1, GA20OX2, and other cell division- and elongation-related genes (e.g., CYCLIN and EXPANSIN) to induce endodormancy release.
ChIP-seq analysis reveals a possible association of H3K4me3 with dormancy in pear
Earlier research indicated that H3K4me3, which is a histone marker for active transcription, is involved in dormancy regulation in peach, sweet cherry, chestnut, and kiwifruit16,17,18,51. Our current data show that H3K4me3 levels are correlated with the expression of specific genes (Fig. 6) during the dormancy cycle. Additionally, these findings support the fact that pear DAM genes are targeted for H3K4me3 modifications during the chilling accumulation period (Fig. 6a, b), which is consistent with the results of related studies on peach, sweet cherry, and kiwifruit16,18,51. Although it is unclear how chilling regulates histone modifications at DAM loci, recent research has revealed the presence of small RNAs and noncoding RNAs at peach DAM loci, implying that they contribute to the regulation of histone modifications at these sites19.
We also observed that the gene expression of NAC88 and CYCJ18 was correlated with H3K4me3 modification (Fig. 6) during the dormancy transition. In poplar trees exposed to drought stress, the expression of genes encoding NAC transcription factors is reportedly controlled by H3K9ac histone modifications mediated by AREB152, while CYCJ18 affects nuclear division cycles during early Drosophila embryogenesis46. In previous investigations, we detected dividing cells as well as an increase in the number of nuclei in buds during maintenance and release of endodormancy35. Moreover, in poplar, SVL inhibited cyclin-D through EARLY BUD BREAK 3 (EBB3) expression, which is epigenetically regulated by H3K27me3 to inhibit endodormancy release and bud break53. Our results suggest that cyclin genes are direct targets for histone modifications during endodormancy release. Indeed, taken together, these observations suggest that H3K4me3 modification is associated with the regulation of endodormancy release and that it does not target only DAM genes.
In conclusion, we assembled the genome of the sand pear cultivar 'Cuiguan', which should prove useful as a reference genome for future molecular studies. Using this new reference genome, we identified five DAM genes tandemly arrayed on chromosomes 8 and 15 and unified the names of the DAM genes according to their chromosomal positions. We established a VIGS technique applicable to pear floral buds, thereby providing researchers with a new method for functionally characterizing genes during the dormancy cycle of floral buds, especially those from fruit trees with long juvenile periods. This new reference genome enables higher-quality RNA-seq and ChIP-seq analyses that can be used in the future to identify additional candidate genes that may be involved in endodormancy release.
Materials and methods
Plant materials and the ACA
The leading Chinese sand pear (P. pyrifolia) cultivar 'Cuiguan' grown in China was used for genome sequencing. Dormancy regulation was investigated using 'Suli' pear (P. pyrifolia White Pear Group; also named P. bretschneideri) samples with known dormancy transition phenotypes and an available sequenced genome.
For samples under natural conditions, shoots were collected starting on September 15, 2016, and 2017, as previously described13,54. ACA treatment was completed using detached branches from 1-year-old shoots to eliminate the effects of the external environment. Shoots were collected starting on October 15, 2019. 'Suli' pear buds entered the endodormancy stage at this time point. The shoots were randomly divided into three groups to form three biological replicates and incubated at 15–18 °C for 3 days. Afterward, they were kept in a phytotron at 4 °C (with 90% humidity) for ACA treatment until endodormancy release (up to 50 days). Lateral flower buds were removed from the detached shoots at 0, 10, 20, 30, 35, 40, 45, and 50 days after initiating the ACA treatment, immediately frozen in liquid nitrogen, and stored at −80 °C until analysis. The dormancy status at each time point was determined by calculating the bud break percentage under forcing conditions (25 °C/23 °C, 16-h day/8-h night cycle) for 21 days.
Genome assembly
Approximately 62.45 Gb of PacBio subreads with a mean length of 12,466 bp were generated for genome assembly. We first used Canu55 to correct the PacBio reads. Canu and WTDBG (https://github.com/ruanjue/wtdbg) were used to independently assemble the high-quality PacBio reads, and five rounds of polishing were carried out by Pilon (https://github.com/broadinstitute/pilon) using Illumina short reads. Self-alignment was performed to remove redundancies with MUMmer56. The Canu and WTDBG draft assemblies were merged using Quickmerge (https://github.com/mahulchak/quickmerge), and Pilon was used to polish the merged assembly. We generated 74.03 Gb of clean data from the Hi-C library for sequence assembly at the chromosome level. BWA57 was used to align reads to the assembly, and only uniquely mapped read pairs were retained. Additionally, invalid read pairs were filtered with HiC-Pro (v2.8.1)58, and valid read pairs that were used for subsequent analysis accounted for 81.33% of the uniquely mapped read pairs. LACHESIS software59 was used to cluster, order, and orient the genome sequences into pseudochromosomes with the parameters CLUSTER_MIN_RE_SITES=43, CLUSTER_MAX_LINK_DENSITY=2, CLUSTER_NONINFORMATIVE_RATIO=2, ORDER_MIN_N_RES_IN_TRUN=25, and ORDER_MIN_N_RES_IN_SHREDS=21.
Genome annotation
Repeat sequences were predicted as follows. A de novo repeat library was constructed with LTR FINDER (v1.05)60, MITE-Hunter61, RepeatScout (v1.0.5)62, and PILER-DF (v2.4)63 using the default parameters. The database was then classified using PASTEClassifier64 and merged with Repbase as the final repetitive sequence database. Finally, RepeatMasker (http://www.repeatmasker.org/) was used to screen any repeats. The LAI was independently calculated using the LTR_retriever pipeline (https://github.com/oushujun/LTR_retriever).
The EVM (v1.1.1) pipeline65 was used to predict genes in the assembled genome sequence. Briefly, the default parameters of GenScan66, Augustus (v2.4)67, Glimmer HMM (v3.0.4)68, Gene ID (v1.4)69, and SNAP (v2006-07-28)70 were applied for de novo gene prediction. The default parameters of GeMoMa (v1.3.1)71 were used to predict homologs in the A. thaliana, F. vesca, and P. bretschneideri databases. The PASA program (v2.0.2)72 was used to predict unigene sequences based on the RNA-seq data for 'Cuiguan' pear. All the acquired data were then combined and revised using EVM and PASA. Protein-coding genes were annotated based on BLAST searches (e-value cutoff of 1e−5) of the information housed in the NR (http://www.ncbi.nlm.nih.gov), KOG (ftp://ftp.ncbi.nih.gov/pub/COG/KOG), GO (http://geneontology.org/), KEGG (http://www.genome.jp/kegg), and TrEMBL (http://www.UniProt.org/) databases.
Phytohormone quantification
We entrusted Shanghai Applied Protein Technology Co., Ltd. (APTBIO, Shanghai, China), to extract and quantify the phytohormones in pear buds as previously described73.
RNA extraction and RNA-seq
Total RNA was extracted from buds using a modified cetyltrimethylammonium bromide method74. We entrusted Novogene Co., Ltd. (Beijing, China), and BGI (Shenzhen, China) to construct and sequence the RNA-seq library of ACA-treated samples and samples under natural conditions, respectively. The raw data were processed during a quality control step to produce clean data.
cDNA synthesis and qRT-PCR
First-strand cDNA was synthesized from 1 μg of total RNA using HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech Co., Ltd., Nanjing, China). The cDNA was subjected to a qRT-PCR assay, which was completed with iTaq Universal SYBR Green SuperMix (Bio-Rad Laboratories, Beijing, China), as previously described13. The pear actin (JN684184) gene was used as a reference control because it is constitutively expressed during the dormancy transition21. The qRT-PCR primers used are listed in Supplementary Table S8.
ChIP-seq
The modified ChIP method used in this study was based on protocols described by Haring et al.75 and Li et al.76. Scales were removed from buds collected at 0, 10, 20, 30, 40, 45, and 50 days after the ACA treatment was started. The buds were treated with 1% (w/v) formaldehyde for 10 min under vacuum at room temperature in the cross-linking step. Afterward, 1.25 M glycine was added, and the tissues were incubated under vacuum for 5 min to terminate the cross-linking reaction. The supernatant was discarded, and the tissues were washed twice with distilled water and then allowed to dry on filter paper. The tissues were then frozen in liquid nitrogen and stored at −80 °C until analysis. After extracting the nuclei, 320 μl of nuclear lysis buffer [50 mM Tris-HCl (pH 8.0), 10 mM ethylenediaminetetraacetic acid, 1% (w/v) sodium dodecyl sulfate, 1 mM phenylmethylsulfonyl fluoride, and proteinase inhibitor cocktail] was added. The chromatin solution was subsequently sonicated 30 times with a bioruptor (30 s each time at 30 s intervals). The solution was centrifuged (16,000 × g at 4 °C for 5 min), after which 300 μl of the supernatant was transferred to a new 2-ml tube. Afterward, 1660 μl of ice-cold ChIP Ab incubation buffer was added to the chromatin solution, which was thoroughly mixed. A 60 μl aliquot was used as the input. ChIP-grade anti-H3K4me3 antibodies (Abcam, Cambridge, UK) and Pierce ChIP-grade Protein A/G magnetic beads (Thermo Fisher Scientific, Waltham, MA, USA) were used for IP. Following a reverse cross-linking step, the chromatin was purified with an Easy Gel Extraction & Clean-up Kit (Zhejiang Easydo Biotechnology Co., Ltd., Hangzhou, China) and eluted with ddH2O for sequencing. After immunoprecipitation of the samples (three biological replicates), the precipitated DNA was combined for deep sequencing analysis. We entrusted Novogene Co., Ltd., to construct and sequence the ChIP-seq library.
Phylogenetic, RNA-seq, and ChIP-seq analyses
Detailed procedures of the phylogenetic, RNA-seq, and ChIP-seq analyses are described in Methods S1, Methods S2, and Methods S3.
VIGS assay of pear buds
A VIGS assay was completed with TRV. A 244-bp reverse complementary fragment (end of the K-box) of DAM4 was cloned and inserted into a pTRV2 vector using the primers listed in Supplementary Table S9. The construct was then inserted into Agrobacterium EHA105 cells. pTRV1 and pTRV2 empty vectors were inserted into separate EHA105 cells, and pTRV2 served as a negative control. The VIGS assay was performed using 'Suli' pear buds. After the bacteria reached the logarithmic growth phase, the culture was diluted with infection buffer [10 mM MgCl2, 10 mM MES (pH 5.6), and 100 μM acetosyringone] until an OD600 of 0.8 was reached. Equal volumes of pTRV1 and pTRV2 bacterial solutions were combined and incubated at room temperature for 1 h. 'Suli' shoots were collected on November 1, 2019 (with buds in the endodormancy stage), from plants growing under natural conditions and then cut into individual nodes. Syringe needles were used to pierce the bud and branch junction. The single-node shoots were subsequently soaked in the bacterial solution for 10 min at room temperature conditions under vacuum. The shoots were then placed on water-soaked floral foam and incubated at 25 °C/23 °C (light/dark) for 10 days. Bud samples were collected 5 days after infection, immediately frozen in liquid nitrogen, and stored at −80 °C until analysis. Bud break percentages were calculated at 10 days after infection when the bud scales were open and green tips were visible.
Data availability
The accession numbers of the genes in this work are listed in Supplementary Tables S3, S4, S5, and S7. The RNA-seq and ChIP-seq data for bud dormancy have been deposited in the NCBI database under BioProject number PRJNA669907. The whole-genome sequence data reported in this paper have been deposited in the Genome Warehouse at the National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, under accession number GWHBAOS00000000 and are publicly accessible at https://bigd.big.ac.cn/gwh.
References
Lang, G. A., Early, J. D., Darnell, R. L. & Martin, G. C. Endo-, para-, and eco-dormancy: physiological terminology and classification for dormancy research. Hortscience 22, 371–377 (1987).
Saito, T. et al. Development of flower buds in the Japanese pear (Pyrus pyrifolia) from late autumn to early spring. Tree Physiol. 35, 653–662 (2015).
Ito, A. et al. Comparative phenology of dormant Japanese pear (Pyrus pyrifolia) flower buds: a possible cause of ‘flowering disorder’. Tree Physiol. 38, 825–839 (2018).
Legave, J. M. et al. A comprehensive overview of the spatial and temporal variability of apple bud dormancy release and blooming phenology in Western Europe. Int. J. Biometeorol. 57, 333–335 (2013).
Bielenberg, D. G. et al. Sequencing and annotation of the evergrowing locus in peach [Prunus persica (L.) Batsch] reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genet. Genomes 4, 495–507 (2008).
Sasaki, R. et al. Functional and expressional analyses of PmDAM genes associated with endodormancy in Japanese apricot. Plant Physiol. 157, 485–497 (2011).
Yamane, H. Regulation of bud dormancy and bud break in Japanese apricot (Prunus mume Siebold & Zucc.) and peach [Prunus persica (L.) Batsch]: a summary of recent studies. J. Jpn. Soc. Hort. Sci. 83, 187–202 (2014).
Wu, R. M. et al. SVP-like MADS box genes control dormancy and bud break in apple. Fronti. Plant Sci. 8, 477 https://doi.org/10.3389/fpls.2017.00477 (2017).
Saito, T. et al. Expression and genomic structure of the dormancy-associated MADS box genes MADS13 in Japanese pears (Pyrus pyrifolia Nakai) that differ in their chilling requirement for endodormancy release. Tree Physiol. 33, 654–667 (2013).
Niu, Q. et al. Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifolia White Pear Group) flower bud. J. Exp. Bot. 67, 239 (2016).
Ubi, B. E. et al. Molecular cloning of dormancy-associated MADS-box gene homologs and their characterization during seasonal endodormancy transitional phases of Japanese pear. J. Am. Soc. Hortic. Sci. 135, 174 (2010).
Li, J. Z. et al. PpCBFs selectively regulate PpDAMs and contribute to the pear bud endodormancy process. Plant Mol. Biol. 99, 575–586 (2019).
Yang, Q. et al. ABA-responsive ABRE-BINDING FACTOR3 activates DAM3 expression to promote bud dormancy in Asian pear. Plant Cell Environ. 43, 1360–1375 (2020).
Singh, R. K. et al. A genetic network mediating the control of bud break in hybrid aspen. Nat. Commun. 9, 4173 (2018).
Tylewicz, S. et al. Photoperiodic control of seasonal growth is mediated by ABA acting on cell-cell communication. Science 360, 212–215 (2018).
Leida, C., Conesa, A., Llácer, G., Badenes, M. L. & Ríos, G. Histone modifications and expression of DAM6 gene in peach are modulated during bud dormancy release in a cultivar-dependent manner. N. Phytol. 193, 67–80 (2012).
Santamaría, M. E., Rodríguez, R., Cañal, M. J. & Toorop, P. E. Transcriptome analysis of chestnut (Castanea sativa) tree buds suggests a putative role for epigenetic control of bud dormancy. Ann. Bot. 108, 485 (2011).
Vimont, N. et al. ChIP-seq and RNA-seq for complex and low-abundance tree buds reveal chromatin and expression co-dynamics during sweet cherry bud dormancy. Tree Genet. Genomes 16, 9 https://doi.org/10.1007/s11295-019-1395-9 (2019).
Zhu, H. et al. Thermal-responsive genetic and epigenetic regulation of DAM cluster controlling dormancy and chilling requirement in peach floral buds. Hort. Res. 7, 114 (2020).
Saito, T. et al. Histone modification and signalling cascade of the dormancy-associated MADS-box gene, PpMADS13-1, in Japanese pear (Pyrus pyrifolia) during endodormancy. Plant Cell Environ. 38, 1157–1566 (2015).
Liu, G. et al. Transcriptomic analysis of ‘Suli’ pear (Pyrus pyrifolia White Pear Group) buds during the dormancy by RNA-Seq. BMC Genom. 13, 700 (2012).
Feng, L., Ma, C., Wu, H., Qi, K. & Zhang, S. Comparative studies of chilling requirement and covering time of forcing cultivation of different pear cultivars in Nanjing. Chin. Agric. Sci. Bull. 29, 94–98 (2013).
Wu, J. et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396 (2013).
Linsmith, G. et al. Pseudo-chromosome-length genome assembly of a double haploid “Bartlett” pear (Pyrus communis L.). Gigascience 8: 1-9, https://doi.org/10.1093/gigascience/giz138 (2019).
Dong, X. et al. De novo assembly of a wild pear (Pyrus betuleafolia) genome. Plant Biotechnol. J. 18, 581–595 (2020).
Ou, C. et al. A de novo genome assembly of the dwarfing pear rootstock Zhongai 1. Sci. Data 6, 281 https://doi.org/10.1038/s41597-019-0291-3 (2019).
Teng, Y. & Tanabe, K. Reconsideration on the origin of cultivated pears nativeto east asia. Acta Hortic. 634, 175–182, https://doi.org/10.17660/ActaHortic.2004.634.21 (2004).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245 (2019).
Zhang, L. et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 10, 1494 (2019).
Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494 (2013).
Raymond, O. et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777 (2018).
Edger, P. P. et al. Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. Gigascience 7, 1–7 (2018).
VanBuren, R. et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. Gigascience 7, giy094, https://doi.org/10.1093/gigascience/giy094 (2018).
Tomato Genome, C. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Yang, Q. et al. PpyGAST1 is potentially involved in bud dormancy release by integrating the GA biosynthesis and ABA signaling in ‘Suli’ pear (Pyrus pyrifolia White Pear Group). Environ. Exp. Bot. 162, 302–312 (2019).
Kumar, L. & Futschik, M. E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Bao, L., Chen, K., Zhang, D., Li, X. & Teng, Y. An assessment of genetic variability and relationships within Asian pears based on AFLP (amplified fragment length polymorphism) markers. Sci. Hortic. 116, 374–380 (2008).
Jiang, S. et al. Primitive genepools of Asian pears and their complex hybrid origins inferred from fluorescent sequence-specific amplification polymorphism (SSAP) markers based on LTR retrotransposons. PLoS ONE 11, e0149192 (2016).
Yue, X. et al. Combined analyses of chloroplast DNA haplotypes and microsatellite markers reveal new insights into the origin and dissemination route of cultivated pears native to East Asia. Front. Plant Sci. 9, 591 (2018).
Jiménez, S., Reighard, G. L. & Bielenberg, D. G. Gene expression of DAM5 and DAM6 is suppressed by chilling temperatures and inversely correlated with bud break rate. Plant Mol. Biol. 73, 157–167 (2010).
Tian, J. et al. Population genomic analysis of gibberellin-responsive long non-coding RNAs in Populus. J. Exp. Bot. 67, 2467–2482 (2016).
Bai, S. et al. BBX16, a B-box protein, positively regulates light-induced anthocyanin accumulation by activating MYB10 in red pear. Plant Biotechnol. J. 17, 1985–1997 (2019).
Hao, L. et al. A constitutive and drought-responsive mRNA undergoes long-distance transport in pear (Pyrus betulaefolia) phloem. Plant Sci. 293, 110419 (2020).
Gabay, G. et al. Transcriptome analysis and metabolic profiling reveal the key role of alpha-linolenic acid in dormancy regulation of European pear. J. Exp. Bot. 70, 1017–1031 (2019).
Kolonin, M. G. & Finley, R. L. Jr. A role for cyclin J in the rapid nuclear division cycles of early Drosophila embryogenesis. Dev. Biol. 227, 661–672 (2000).
Rinne, P. L. et al. Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-beta-glucanases to reopen signal conduits and release dormancy in Populus. Plant Cell 23, 130–146 (2011).
Ahmad, M. et al. Genome wide identification and predicted functional analyses of NAC transcription factors in Asian pears. BNC Plant Biol. 18, 214 (2018).
Ferrero, L. V., Viola, I. L., Ariel, F. D. & Gonzalez, D. H. Class I TCP transcription factors target the gibberellin biosynthesis gene GA20ox1 and the growth-promoting genes HBI1 and PRE6 during thermomorphogenic growth in Arabidopsis. Plant Cell Physiol. 60, 1633–1645 (2019).
Lee, S. et al. Overexpression of PRE1 and its homologous genes activates gibberellin-dependent responses in Arabidopsis thaliana. Plant Cell Physiol. 47, 591–600 (2006).
Wu, R. et al. Histone modification and activation by SOC1-like and drought stress-related transcription factors may regulate AcSVP2 expression during kiwifruit winter dormancy. Plant Sci. 281, 242–250 (2019).
Li, S. et al. Histone acetylation cooperating with AREB1 transcription factor regulates drought response and tolerance in Populus trichocarpa. Plant Cell 31, 663–686 (2018).
Azeez, A. et al. EARLY BUD-BREAK 1 and EARLY BUD-BREAK 3 control resumption of poplar growth after winter dormancy. Nat. Commun. 12, 1123, https://doi.org/10.1038/s41467-021-21449-0 (2021).
Li, J. et al. Abscisic Acid (ABA) promotes the induction and maintenance of pear (Pyrus pyrifolia White Pear Group) flower bud endodormancy. Int. J. Mol. Sci. 19, 310 (2018).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive -mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982 (2007).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Curr. Protoc. Bioinforma. 18, 4.3.1–4.3.28 (2007).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Shao, Y. et al. OsSPL3, an SBP-domain protein, regulates crown root development in rice. Plant Cell 31, 1257–1275 (2019).
Chang, S., Puryear, J. & Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Report. 11, 113–116 (1993).
Haring, M. et al. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant methods 3, 11 (2007).
Li, W. et al. A robust chromatin immunoprecipitation protocol for studying transcription factor-DNA interactions and histone modifications in wood-forming tissue. Nat. Protoc. 9, 2180–2193 (2014).
Acknowledgements
We thank Ms. Juan Tian for collecting 'Suli' pear samples from Dangshan County and Mr. Zhican He for collecting 'Cuiguan' pear samples from Hangzhou.
Funding
This work was supported by the National Key Research and Developmental Program of China (2018YFD1000104), the Earmarked Fund for China Agriculture Research System (CARS-28), and the Specialized Research Fund for Major Science and Technique of Zhejiang Province of China (2016C02052-4 and 2018C02011).
Author information
Authors and Affiliations
Contributions
S.B., Y.G., Q.Y. and Y.T. designed the study. S.B. and Q.Y. collected the samples. Q.Y. conducted most of the experiments. S.B., Y.G. and Q.Y. analyzed the genomic, RNA-seq, and ChIP-seq data. Y.G., X.Y., X.W., F.Y., J.L., J.W., J.N. and M.A. conducted some of the experiments. S.B., Y.G. and Q.Y. analyzed the data and drafted the manuscript. Y.T. revised and verified the manuscript. Y.G. and Q.Y. contributed equally to this study.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, Y., Yang, Q., Yan, X. et al. High-quality genome assembly of 'Cuiguan' pear (Pyrus pyrifolia) as a reference genome for identifying regulatory genes and epigenetic modifications responsible for bud dormancy. Hortic Res 8, 197 (2021). https://doi.org/10.1038/s41438-021-00632-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41438-021-00632-w
This article is cited by
-
Increased DNA methylation contributes to the early ripening of pear fruits during domestication and improvement
Genome Biology (2024)
-
Molecular regulation of bud dormancy in perennial plants
Plant Growth Regulation (2024)
-
Genome-wide analysis of the PME gene family reveals its role in suppressing fruit lignification in pear
Horticulture Advances (2024)
-
The pear genomics database (PGDB): a comprehensive multi-omics research platform for Pyrus spp.
BMC Plant Biology (2023)
-
Genome-wide identification and molecular characterization of the AP2/ERF superfamily members in sand pear (Pyrus pyrifolia)
BMC Genomics (2023)
